

Abstract
The phenomenon of ischemic preconditioning was initially observed over 20 years ago. The basic tenant is that if stimuli are applied at a subtoxic level, cells upregulate endogenous protective mechanisms to block injury induced by subsequent stress. Since this discovery, many conserved signaling mechanisms that contribute to activation of this potent protective program have been identified in the brain. A clinical correlate of this basic research finding can be found in patients with a history of transient ischemic attack (TIA), who have a decreased morbidity after stroke. In spite of multidisciplinary efforts to design safer, more effective stroke therapies, we have thus far failed to translate our understanding of endogenous protective pathways to treatments for neurodegeneration. This review is designed to provide clinicians and basic scientists with an overview of stress biology after TIA and preconditioning, discuss new therapeutic strategies to target the protein dysfunction that follows ischemic injury, and propose enhanced biochemical profiling to identify individuals at risk of stroke after TIA. We pay particular attention to the unanticipated consequences of overly aggressive intervention after TIA in which we have found that traditional cytotoxic agents such as free radicals and apoptosis associated proteases is essential for neuroprotection and communication in the stressed brain. These data emphasize the importance of understanding the complex interplay between chaperones, apoptotic proteases including caspases, and the proteolytic degradation machinery in adaptation to neurological injury.
Keywords: caspases, neuroprotection, preconditioning, stroke, transient ischemic attack, tolerance
Introduction
Nihilist philosopher Friedrich Nietzsche brought us the idea of ‘that which doesn’t kill you makes you stronger’, and modern day findings of molecular medicine show there is great truth in this statement. Indeed, the brain is capable of withstanding enormous stress, but even more impressive than its durability is its capacity for adaptation to stress. Ischemic preconditioning is an evolutionarily conserved cellular defense program in which exposure to a subtoxic insult results in subsequent resistance to a host of otherwise lethal stressors. This phenomenon was initially identified in the heart (Murry et al, 1986) and has since been observed in liver, kidney, muscle, intestines, brain, and other tissue (Gross and Auchampach, 1992; Kitagawa et al, 1990; Kukreja et al, 1994; Lee et al, 2003).
The phenomenon of preconditioning offers a unique window into the endogenous protective mechanisms cells use to avert irreversible injury. These defenses are remarkably rigorous and can block over 50% of the cell death elicited by an insult that would otherwise induce 100% lethality (Cohen et al, 2000; McLaughlin et al, 2003; Yellon and Dana, 2000). Moreover, the repertoire of cell defenses is quite versatile. Small hypoxic stressors provide protection against a wide variety of subsequent injuries including more intense hypoxia, aβ peptide and ceramide exposure, cytokine activation, excitotoxicity, energetic dysfunction, and other stressors (Liu et al, 2000; Riepe et al, 1997; Tremblay et al, 2000; Weih et al, 1999). These findings support an appealing therapeutic possibility that a common target or pathway could be manipulated to provide protection from a host of neuropathological conditions including stroke, Parkinson’s disease, Alzheimer’s disease, head injury, and other insults.
In addition to identifying novel therapeutic targets, understanding preconditioning responses in chronically stressed cells is essential, given that the long-term upregulation of protective programs on normal cell functioning is not well understood. For instance, genetic mutations associated with Alzheimer’s and Huntington’s disease can actually render cells less vulnerable to stressors such as excitotoxicity and ischemia (Dargusch and Schubert, 2002; Hansson et al, 1999, 2001; Hickey and Morton, 2000; Morton and Leavens, 2000; Petersen et al, 2001; Sagara et al, 1998; Schubert and Behl, 1993). That is, rather than enhancing vulnerability to acute stressors, genetic disorders can provide resistance to other injuries. These effects are likely caused by aberrant gene products upregulating expression of defensive proteins and pathways and, in essence, providing preconditioning. Therefore, a clinically important problem then becomes designing effective therapeutics that mimic endogenous cell responses to low-level stress in acute conditions such as stroke as well as enhancing existing defenses in chronic disorders such as Alzheimer’s disease.
Transient Ischemic Attacks Recapitulate the Findings of Neuroprotection in Preconditioned Cells
The clinical parallel to preconditioning neuroprotection is transient ischemic attacks (TIAs) (Figure 1). Transient ischemic attack has recently been redefined by the TIA Working Group as: ‘a brief episode of neurologic dysfunction caused by focal brain or retinal ischemia, with clinical symptoms typically lasting less than 1 h, and without evidence of acute infarction’ (Albers et al, 2002). At least 700,000 strokes and 300,000 TIAs occur annually in the United States although the true incidence of TIA may be much higher due to lack of self-reporting. In spite of this caveat, TIAs occur in one in 15 people over the age of 65, and an estimated 4.9 million people in the United States document having been diagnosed with a TIA (Mitka, 2005). Our failure to understand the genetic and biochemical factors that render individuals at higher risk for stroke after TIA is startlingly evidenced by the estimate that 10% of emergency room patients diagnosed with TIAs returned within ninety days having suffered a stroke. Half of these return visits occurred within 48 h (Johnston et al, 2000).
Figure 1.
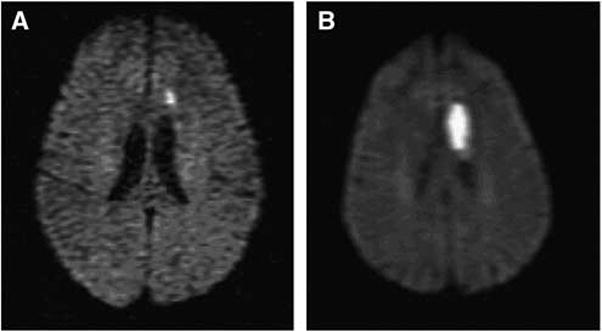
Diffusion weighted imaging of patient with TIA (A) who returned to ED 4 days later with stroke. (B) A 61-year-old woman with history of hypertension and prior basal ganglia lacunar stroke, presented initially with mild expressive aphasia and unsteadiness. Symptoms improved, she was discharged on antiplatelet agent, adjusted antihypertensive regimen, but returned 4 days later with worsened aphasia, right hemiparesis, and slow unsteady gait. Diffusion magnetic resonance imaging initially showed small left frontal subcortical infarction, and repeat scan shows significant extension of stroke in this region.
Transient ischemic attack patients generally fall into three treatment categories: (1) those identified with modifiable risk factors, including hypertension, diabetes mellitus, hyperlipidemia, atrial fibrillation, and cigarette smoking; (2) individuals who have greater than 70% carotid stenosis and are candidates for neurointerventional or surgical procedures such as stenting or endarterectomy (Barnett et al, 1998; Mayberg et al, 1991); and (3) those who fall into neither of these categories and are released with a standard course of antiplatelet therapy.
Surgical Intervention After Transient Ischemic Attack: A Prime Opportunity to Advance Understanding of Preconditioning and Enhance Protection
Surgical removal of fat and cholesterol laden atherosclerotic plaques in the carotid artery (i.e. carotid endarterectomy) has become the most powerful form of stroke prevention for individuals with symptomatic and asymptomatic high-grade carotid artery stenosis (Figure 2). Indeed, the 3 year risk of stroke and death in individuals is reduced by half in individuals who received endarterectomy compared with aspirin therapy alone (Mayberg et al, 1991). An alternative strategy for restoring blood flow to the brain is percutaneous transluminal angioplasty with stent (or ‘carotid stenting’), which is becoming a desirable alternative to endarterectomy particularly in patients with restenosis after endarterectomy, those who have serious angina, chronic obstructive pulmonary disease, heart failure or other circumstances where anesthesia and surgery pose high risks (Yadav et al, 2004).
Figure 2.
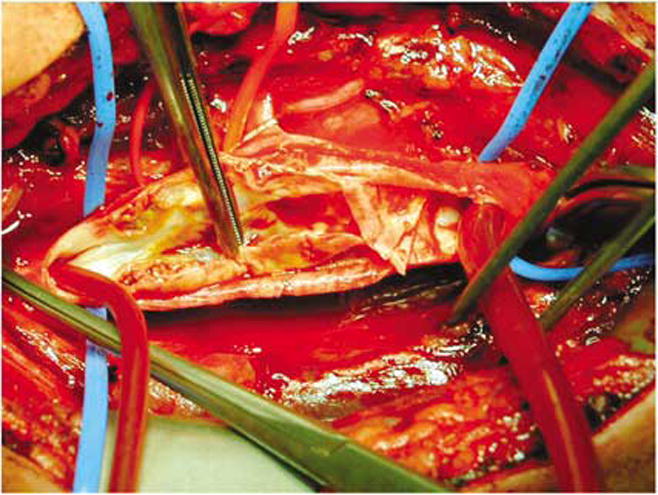
Carotid endarterectomy offers a prime opportunity to deliver pharmacological therapies to increase angiogenesis, retard cell death, and decrease inflammation as well as understand neuroadaptation to stress. Pictured is the surgical removal of plaque from the carotid artery performed to increase blood flow to the brain and prevent strokes. Image shows the exposed common carotid artery and its branches. A cleavage plane is achieved between the plaque and the healthier remaining parts of the artery. The plaque, a pale yellow sheath which is in the grip of the forceps is removed. Plastic tubing bridges between the internal and common carotid artery to provide a detour for blood and provide blood flow to the brain while the operation is preformed. As evidenced in this figure, the CNS access granted by a relatively minimally invasive procedure has proved to be the most useful means of surgical stroke prevention and offers an outstanding opportunity to assess neurochemical, physiological, and molecular changes that accompany impaired blood flow to the human brain. Image courtesy of the Vascular Disease Centre, Vascular Surgery, University of Hong Kong.
One of the ironies of performing either stenting or endarterectomy is the induction of a period of intensified ischemia and activation of potentially cytotoxic pathways as a consequence of these procedures, which can include guide wire insertion, catheter manipulation, stent placement, postdilation, balloon deflation, and other manipulations (Jodicke et al, 2003; Poppert et al, 2004). Indeed, preliminary data suggest that stenting causes a three-fold increase in the release of the excitatory neurotransmitter glutamate within 24h accompanied by induction of markers of cell death and inflammation more commonly associated with protracted periods of ischemia (Nombela et al, 2006; Poppert et al, 2004). Doppler monitoring confirms the repercussions of interventional procedures for stenosis where an increase in the emboli during vascular intervention has been identified. As the majority of these emboli are thought to be air bubbles, they may be less damaging than emboli containing particulate matter, which have been closely linked to neuropsychometric deterioration (Crawley et al, 2000). Moreover, new diffusion weighted imaging focal lesions are detectable in 30% to 50% of stenting patients although the majority of these lesions are commonly referred to as ‘clinically silent’ (Poppert et al, 2004; Roh et al, 2005).
Both endarterectomy and stenting offer prime opportunities to couple intervention with administration of small molecule neuroprotective agents (Figure 2). The question then becomes what level of stress is acceptable, and possibly even desirable, to precondition CNS tissue compared with that which has long-term consequences on executive function. There is a growing body of evidence that supports the hypothesis that TIAs may indeed precondition the brain against subsequent injury. That is, patients with previous TIAs have a more favorable outcome on subsequence ischemic challenge than those without TIAs (Moncayo et al, 2000). This is particularly important in light of the fact that one or more TIA increases a patient’s risk of stroke approximately 10-fold (Johnston et al, 2000). Even TIAs lasting 10 to 20 min are still associated with a more favorable outcome when confounding variables, such as diabetes, are taken into account.
Moncayo and co-workers found that the interval between TIA and cerebral infarction greatly influenced long-term prognosis, a factor that is critical in ischemic preconditioning. This study evaluated a cohort of 65 patients with territorial strokes and final infarct size was significantly less in the 16 patients with prodromal TIAs. Decrease in infarct volumes were even more pronounced among patients where TIAs occurred fewer than 4 weeks from the onset of stroke compared with patients without prodromal TIAs (Moncayo et al, 2000). Patients with prodromal TIAs also had milder final clinical deficits than those without and, in fact, prodromal TIA was the only predictive parameter of smaller final infarct volume (Moncayo et al, 2000; Weih et al, 1999).
Recent imaging studies suggest that the protection and increased function associated with TIAs preceding stroke is not dependent on variation in blood flow and collateral development (Wegener et al, 2004). That is, no observable changes between individuals with or without a history of TIA were observed on T2 lesion volumes, apparent diffusion coefficient maps or perfusion maps in imaging performed in the first 12 h after stroke. Therefore, despite having equivalent severities of ischemia initially, the final stroke lesion volume was smaller in patients with a prodromal TIA, implicating the important contribution of cellular neuroprotective mechanisms. An important caveat to these observations is that conditions such as diabetes or genetic risk of Alzheimer’s disease can dramatically alter the predicted ‘neuroprotective’ outcome trajectory of prodromal TIA. These data speak to the need to rigorously assess individual risk factors and enhance genetic testing of patients before surgical intervention when possible.
The Clinical Realities of Diabetes and Ischemia Vulnerability
Convincing evidence now exists that diabetes mellitus is a strong independent risk factor of stroke with an estimated attributable risk of 21%; further, stroke is the second leading diabetes-related complication (Bertoni et al, 2002; Folsom et al, 1999; Pulsinelli et al, 1983). The great majority of strokes that occur in the diabetic population are ischemic in nature and of these small deep ‘lacunar’ strokes are more common than large artery atherosclerosis or cardioembolic sources. In addition, TIA has been found to be three times more common in patients with diabetes compared with a control population (Palumbo et al, 1978) moreover, Johnston et al (2000) reported that a history of diabetes increased the risk of stroke within 90 days of a TIA. Perhaps most important, diabetes may be associated with a worse prognosis and mortality after stroke (Bertoni et al, 2002; Pulsinelli et al, 1983).
There are several theories for the causative mechanisms behind diabetes worsening stroke outcomes. One possibility is that blockade of ischemic preconditioning may be a consequence of drug treatments for diabetes. Sulfonylureas (such as glyburide and glipizide) have become the leading oral antihyperglycemic agents over the past half century (Gerich, 1989; Groop, 1992). The mechanism of action of these compounds is to increase insulin release by pancreatic islet cells by blocking ATP-sensitive potassium channels (KATP) resulting in the opening of calcium channels and causing insulin release. Opening of the KATP channel is considered a hallmark feature of ischemic preconditioning in both heart and brain (Cohen et al, 2000; Gross and Auchampach, 1992; McLaughlin et al, 2003; O’Rourke, 2000, 2004). Cell membrane KATP channels are the targets of these glycemic control agents although KATP channels are also present on mitochondrial membranes where they are critical determinants of cell fate. Here, they couple the ionic and biochemical machinery to detect changes in redox and energetic status with a potent mechanism to induce cell death under unfavorable conditions. While controversial (Meier et al, 2004), sulfonylureas have been shown to block neuroprotective and cardioprotective effects of preconditioning in animal and culture models and have been implicated as a causative factor in the increased mortality observed in stroke patients (Forlani et al, 2004).
Conversely, the neuroprotective potential of KATP channel activation by agents such as diazoxide has also been demonstrated in animal models of brain ischemia (Ballanyi, 2004). It has been hypothesized that alterations in cellular redox status and generation of reactive oxygen species (ROS) elicit preconditioning by contributing to the opening of KATP channels within the mitochondria (Avshalumov and Rice, 2003). Channel opening decreases the mitochondrial membrane potential and accelerates electron transfer resulting in a net oxidation of the mitochondria (Liu et al, 1998). Reactive oxygen species produced by an oxidizing environment likely increase the opening of KATP channels via modification of thiol residues within the channels and thus establishing a positive feedback loop for protection (Forbes et al, 2001; Grigoriev et al, 1999; Pain et al, 2000). Preconditioning protection can be blocked with antioxidants and KATP channel blockers (Baines et al, 1997; Das et al, 1999; Forbes et al, 2001; Lebuffe et al, 2003; Ravati et al, 2001). We hypothesize the major importance of KATP channel opening during mild ischemia is to enhance the generation of ROS (McLaughlin, 2004, 2003).
The contribution of ROS and other typically ‘prodeath’ molecules to preconditioning neuroprotection appears, however, to be significantly more complex than altering mitochondrial ion homeostasis (Figure 3). Indeed, application of KATP agonists blocks cytochrome c release, poly-ADP ribose polymerase cleavage and caspase activation after major hypoxic insults (Xu et al, 2001). Notably, each of these events is required for preconditioning-induced protection. It is possible, even probable, that the ultimate fate of neurons is not dependent on which of these molecules are activated, but rather the extent and duration of their activation. There is a clear need for combining pharmacological and toxicological approaches to understanding mitochondrial dysfunction and compensation in neural cells that have undergone low-level ischemic injury akin to TIA to provide a framework to identify potential therapeutic targets. These observations also speak to the need to carefully assess any potential antioxidant strategies during TIA, which may have the undesirable consequence of blocking intrinsic protection pathways activated by mild ischemia.
Figure 3.
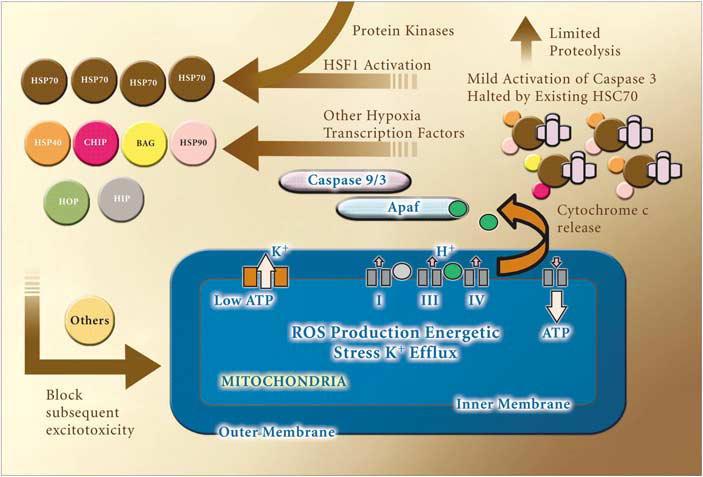
Model of neuronal ischemic preconditioning. Based on our observations that preconditioning elicits caspase cleavage and ROS generation, which are required for expression of protection and that this protection requires new protein synthesis, we propose the following pathway mediates protection. We hypothesize that the initial energetic stress put on cells generates ROS and activation of mitochondrial KATP channels. These events likely contribute to limited cytochrome c redistribution and caspase 3 activation. Cleaved caspases are likely held in check by preexisting proteins such as HSC 70. When these proteins are depleted, this results in the activation of a positive feedback cycle leading to increased production of HSPs and other neuroprotective proteins. Signal transduction cascades such as protein kinases of the MAPK family and PKC family as well as others, are likely critical components of this protective pathway. The upregulation of HSP 70 is able to block normally lethal exposure to subsequent injuries. Many of the kinases, proteases, chaperones and transcription factors in this pathway could be exploited for development of neurotherapeutics to recapitulate endogenous neuroprotection.
Activation of Stress and ‘Death’ Proteins is Essential for Ischemic Preconditioning
In vivo and in vitro models of preconditioning have been allowed for the identification of neurotherapeutic targets downstream of KATP channels. Recent observations by several laboratories, including our own, suggest that cytoprotective agents such as antioxidants and protease inhibitors paradoxically increase vulnerability to subsequent injury if administered during the preconditioning period (McLaughlin et al, 2003). This calls into question the wisdom of overly aggressive intervention against mild ischemic injury. We have learned that low-level preconditioning stressors are capable of co-opting the apoptotic machinery to evoke neuroprotection in preconditioned cells. Metabolic dysfunction induced by preconditioning evokes not only the opening of KATP channels and production of ROS but also the induction of heat-shock proteins (HSPs) and cleavage of the killer protease caspase 3. Indeed, appreciable caspase activation is present following preconditioning, but remarkably apoptotic cell death is not observed following preconditioning. Caspases are traditionally considered to be ‘executioner’ proteases, activated only in the terminal stages of apoptotic cell death and caspase cleavage has been synonymous with cell death until these observations were made. In preconditioning models, caspase activation is dependent upon ROS generation and energetic dysfunction, and prevention of caspase 3 cleavage blocks induction of the neuroprotective protein HSP 70 (Figure 3). The clinical relevance of this finding is that developing caspase inhibitors for the treatment of neurodegeneration is a major area of neurotherapeutic interest. While these agents no doubt block cell death in models of stroke, they may increase stroke induced cell death if given prophylactically during TIA.
Heat-Shock Proteins as Modulators of Preconditioning: Targeting the Chaperones
The upregulation of HSP 70 has been observed in the majority of models of preconditioning (for a discussion of other pathways activated in ‘kindling’ type protection, the reader is referred to Cendes (2005)). This multifaceted protein is essential to neuroprotection in a host of chronic diseases such as Alzheimer’s and Parkinson’s. Molecular chaperones provide a vital link between protein trafficking, folding, and targeting for proteasomal destruction (Figure 4). The HSPs are highly conserved, abundantly expressed proteins with diverse functions including the assembly of multiprotein complexes, transportation of nascent polypeptides, and regulation of protein folding. Heat-shock protein 70 is the major inducible HSP found in interacts with many of the same binding partners and client proteins as HSC 70. While HSP 70 has been shown to be upregulated after preconditioning stress, the mechanism by which it affords protection is not known, as its activity is dependent on its association with different binding partners. For instance, the C-terminal HSC 70 interacting protein (CHIP) competes for C-terminal binding to HSP 70 with HOP. Similarly, BAG-1 competes with HIP for N-terminal binding. Formation of BAG-1/HSP 70/CHIP complexes is thought to redirect HSP activity away from protein refolding and towards ubiquitination and proteasomal degradation (Hohfeld et al, 2001). Resurgent interest in HSP 70 complex formation and function followed the 2002 observation that overexpression of CHIP could block neuropathological aggregate formation in vitro associated with a mutation in the Parkin gene (Imai et al, 2002). CHIP is a member of the family of E3 ligases that perform essential roles in docking Ub proteins with the proteasome.
Figure 4.
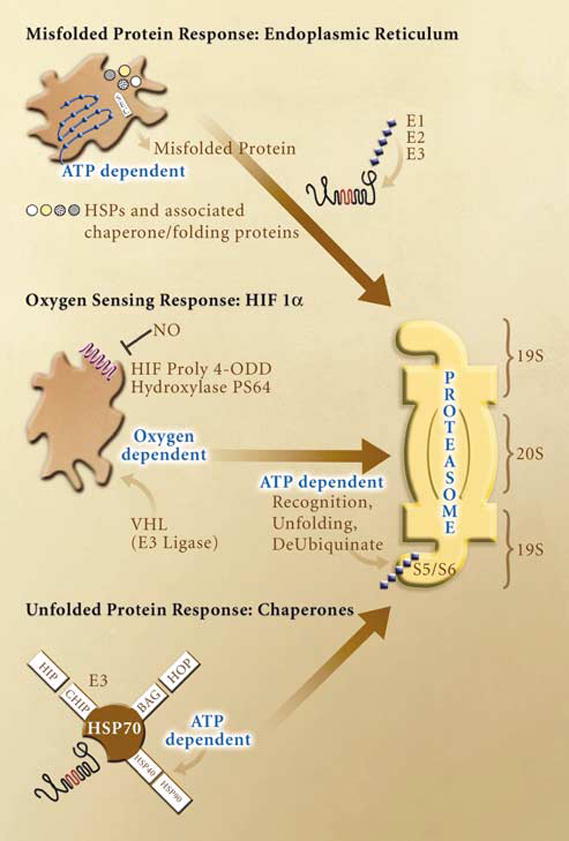
The UPS is essential to the cellular response to ischemic stress. Multiple energy and oxygen dependent pathways converge on the proteasome to remove damaged proteins and activate transcription factors with roles in increasing oxygenation, vascularization and survival. Examples of these pathways are shown for the unfolded protein response in the ER, the stabilization of hypoxia inducible factor 1α (HIF1α) and the chaperone triage system. Protein trafficking and assembly in the endoplasmic reticulum is responsive to multiple energetic and ionic perturbations including calcium buffering, redox regulation, caspase activation, and post-translation modification of proteins. These functions are dependent on ATP content and redox sensitive proteins including the Sec ATPases. Misfolded proteins are targeted to the proteasome thru ubiquitination (E1, E2, and E3 enzymes). Oxygen sensitivity is showed in the HIF1α subunit of the HIF transcription factor. Under normoxia, HIF hydroxylases modify proline residues within the oxygen dependent domain (ODD) of HIF allowing for rapid ubiquitination by the von Hippel–Lindau (vHL) E3 ligase. Anerobic conditions block these modifications leading to enhance HIF 1a expression and transcriptional activity. Finally, chaperones play an essential role in the stress response to ischemia. Some of the interactions of the stress inducible chaperone HSP 70 are depicted in this figure. Competition for HSP 70 binding leads to altered chaperone activity in which the protein moves from a primary role in refolding to one of targeting proteins to the proteasome to be degraded. Again, the function of this protein is dependent on ATPase activity of interacting chaperones.
For preconditioning, we have shown that the protein refolding function of HSPs is less important than their ability to sequester activated caspases and other cell death proteins (McLaughlin et al, 2003). Data from preconditioning experiments suggest that cleaved caspase 3 binds to the HSP 70 homologue HSC 70 which is constitutively expressed, and this binding prevents caspase activation from leading to apoptosis. A delicate balance clearly exists in which caspase 3 can be activated in a subthreshold manner to enhance cellular defenses whereas once sufficient intensity, duration, or localization of activated caspases is reached, cells undergo apoptosis. The feedback loop by which depletion of HSC 70 results in HSP 70 upregulation and protection from subsequent stress has not been identified nor have the factors that temporally and spatially limit caspases from cleaving all intracellular substrates and evoking death.
Strong support of the biological importance of low level, spatially restricted caspase activation to neuro-adaptive processes comes from work on synaptic reorganization. A compelling series of studies by Campbell and Holt have shown that caspase 3 is activated in developing growth cones where a dynamic process of protein synthesis, cleavage, and degradation is essential for neurite formation (Campbell and Holt, 2001, 2003). Indeed, regional and cellular differences in the abundance of proteins, which govern these processes, may limit the ability of neurons in the CNS to sprout and form appropriate connections after injury (Verma et al, 2005).
Calcium entry and calcium activated proteases such as calpains are also essential to neural growth cone expansion (Robles et al, 2003). This suggests that calcium-buffering organelles, such as the endoplasmic reticulum and mitochondria that translate changes in ion homeostasis to molecular signaling pathways, likely play an important role in caspase 3 cleavage. Compartmentalization of caspase activity has been observed in nuclear and mitochondrial fractions in the absence of widespread proteolytic activity (Chandler et al, 1998; Chandra and Tang, 2003; Mancini et al, 1998; Nakagawa et al, 2000). However, caspase activity is not likely limited in this manner in preconditioned cells as cleavage of cytosolic substrates has been observed (Yang and Du, 2004). The role of caspases in CNS remodeling has clear implications for neurotherapeutics as well. It remains to be determined if cells that are ‘saved’ by caspase inhibitors are capable of forming and maintaining viable synapses in vivo.
The Ubiquitin Proteasome System Plays a Key Role in Ischemic Injury and Neuronal Survival
The interplay between protein cleavage and degradation by caspases and calpains is an excellent example of the fine balance between adaptive and destructive pathways in ischemic tissue. Central to mediating these responses is the ubiquitin–proteasome system (Stampfer et al, 1992), another important therapeutic target widely cited for its potential to block neurodegeneration. Under non-stressed conditions, an estimated 30% of newly synthesized proteins are degraded because of folding errors and this process is largely controlled by chaperones such as HSPs (Schubert et al, 2000). When protein refolding becomes untenable because of excessive denaturation, the majority of proteins become ubiquinated and are degraded. Rapid elimination of denatured, misfolded, and damaged proteins by the proteasome is a critical determinant of cell fate, which is achieved by covalent attachment of multiple ubiquitin (Ub) molecules to generate the poly-Ub chain that serves as a recognition marker for the proteasome. Multi-Ub chains are formed by carboxy-terminal glycine linkage to Ub on Lys48 of the preceding Ub (Chau et al, 1989). The fate of ubiquitinated proteins in part depends on the length and linkage type of the Ub chain. In general, substrates with four or more Ub moieties linked via Lys29 or Lys48 are targeted for degradation by the 26S proteasome (Hershko and Ciechanover, 1998). Ubiquitination is also a powerful mechanism to redirect protein activity and dampen cellular communication. For example, Ub linkage to Lys63 of a receptor protein can cause protein internalization (Hicke and Dunn, 2003). Alternatively, mono-ubiquitination can lead to nuclear export and translocation of proteins into the cytoplasm.
High-resolution confocal microscopy has been used to document the presence of protein aggregates surrounding nuclei and along dendrites in post-ischemic neurons (Liu et al, 2004; Vannucci et al, 1998). These aggregates contain ubiquinated proteins, which can also be found in the neuronal soma, dendrites, and axons. Ub-proteins in neuronal lysosomal vesicles and in late endosome-like organelles in the ischemic area (Colbourne et al, 1999) may result from an attempt to eliminate accumulating Ub-proteins by autophagy. Biochemical characterization of the Ub-immunoreactive material from CNS disorders is an area of active research. As in many chronic neurodegenerative diseases, HSPs co-localize with Ub-proteins in the inclusions after ischemic injury (Domorakova et al, 2004; Giffard et al, 2004; Hu et al, 2000, 2001; Kumar et al, 2003; Liu et al, 2004). Not surprisingly, the expression of HSP 70 is increased after ischemia in vivo and in vitro, and these protein aggregates are rich in chaperones (Aoki et al, 1993; Chen et al, 1996; Currie et al, 2000; Mailhos et al, 1994; Rajdev et al, 2000; Tsuchiya et al, 2003a, b).
The 26S proteasome itself is a multicomponent complex composed of a 20S catalytic core particle and a regulatory 19S regulatory particle. The 19S regulatory particle recognizes ubiquitinated proteins and other potential substrates via two subunits Rpn10 (S5a) and Rpt5 (S6′). The 19S regulatory particle allows substrate entry into the proteolytic chamber where ATPases provide energy for both the assembly of the 26S proteasome and the injection of substrates into the catalytic chamber. Decreases in proteasome activity associated with pathophysiological events (Canu et al, 2000) is likely due in part to caspase mediated cleavage of S6′, S5a, as well as S1 proteasome subunits, which cooperate with other subunits to hold together the 19 and 20S components (Adrain et al, 2004). Caspases thus inhibit proteasomal degradation of both Ub-dependent and -independent cellular substrates, including proapoptotic molecules such as Smac, p53, and caspases. In this way, proteasomal uncoupling is thought to enhance the execution of the apoptotic program as part of a feed-forward amplification loop (Adrain et al, 2004). Another consequence of this proteolysis is the partial dissociation of the 19S regulatory complex from the 20S catalytic core, which is caused by caspase mediated degradation. This dissociation process may also preserve high-energy phosphate used to maintain ion homeostasis or promotion of apoptosis depending upon the extent of uncoupling.
Proteasomal Dampening of Excitotoxic Transmission as a Means to Block Ischemic Injury
To survive hypoxia and reperfusion, neurons must adapt to the excessive stimulation of glutamate receptors and the subsequent influx of calcium and production of ROS. Proteasomal regulation of these receptors provides an excellent means of dampening excitotoxic signal transduction. Loss of blood flow during myocardial infarction, hemorrhagic, or ischemic stroke causes energetic stress exacerbated by the influx of sodium, chloride and water. Resulting membrane depolarization causes massive glutamate efflux which, when coupled with transport failure, causes neuronal cell death via overstimulation of glutamate receptors. The NMDA receptor is normally blocked by magnesium, but membrane depolarization during stress relieves this block and calcium influx initiates a cascade commonly referred to as excitotoxicity. Indeed, targeted blockade of the NMDA receptor has been the most actively pursued therapeutic target for the treatment of stroke until multiple trials were halted because of untoward side effects of the compounds, poor pharmacokinetic profile, and the short neuroprotective window these reagents provided (Albers et al, 2001; Lee et al, 1999).
Other mechanisms exist to modulate excitatory neurotransmission and the signaling pathways that are neurotoxic to cells after stroke. Overstimulation of glutamate receptors by NMDA application leads to ubiquitination of the scaffolding protein PSD-95 via the E3 ligase Mdm2, resulting in receptor degradation. Thus, targeting PSD-95 for more rapid degradation by upregulating Mdm2 activity or PSD-95 recognition could provide a therapeutic strategy for acute insults such as stroke, which involve excessive glutamate release and receptor activation. PSD-95 degradation likely has important ramifications on synaptic transmission, stability, and plasticity particularly given that PSD-95 also associates with glutamatergic AMPA receptors, which are endocytosed in an NMDA receptor-dependent manner. Indeed, Tymianski and co-workers (Arundine and Tymianski, 2003) have shown that an HIV-Tat fusion protein containing C-terminal NMDA residues designed to interfere with PSD-95/receptor interactions blocks excitotoxic cell death in vitro as well as ischemia in vivo. This cycle may also have a feed-forward loop as glutamatergic stimulation has been shown to upregulate the expression of the p112 proteasome subunit.
In addition to the widely appreciated consequences of NMDA receptor overactivation, glutamateric AMPA receptors have become desirable modulatory targets to change excitatory synaptic tone (Weiser, 2002). The activity-dependent endocytosis of AMPA receptors is sensitive to proteasome inhibition (Colledge et al, 2003; Patrick et al, 2003). Direct ubiquitination of GluR1 AMPA receptors contributes to receptor accumulation in invertebrates (Burbea et al, 2002) although this has yet to be confirmed in mammalian systems. PSD-95 also promotes the synaptic accumulation of AMPA receptors and provides another site for neurotherapeutic development.
The oxidative injury caused by overstimulation of glutamatergic synapses has also been linked to both acute and chronic neurodegeneration. Oxidative injury has been observed to some extent in virtually all neurodegenerative conditions. The immediate consequences of oxidative stress include the modification of disulfide bonds within ion gated channels, transcription factors, and metal binding proteins that exasserbate ionic dysfunction associated with energetic failure resulting in free transition metals potentially furthering proteasomal dysfunctions (Conconi et al, 1998; Kim et al, 2004; Kiss et al, 2005).
20S proteasomes can degrade mildly oxidized proteins without previous ubiquitination; they are however unable to degrade extensively oxidized proteins (Davies, 2001; Ding et al, 2003; Ferrington and Kapphahn, 2004; Figueiredo-Pereira and Cohen, 1999; Grune et al, 2004). Disulfide oxidation can also result in decreased protein solubility and aggregation, loss of proteasome function and alterations in the composition of the proteasome (Ferrington et al, 2005; Branza-Nichita et al, 2002). Long-term consequences of extended oxidative stress may include decrease in total number of proteasome complexes during aging (reviewed by Bulteau et al, 2001; Ferrington and Kapphahn, 2004). Specifically, ROS modification of proteasomal subunits α1, α2, and α4 (Bulteau et al, 2001; Keller et al, 2000a) impairs protease degradative capacity although the extent of oxidation induced proteasome inhibition is dependent on the nature of the oxidative modification.
Loss of ATP resulting from oxygen deprivation also has profound and immediate consequences on a host of cellular functions including proteasome function (Asai et al, 2002; Bulteau et al, 2001; Grune, 2000; Hayashi et al, 1993; Kamikubo and Hayashi, 1996; Mehlhase and Grune, 2002; Tanaka, 1995). One hour transient focal cerebral ischemia induces marked depletion of the E3 ligase Parkin but does not affect the levels of E2 conjugating enzymes (Mengesdorf et al, 2002). Parkin upregulation has been shown to protect cells from injury induced by ER stress. This suggests that parkin depletion may increase the sensitivity of neurons to stimuli managed by the ER including the unfolded protein response and calcium dysregulation, which are already impaired by loss of cellular ATP levels (DeGracia and Montie, 2004; Kumar et al, 2003; Ryu et al, 2002). Interestingly, in the ischemic core, ATP- and Ub-dependent degradation mediated by the 26S proteasome is impaired, while the ATP- and Ub-independent degradation mediated by the 20S proteasome is unimpeded (Kamikubo and Hayashi, 1996). This is likely because of the dissociation of 26S proteasomes, which occurs under stress, into 20S proteasomes and PA700 caps. While the 26S proteasome activity recovers in many regions after reperfusion, in particularly vulnerable areas including the CA1 region of the hippocampus, PA700 and 20S proteasomes do not fully reassociate, a problem that may underlie the delayed neuronal cell death in these regions (Asai et al, 2002). Understanding the factors that underlie proteasome complex dissociation may prove beneficial in identifying mechanisms that re-couple 26S proteasomes and enhance survival.
Temporal and Spatial Regulation of Proteolysis: A Critical Determinate of Neuronal Survival
We have hypothesized that limited proteolytic cleavage of some structural and repair proteins may be adaptive in preconditioned cells by limiting energetic output as well as aiding in cytoskeletal flexibility and reestablishment of appropriate neural connections (McLaughlin, 2004). This theory is supported by work in which the DNA repair enzyme poly-ADP ribose polymerase, which consumes cellular NADH, is cleaved by caspase 3 in preconditioned cells (Garnier et al, 2003). Moreover, RasGAP, which regulates both Ras- and Rho-dependent pathways, is partially cleaved resulting in the generation of an N-terminal fragment, which induces a potent Ras–PI3K–Akt-dependent survival pathway. Full cleavage of this protein does not occur in this preconditioning model and is another example of limited temporal and spatial proteolysis of substrates contributing to preconditioning protection. Indeed, neurons exposed to apoptotic triggers undergo extensive RasGAP cleavage (Yang et al, 2004). The Ras–Raf pathway has also been implicated in the expression of ischemic tolerance by the Dawson’s who reported that dominant-negative Ras mutants cannot be preconditioned (Gonzalez-Zulueta et al, 2000).
Because proteolytic degradation includes a large variety of proteins with crucial function(s) in regulation of cell growth, the proteasome may seem to be a unwieldy therapeutic target. However, potent and specific chemical inhibitors of the proteasome have been created for cancer treatment and may have application to inflammatory and immune diseases offering the potential cross purposing of these agents to increase neuronal survival during short-term stress such as that caused by neurovascular procedures (McLaughlin and DiNapoli, 2005). One of the most well understood inhibitors is MLN-519, which consistently reduces cerebral infarct volume after middle cerebral artery occlusion in a dose-dependent manner with a therapeutic window of up to 6 h after the onset of ischemia. MLN-519 also exhibits a neuroprotective effect after focal brain ischemia with a 50% to 60% reduction of infarct volume and decreased leukocyte infiltration, as well as prevention of NFκB activation after reperfusion (Zhang and Stanimirovic, 2002). When MLN-519 was used in combination with tissue plasminogen activator in an embolic stroke model, it decreased infarct volume and improved neurological outcome 1 week after the ischemic episode, as well as eliminating hemorrhage associated with tissue plasminogen activator treatment, even when administered 6 h after vessel occlusion. These neuroprotective effects were also replicated in models of cerebral hemorrhage (Al-Senani et al, 2001).
Indeed, temporally and spatially controlled proteasome inhibition may promote neuronal survival after stroke by helping neurons evoke chaperones, dampen inflammation, and promote revascularization and energetic repletion. However, neurons subjected to prolonged and/or complete proteasome inhibition are likely to be severely damaged, as a result of an inability to resume requisite communication, protein turnover, and trafficking. The problem of defining the temporal window during which proteasome inhibitors are efficacious is particularly important, given that protracted use of these compounds results in neuronal cell death (Canu et al, 2000; Ding et al, 2003; Qiu et al, 2000; Rideout and Stefanis, 2002; Rockwell et al, 2000). This hypothesis is supported by the observation that the temporal window of decreased proteasome activity is coincident with the therapeutic window during which proteasome inhibitors promote neuronal survival after stroke (Asai et al, 2002; Kamikubo and Hayashi, 1996; Keller et al, 2000b). Postischemic impairment of proteasome activity likely leads to accumulation of Ub conjugates, contributing to loss of neuronal function.
In addition to temporal constraints, important spatial considerations must also be addressed such as the means to deliver a proteasome inhibitory cocktail, since the specific neurotransmitter profile and metabolic activity of cells alter proteasome inhibitor efficacy as evidenced by the observation that hypoxic endothelia showed a > 10-fold increase in sensitivity to proteasome inhibitors (Zund et al, 1997). Under conditions where ATP is limiting, such as during hypoxia, the kinetics of drugs targeting the proteasome may significantly alter the actions of these drugs. Further, the unique neurochemical and transcriptional identity of neurons is particularly important for eliciting proteasome-dependent inflammation pathways. While neuronal culture work has shown that activation of NFκB protects against excitotoxic and metabolic insults (Dudek et al, 2001), these effects differ between cell types. Indeed, activation of NFκB in microglia promotes ischemic neuronal degeneration (Mattson, 2005) and the net consequences of modulating NFκB by way of the proteasome may be unique to each species, and possibly even strains within species (Perkins and Gilmore, 2006). Recent work with more specific regulation of turnover of the pathway has shown tremendous promise in blocking ischemic cell death by decreasing NFκB activation (Herrmann et al, 2005). As the roles of the ubiquitination machinery, the factors that dictate substrate specificity and the interactions of ischemia-induced biochemical changes on discreet regions of the proteasome become known, we will likely be capable of designing improved therapeutic agents with which to selectively target components of the ubiquitin–proteasome system.
Conclusions
Our failure to develop accurate models of stroke while having made great strides in understanding the cellular and molecular events that contribute to neurodegeneration has become a clinical liability, as illustrated by our lack of good predictors of which patients who present with TIAs will experience severe hypoxic injury in the hours, days, and weeks after TIA. Individuals who have had a TIA can fair better than those who have not had an ischemic episode. This allows us the opportunity to identify the cellular and molecular mechanisms that underlie this powerful form of neuroprotection and design therapies that mimic this effect. The operative part of this statement is that the patient with a TIA fairs better, not that they are entirely immune to secondary stroke damage. The ideal situation would be one in which genetic, epigenetic, and individual risk factors could be used to develop a more accurate profile of stroke risk and secondary hypoxic events could be avoided entirely.
Transient ischemic attack and stroke are part of a continuum of pathology caused by cerebral ischemia although we are not in a position to determine when tissue hypoxia in humans transitions from a perturbation which is defensible to one which leads to cell death. The ongoing challenge to physicians and physician-scientists is to continue to press the detection limits of imaging technology to parallel our understanding of the cellular biology of ischemia; clearly, our knowledge of the cellular and molecular mechanisms that contribute to acute neuroprotection in vitro and in vivo has vastly outpaced our understanding of the factors that are evoked in individuals who have experienced TIAs. As microelectrode assessment of metabolic and energetic status during surgical intervention for stenosis becomes more routine, clinical researchers will be poised to understand the utility of local administration of neuroprotective agents, allow for physiological adaptation such as ‘permissive hypertension’ to increase oxygen delivery and evaluate the biological stress of new devices to increase CNS blood flow.
Preconditioning is highly relevant not only to acute degenerative events such as stroke, head injury, and myocardial infarction but also for chronic neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. A growing body of literature supports the theory that cells chronically stressed by long-term exposure to pathological protein aggregates or fragments, energetic dysfunction, and oxidative injury have already exploited preconditioning signaling mechanisms such as upregulation of chaperones and energetic overcompensation. The continued identification of proteins and signaling pathways employed by neurons with genetic mutations associated with neurodegeneration will allow us to provide more effective complementary therapies to re-enforce those already employed by cells. In the interim, advances in detection of TIAs by developing peripheral biomarkers as well as building on imaging technology will enable us to more rapidly assess and serve our clinical populations.
Acknowledgments
This work was supported in part by NICHD Grant P30HD15052 and by 1R01NS050396-01.
The authors would like to thank Dr Robert Mericle and Dr Howard Kirshner for helpful suggestions on interventional strategies as well as Mr Joshua Parlaman and Dr Gregg Stanwood for their thoughtful comments regarding signaling systems. We gratefully acknowledge the help of Ms Kylie Beck of the Vanderbilt Kennedy Center for her exceptional graphic artistry.
Footnotes
Conflict of Interest
The authors do not have any commercial or other associations that might pose a conflict of interest in connection with the submitted article.
References
- Adrain C, Creagh EM, Cullen SP, Martin SJ. Caspase-dependent inactivation of proteasome function during programmed cell death in Drosophila and man. J Biol Chem. 2004;279:36923–30. doi: 10.1074/jbc.M402638200. [DOI] [PubMed] [Google Scholar]
- Albers GW, Caplan LR, Easton JD, Fayad PB, Mohr JP, Saver JL, Sherman DG the TIAWG. Transient ischemic attack—proposal for a new definition. N Engl J Med. 2002;347:1713–6. doi: 10.1056/NEJMsb020987. [DOI] [PubMed] [Google Scholar]
- Albers GW, Goldstein LB, Hall D, Lesko LM for the Aptiganel Acute Stroke I. Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA. 2001;286:2673–82. doi: 10.1001/jama.286.21.2673. [DOI] [PubMed] [Google Scholar]
- Al-Senani FM, Shirzadi A, Strong R, Elliott P, Grotta JC, Aronowski J. Inhibition of proteasome reduces iNOS expression and behavioral dysfunction after intracerebral hemorrhage in rat. Neurology. 2001;56:A368. [Google Scholar]
- Aoki M, Abe K, Kawagoe J, Nakamura S, Kogure K. Acceleration of HSP70 and HSC70 heat shock gene expression following transient ischemia in the preconditioned gerbil hippocampus. J Cereb Blood Flow Metab. 1993;13:781–8. doi: 10.1038/jcbfm.1993.99. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–37. doi: 10.1016/s0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- Asai A, Tanahashi N, Qiu JH, Saito N, Chi S, Kawahara N, Tanaka K, Kirino T. Selective proteasomal dysfunction in the hippocampal CA1 region after transient forebrain ischemia. J Cereb Blood Flow Metab. 2002;22:705–10. doi: 10.1097/00004647-200206000-00009. [DOI] [PubMed] [Google Scholar]
- Avshalumov MV, Rice ME. Activation of ATP-sensitive K+ (KATP) channels by H2O2 underlies glutamate-dependent inhibition of striatal dopamine release. Proc Natl Acad Sci. 2003;100:11729–34. doi: 10.1073/pnas.1834314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Goto M, Downey JM. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J Mol Cell Cardiol. 1997;29:207–16. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- Ballanyi K. Protective role of neuronal K-ATP channels in brain hypoxia. J Exp Biol. 2004;207:3201–12. doi: 10.1242/jeb.01106. [DOI] [PubMed] [Google Scholar]
- Barnett HJM, Taylor DW, Eliasziw M, Fox AJ, Ferguson GG, Haynes RB, Rankin RN, Clagett GP, Hachinski VC, Sackett DL, Thorpe KE, Meldrum HE. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med. 1998;339:1415. doi: 10.1056/NEJM199811123392002. [DOI] [PubMed] [Google Scholar]
- Bertoni AG, Anderson GF, Krop JS, Brancati FL. Diabetes-related morbidity and mortality in a national sample of US elders. Diabetes Care. 2002;25:471–5. doi: 10.2337/diacare.25.3.471. [DOI] [PubMed] [Google Scholar]
- Branza-Nichita N, Lazar C, Durantel D, Dwek RA, Zitzmann N. Role of disulfide bond formation in the folding and assembly of the envelope glycoproteins of a pestivirus. Biochem Biophys Res Commun. 2002;296:470–6. doi: 10.1016/S0006-291X(02)00907-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–63. doi: 10.1074/jbc.M100142200. [DOI] [PubMed] [Google Scholar]
- Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C-elegans. Neuron. 2002;35:107–20. doi: 10.1016/s0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–26. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron. 2003;37:939–52. doi: 10.1016/s0896-6273(03)00158-2. [DOI] [PubMed] [Google Scholar]
- Canu N, Barbato C, Ciotti MT, Serafino A, Dus L, Calissano P. Proteasome involvement and accumulation of ubiquitinated proteins in cerebellar granule neurons undergoing apoptosis. J Neurosci. 2000;20:589–99. doi: 10.1523/JNEUROSCI.20-02-00589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cendes F. Progressive hippocampal and extrahippocampal atrophy in drug resistant epilepsy. Curr Opin Neurol. 2005;18:173–7. doi: 10.1097/01.wco.0000162860.49842.90. [DOI] [PubMed] [Google Scholar]
- Chandler JM, Cohen GM, MacFarlane M. Different subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver. J Biol Chem. 1998;273:10815–8. doi: 10.1074/jbc.273.18.10815. [DOI] [PubMed] [Google Scholar]
- Chandra D, Tang DG. Mitochondrially localized active caspase-9 and caspase-3 result mostly from translocation from the cytosol and partly from caspase- mediated activation in the organelle. J Biol Chem. 2003;278:17408–20. doi: 10.1074/jbc.M300750200. [DOI] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- Chen J, Graham SH, Zhu RL, Simon RP. Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:566–77. doi: 10.1097/00004647-199607000-00006. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: from adenosine receptor to KATP channel. Annu Rev Physiol. 2000;62:79–109. doi: 10.1146/annurev.physiol.62.1.79. [DOI] [PubMed] [Google Scholar]
- Colbourne F, Sutherland GR, Auer RN. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 1999;19:4200–10. doi: 10.1523/JNEUROSCI.19-11-04200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin YT, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conconi M, Petropoulos I, Emod I, Turlin E, Biville F, Friguet B. Protection from oxidative inactivation of the 20 S proteasome by heat-shock protein 90. Biochemical J. 1998;333:407–15. doi: 10.1042/bj3330407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley F, Stygall J, Lunn S, Harrison M, Brown MM, Newman S. Comparison of microembolism detected by transcranial Doppler and neuropsychological sequelae of carotid surgery and percutaneous transluminal angioplasty. Stroke. 2000;31:1329–34. doi: 10.1161/01.str.31.6.1329. [DOI] [PubMed] [Google Scholar]
- Currie RW, Ellison JA, White RF, Feuerstein GZ, Wang X, Barone FC. Benign focal ischemic preconditioning induces neuronal Hsp70 and prolonged astrogliosis with expression of Hsp27. Brain Res. 2000;863:169–81. doi: 10.1016/s0006-8993(00)02133-8. [DOI] [PubMed] [Google Scholar]
- Dargusch R, Schubert D. Specificity of resistance to oxidative stress. J Neurochem. 2002;81:1394. doi: 10.1046/j.1471-4159.2002.00950.x. [DOI] [PubMed] [Google Scholar]
- Das DK, Engelman RM, Maulik N. Oxygen free radical signaling in ischemic preconditioning. Ann NY Acad Sci. 1999;874:49–65. doi: 10.1111/j.1749-6632.1999.tb09224.x. [DOI] [PubMed] [Google Scholar]
- Davies KJA. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–10. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. 2004;91:1–8. doi: 10.1111/j.1471-4159.2004.02703.x. [DOI] [PubMed] [Google Scholar]
- Ding QX, Reinacker K, Dimayuga E, Nukala V, Drake J, Butterfield DA, Dunn JC, Martin S, Bruce-Keller AJ, Keller JN. Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. Febs Lett. 2003;546:228–32. doi: 10.1016/s0014-5793(03)00582-9. [DOI] [PubMed] [Google Scholar]
- Domorakova I, Mechirova E, Burda J, Ferikova M. Ubiquitin activity following forebrain ischemia/reperfusion in the rat. Acta Veterinaria Brno. 2004;73:45. [Google Scholar]
- Dudek EJ, Shang F, Taylor A. H2O2-mediated oxidative stress activates NF-kappa B in lens epithelial cells. Free Radical Biol Med. 2001;31:651–8. doi: 10.1016/s0891-5849(01)00634-7. [DOI] [PubMed] [Google Scholar]
- Ferrington DA, Husom AD, Thompson LV. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–6. doi: 10.1096/fj.04-2578fje. [DOI] [PubMed] [Google Scholar]
- Ferrington DA, Kapphahn RJ. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. Febs Lett. 2004;578:217–23. doi: 10.1016/j.febslet.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Figueiredo-Pereira ME, Cohen G. The ubiquitin/proteasome pathway: friend or foe in zinc-, cadmium-, and H2O2-induced neuronal oxidative stress. Mol Biol Rep. 1999;26:65. doi: 10.1023/a:1006909918866. [DOI] [PubMed] [Google Scholar]
- Folsom AR, Rasmussen ML, Chambless LE, Howard G, Cooper LS, Schmidt MI, Heiss G. Prospective associations of fasting insulin, body fat distribution, and diabetes with risk of ischemic stroke. Diabetes Care. 1999;22:1077–83. doi: 10.2337/diacare.22.7.1077. [DOI] [PubMed] [Google Scholar]
- Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ Res. 2001;88:802–9. doi: 10.1161/hh0801.089342. [DOI] [PubMed] [Google Scholar]
- Forlani S, Tomai F, De Paulis R, Turani F, Colella DF, Nardi P, De Notaris S, Moscarelli M, Magliano G, Crea F, Chiariello L. Preoperative shift from glibenclamide to insulin is cardioprotective in diabetic patients undergoing coronary artery bypass surgery. J Cardiovasc Surg. 2004;45:117–22. [PubMed] [Google Scholar]
- Garnier P, Ying WH, Swanson RA. Ischemic preconditioning by caspase cleavage of poly(ADP-ribose) polymerase-1. J Neurosci. 2003;23:7967–73. doi: 10.1523/JNEUROSCI.23-22-07967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerich JE. Oral hypoglycemic agents. N Engl J Med. 1989;321:1231–45. doi: 10.1056/NEJM198911023211805. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Xu LJ, Heng Z, Carrico W, Ouyang YB, Qiao YL, Sapolsky R, Steinberg G, Hu BR, Yenari MA. Chaperones, protein aggregation, and brain protection from hypoxic/ischemic injury. J Exp Biol. 2004;207:3213–20. doi: 10.1242/jeb.01034. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci USA. 2000;97:436–41. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev SM, Skarga YY, Mironova GD, Marinov BS. Regulation of mitochondrial KATP channel by redox agents. Biochim Biophys Acta. 1999;1410:91–6. doi: 10.1016/s0005-2728(98)00179-0. [DOI] [PubMed] [Google Scholar]
- Groop LC. Sulfonylureas in NIDDM. Diabetes Care. 1992;15:737–54. doi: 10.2337/diacare.15.6.737. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Auchampach JA. Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res. 1992;70:223–33. doi: 10.1161/01.res.70.2.223. [DOI] [PubMed] [Google Scholar]
- Grune T, Jung T, Merker K, Davies KJA. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Grune T. Oxidative stress, aging and the proteasomal system. Biogerontology. 2000;1:31–40. doi: 10.1023/a:1010037908060. [DOI] [PubMed] [Google Scholar]
- Hansson O, Castilho RF, Korhonen L, Lindholm D, Bates GP, Brundin P. Partial resistance to malonate-induced striatal cell death in transgenic mouse models of Huntington’s disease is dependent on age and CAG repeat length. J Neurochem. 2001;78:694–703. doi: 10.1046/j.1471-4159.2001.00482.x. [DOI] [PubMed] [Google Scholar]
- Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington’s disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci. 1999;96:8727–32. doi: 10.1073/pnas.96.15.8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Tanaka J, Kamikubo T, Takada K, Matsuda M. Increase in ubiquitin conjugates dependent on ischemic damage. Brain Res. 1993;620:171–3. doi: 10.1016/0006-8993(93)90288-x. [DOI] [PubMed] [Google Scholar]
- Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, Malfertheiner M, Kohemann M, Potrovita I, Maegele I, Beyer C, Burke JR, Hasan MT, Bujard H, Wirth T, Pasparakis M, Schwaninger M. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–9. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Ann Rev Cell Dev Biol. 2003;19:141–72. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Morton AJ. Mice transgenic for the Huntington’s disease mutation are resistant to chronic3-nitropropionic acid-induced striatal toxicity. J Neurochem. 2000;75:2163–71. doi: 10.1046/j.1471-4159.2000.0752163.x. [DOI] [PubMed] [Google Scholar]
- Hohfeld J, Cyr DM, Patterson C. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Reports. 2001;10:885–90. doi: 10.1093/embo-reports/kve206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjo BK, Liu CL. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21:865–75. doi: 10.1097/00004647-200107000-00012. [DOI] [PubMed] [Google Scholar]
- Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20:3191–9. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama K-I, Takahashi R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol Cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- Jodicke A, Hubner F, Boker DK. Monitoring of brain tissue oxygenation during aneurysm surgery: prediction of procedure-related ischemic events. J Neurosurg. 2003;98:515–23. doi: 10.3171/jns.2003.98.3.0515. [DOI] [PubMed] [Google Scholar]
- Johnston SC, Gress DR, Browner WS, Sidney S. Short-term prognosis after emergency department diagnosis of TIA. JAMA. 2000;284:2901–6. doi: 10.1001/jama.284.22.2901. [DOI] [PubMed] [Google Scholar]
- Kamikubo T, Hayashi T. Changes in proteasome activity following transient ischemia. Neurochem Int. 1996;28:209–12. doi: 10.1016/0197-0186(95)00071-2. [DOI] [PubMed] [Google Scholar]
- Keller JN, Huang FF, Dimayuga ER, Maragos WF. Dopamine induces proteasome inhibition in neural PC12 cell line. Free Radical Biol Med. 2000a;29:1037–42. doi: 10.1016/s0891-5849(00)00412-3. [DOI] [PubMed] [Google Scholar]
- Keller JN, Huang FF, Zhu H, Yu J, Ho YS, Kindy MS. Oxidative stress-associated impairment of proteasome activity during ischemia–reperfusion injury. J Cereb Blood Flow Metab. 2000b;20:1467–73. doi: 10.1097/00004647-200010000-00008. [DOI] [PubMed] [Google Scholar]
- Kim I, Kim CH, Kim JH, Lee J, Choi JJ, Chen ZA, Lee MG, Chung KC, Hsu CY, Ahn YS. Pyrrolidine dithiocarbamate and zinc inhibit proteasome-dependent proteolysis. Exp Cell Res. 2004;298:229–38. doi: 10.1016/j.yexcr.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Kiss P, Szabo A, Hunyadi-Gulyas E, Medzihradszky KF, Lipinszki Z, Pal M, Udvardy A. Zn2+-induced reversible dissociation of subunit Rpn10/p54 of the Drosophila 26 S proteasome. Biochem J. 2005;391:301–10. doi: 10.1042/BJ20050523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K. Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528:21–4. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- Kukreja RC, Kontos MC, Loesser KE, Batra SK, Qian YZ, Gbur CJ, Jr, Naseem SA, Jesse RL, Hess ML. Oxidant stress increases heat shock protein 70 mRNA in isolated perfused rat heart. Am J Physiol. 1994;267:H2213–9. doi: 10.1152/ajpheart.1994.267.6.H2213. [DOI] [PubMed] [Google Scholar]
- Kumar R, Krause GS, Yoshida H, Mori K, DeGracia DJ. Dysfunction of the unfolded protein response during global brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2003;23:462–71. doi: 10.1097/01.WCB.0000056064.25434.CA. [DOI] [PubMed] [Google Scholar]
- Lebuffe G, Schumacker PT, Shao ZH, Anderson T, Iwase H, Vanden Hoek TL. ROS and NO trigger early preconditioning: relationship to mitochondrial K-ATP channel. Am J Physiol. 2003;284:H299–308. doi: 10.1152/ajpheart.00706.2002. [DOI] [PubMed] [Google Scholar]
- Lee HT, Xu H, Ota-Setlik A, Emala CW. Oxidant preconditioning protects human proximal tubular cells against lethal oxidant injury via p38 MAPK and heme oxygenase-1. Am J Nephrol. 2003;23:324–33. doi: 10.1159/000072914. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Liu CL, Martone ME, Hu BR. Protein ubiquitination in postsynaptic densities after transient cerebral ischemia. J Cereb Blood Flow Metab. 2004;24:1219–25. doi: 10.1097/01.WCB.0000136706.77918.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ginis I, Spatz M, Hallenbeck JM. Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. Am J Physiol Cell Physiol. 2000;278:C144–53. doi: 10.1152/ajpcell.2000.278.1.C144. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–9. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- Mailhos C, Howard MK, Latchman DS. Heat shock proteins hsp90 and hsp70 protect neuronal cells from thermal stress but not from programmed cell death. J Neurochem. 1994;63:1787–95. doi: 10.1046/j.1471-4159.1994.63051787.x. [DOI] [PubMed] [Google Scholar]
- Mancini M, Nicholson DW, Roy S, Thornberry NA, Peterson EP, Casciola-Rosen LA, Rosen A. The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling. J Cell Biol. 1998;140:1485–95. doi: 10.1083/jcb.140.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. NF-kappa B in the survival and plasticity of neurons. Neurochem Res. 2005;30:883–93. doi: 10.1007/s11064-005-6961-x. [DOI] [PubMed] [Google Scholar]
- Mayberg MR, Wilson SE, Yatsu F, Weiss DG, Messina L, Hershey LA, Colling C, Eskridge J, Deykin D, Winn HR. Carotid endarterectomy and prevention of cerebral ischemia in symptomatic carotid stenosis. JAMA. 1991;266:3289. [PubMed] [Google Scholar]
- McLaughlin B. The kinder side of killer proteases: caspase activation contributes to neuroprotection and CNS remodeling. Apoptosis. 2004;9:111–21. doi: 10.1023/B:APPT.0000018793.10779.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin BA, DiNapoli M. The proteasome ubiquitin system as a drug target in cerebrovascular disease: The therapeutic potential of proteasome inhibitors. Curr Opin Invest Drugs. 2005;6:686–99. [PMC free article] [PubMed] [Google Scholar]
- McLaughlin BA, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in ischemic preconditioning. Proc Natl Acad Sci. 2003;100:715–20. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlhase J, Grune T. Proteolytic response to oxidative stress in mammalian cells. Biol Chem. 2002;383:559–67. doi: 10.1515/BC.2002.057. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Gallwitz B, Schmidt WE, Mugge A, Nauck MA. Is impairment of ischaemic preconditioning by sulfonylurea drugs clinically important? Heart. 2004;90:9–12. doi: 10.1136/heart.90.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengesdorf T, Jensen PH, Mies G, Aufenberg C, Paschen W. Down-regulation of parkin protein in transient focal cerebral ischemia: a link between stroke and degenerative disease? Proc Natl Acad Sci. 2002;99:15042–7. doi: 10.1073/pnas.232588799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitka M. TIA time bombs remain undertreated. JAMA. 2005;293:1435. doi: 10.1001/jama.293.12.1435. [DOI] [PubMed] [Google Scholar]
- Moncayo J, de Freitas GR, Bogousslavsky J, Altieri M, van Melle G. Do transient ischemic attacks have a neuroprotective effect? Neurology. 2000;54:2089–94. doi: 10.1212/wnl.54.11.2089. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Leavens W. Mice transgenic for the human Huntington’s disease mutation have reduced sensitivity to kainic acid toxicity. Brain Res Bull. 2000;52:51–9. doi: 10.1016/s0361-9230(00)00238-0. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia—a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nombela F, Sobrino T, Blanco M, Castellanos M, Escudero D, Moro MA, Silva Y, Castillo J, Davalos A, Mora JV. Molecular changes after carotid angioplasty and stenting mimic signatures of acute cerebral ischemia. Stroke. 2006;37:684–684. [Google Scholar]
- O’Rourke B. Pathophysiological and protective roles of mitochondrial ion channels. J Physiol. 2000;529:23–36. doi: 10.1111/j.1469-7793.2000.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–32. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–6. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- Palumbo PJ, Elveback LR, Whisnant JP. Neurologic Complications of Diabetes Mellitus: Transient Ischemic Attacks, Stroke and Peripheral Neuropathy. New York: Raven Press; 1978. [PubMed] [Google Scholar]
- Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol. 2003;13:2073–81. doi: 10.1016/j.cub.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 2006;13:759–72. doi: 10.1038/sj.cdd.4401838. [DOI] [PubMed] [Google Scholar]
- Petersen A, Hansson O, Puschban Z, Sapp E, Romero N, Castilho RF, Sulzer D, Rice M, DiFiglia M, Przedborski S, Brundin P. Mice transgenic for exon 1 of the Huntington’s disease gene display reduced striatal sensitivity to neurotoxicity induced by dopamine and 6-hydroxydopamine. Eur J Neurosci. 2001;14:1425–35. doi: 10.1046/j.0953-816x.2001.01765.x. [DOI] [PubMed] [Google Scholar]
- Poppert H, Wolf O, Resch M, Theiss W, Schmidt-Thieme T, Graefin von Einsiedel H, Heider P, Martinoff S, Sander D. Differences in number, size and location of intracranial microembolic lesions after surgical versus endovascular treatment without protection device of carotid artery stenosis. J Neurol. 2004;251:1198. doi: 10.1007/s00415-004-0502-4. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Levy DE, Sigsbee B, Scherer P, Plum F. Increased damage after ischemic stroke in patients with hyperglycemia with or without established diabetes-mellitus. Am J Med. 1983;74:540–4. doi: 10.1016/0002-9343(83)91007-0. [DOI] [PubMed] [Google Scholar]
- Qiu JH, Asai A, Chi S, Saito N, Hamada H, Kirino T. Proteasome inhibitors induce cytochrome c-caspase-3-like protease-mediated apoptosis in cultured cortical neurons. J Neurosci. 2000;20:259–65. doi: 10.1523/JNEUROSCI.20-01-00259.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajdev S, Hara K, Kokubo Y, Mestril R, Dillmann W, Weinstein PR, Sharp FR. Mice overexpressing rat heat shock protein 70 are protected against cerebral infarction. Ann Neurol. 2000;47:782–91. [PubMed] [Google Scholar]
- Ravati A, Ahlemeyer B, Becker A, Klumpp S, Krieglstein J. Preconditioning-induced neuroprotection is mediated by reactive oxygen species and activation of the transcription factor nuclear factor-kappa B. J Neurochem. 2001;78:909–19. doi: 10.1046/j.1471-4159.2001.00463.x. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Stefanis L. Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol Cell Neurosci. 2002;21:223–38. doi: 10.1006/mcne.2002.1173. [DOI] [PubMed] [Google Scholar]
- Riepe MW, Esclaire F, Kasischke K, Schreiber S, Nakase H, Kempski O, Ludolph AC, Dirnagl U, Hugon J. Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: ‘chemical preconditioning’. J Cereb Blood Flow Metab. 1997;17:257–64. doi: 10.1097/00004647-199703000-00002. [DOI] [PubMed] [Google Scholar]
- Robles E, Huttenlocher A, Gomez TM. Filopodial calcium transients regulate growth cone motility and guidance through local activation of calpain. Neuron. 2003;38:597–609. doi: 10.1016/s0896-6273(03)00260-5. [DOI] [PubMed] [Google Scholar]
- Rockwell P, Yuan H, Magnusson R, Figueiredo-Pereira ME. Proteasome inhibition in neuronal cells induces a proinflammatory response manifested by upregulation of cyclooxygenase-2, its accumulation as ubiquitin conjugates, and production of the prostaglandin PGE(2) Arch Biochem Biophys. 2000;374:325. doi: 10.1006/abbi.1999.1646. [DOI] [PubMed] [Google Scholar]
- Roh HG, Byun HS, Ryoo JW, Na DG, Moon W-J, Lee BB, Kim D-I. Prospective analysis of cerebral infarction after carotid endarterectomy and carotid artery stent placement by using diffusion-weighted imaging. AJNR Am J Neuroradiol. 2005;26:376–84. [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci. 2002;22:10690–8. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara Y, Tan S, Maher P, Schubert D. Mechanisms of resistance to oxidative stress in Alzheimer’s disease. BioFactors (Oxford, England) 1998;8:45–50. doi: 10.1002/biof.5520080109. [DOI] [PubMed] [Google Scholar]
- Schubert D, Behl C. The expression of amyloid beta protein precursor protects nerve cells from beta-amyloid and glutamate toxicity and alters their interaction with the extracellular matrix. Brain Res. 1993;629:275–82. doi: 10.1016/0006-8993(93)91331-l. [DOI] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–4. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- Stampfer MJ, Malinow MR, Willett WC, Newcomer LM, Upson B, Ullmann D, Tishler PV, Hennekens CH. A prospective study of plasma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA. 1992;268:877–81. [PubMed] [Google Scholar]
- Tanaka K. Molecular-biology of proteasomes. Mol Biol Rep. 1995;21:21–6. doi: 10.1007/BF00990966. [DOI] [PubMed] [Google Scholar]
- Tremblay R, Chakravarthy B, Hewitt K, Tauskela J, Morley P, Atkinson T, Durkin JP. Transient NMDA receptor inactivation provides long-term protection to cultured cortical neurons from a variety of death signals. J Neurosci. 2000;20:7183–92. doi: 10.1523/JNEUROSCI.20-19-07183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya D, Hong S, Matsumori Y, Kayama T, Swanson RA, Dillman WH, Liu JL, Panter SS, Weinstein PR. Overexpression of rat heat shock protein 70 reduces neuronal injury after transient focal ischemia, transient global ischemia, or kainic acid-induced seizures. Neurosurgery. 2003a;53:1179–87. doi: 10.1227/01.neu.0000090341.38659.cf. [DOI] [PubMed] [Google Scholar]
- Tsuchiya D, Hong S, Matsumori Y, Shiina H, Kayama T, Swanson RA, Dillman WH, Liu JL, Panter SS, Weinstein PR. Overexpression of rat heat shock protein 70 is associated with reduction of early mitochondrial cytochrome c release and subsequent DNA fragmentation after permanent focal ischemia. J Cereb Blood Flow Metab. 2003b;23:718–27. doi: 10.1097/01.WCB.0000054756.97390.F7. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Mummery R, Hawkes RB, Rider CC, Beesley PW. Hypoxia–ischemia induces a rapid elevation of ubiquitin conjugate levels and ubiquitin immuno-reactivity in the immature rat brain. J Cereb Blood Flow Metab. 1998;18:376–85. doi: 10.1097/00004647-199804000-00005. [DOI] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, Fawcett JW. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25:331–42. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, Kucinski T, Jungehulsing GJ, Brunecker P, Muller B, Banasik A, Amberger N, Wernecke KD, Siebler M, Rother J, Villringer A, Weih M for the MRI in Acute Stroke Study Group of the German Competence Network Stroke. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A Multicenter Magnetic Resonance Imaging Study. Stroke. 2004;35:616–21. doi: 10.1161/01.STR.0000115767.17923.6A. [DOI] [PubMed] [Google Scholar]
- Weih M, Bergk A, Isaev NK, Ruscher K, Megow D, Riepe M, Meisel A, Victorov IV, Dirnagl U, Dirnagi U. Induction of ischemic tolerance in rat cortical neurons by 3-nitropropionic acid: chemical preconditioning. Neurosci Lett. 1999;272:207–10. doi: 10.1016/s0304-3940(99)00594-7. [DOI] [PubMed] [Google Scholar]
- Weiser T. AMPA receptor antagonists with additional mechanisms of action: new opportunities for neuroprotective drugs? Curr Pharmaceut Des. 2002;8:941–51. doi: 10.2174/1381612024607135. [DOI] [PubMed] [Google Scholar]
- Xu M, Wang Y, Ayub A, Ashraf M. Mitochondrial K(ATP) channel activation reduces anoxic injury by restoring mitochondrial membrane potential. Am J Physiol Heart Circ Physiol. 2001;281:H1295–303. doi: 10.1152/ajpheart.2001.281.3.H1295. [DOI] [PubMed] [Google Scholar]
- Yadav JS, Wholey MH, Kuntz RE, Fayad P, Katzen BT, Mishkel GJ, Bajwa TK, Whitlow P, Strickman NE, Jaff MR, Popma JJ, Snead DB, Cutlip DE, Firth BG, Ouriel K. Protected carotid-artery stenting versus endarterectomy in high-risk patients. N Engl J Med. 2004;351:1493–501. doi: 10.1056/NEJMoa040127. [DOI] [PubMed] [Google Scholar]
- Yang JY, Michod D, Walicki J, Murphy BA, Kasibhatla S, Martin SJ, Widmann C. Partial cleavage of RasGAP by caspases is required for cell survival in mild stress conditions. Mol Cell Biol. 2004;24:10425–36. doi: 10.1128/MCB.24.23.10425-10436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang QH, Du C. Smac/DIABLO selectively reduces the levels of c-IAP1 and c-IAP2 but not thatof XIAP and livin in HeLa cells. J Biol Chem. 2004;279:16963. doi: 10.1074/jbc.M401253200. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Dana A. The preconditioning phenomenon: a tool for the scientist or a clinical reality? Circ Res. 2000;87:543–50. doi: 10.1161/01.res.87.7.543. [DOI] [PubMed] [Google Scholar]
- Zhang W, Stanimirovic D. Current and future therapeutic strategies to target inflammation in stroke. Current Drug Targets. Inflammation Allergy. 2002;1:151–66. doi: 10.2174/1568010023344689. [DOI] [PubMed] [Google Scholar]
- Zund G, Uezono S, Stahl GL, Dzus AL, McGowan FX, Hickey PR, Colgan SP. Hypoxia enhances induction of endothelial ICAM-1: role for metabolic acidosis and proteasomes. Am J Physiol. 1997;273:C1571–80. doi: 10.1152/ajpcell.1997.273.5.C1571. [DOI] [PubMed] [Google Scholar]