

Abstract
Sulfur is an essential element found ubiquitously in living systems. However, there exist only a few sulfur-containing sugars in nature and their biosyntheses have not been studied. BE-7585A produced by Amycolatopsis orientalis subsp. vinearia BA-07585 has a 2-thiosugar and is a member of the angucycline class of compounds. We report herein the results of our initial efforts to study the biosynthesis of BE-7585A. Spectroscopic analyses verified the structure of BE-7585A, which is closely related to rhodonocardin A. Feeding experiments using 13C-labeled acetate were carried out to confirm that the angucycline core is indeed polyketide-derived. The results indicated an unusual manner of angular tetracyclic ring construction, perhaps via a Baeyer-Villiger type rearrangement. Subsequent cloning and sequencing led to the identification of the bex gene cluster spanning ~30 kbp. A total of 28 open reading frames, which are likely involved in BE-7585A formation, were identified in the cluster. In view of the presence of a homologue of a thiazole synthase gene (thiG), bexX, in the bex cluster, the mechanism of sulfur incorporation into the 2-thiosugar moiety could resemble that found in thiamin biosynthesis. A glycosyltransferase homologue, BexG2, was heterologously expressed in E. coli. The purified enzyme successfully catalyzed the coupling of 2-thioglucose 6-phoshate and UDP-glucose to produce 2-thiotrehalose 6-phosphate, which is the precursor of the disaccharide unit in BE-7585A. On the basis of these genetic and biochemical experiments, a biosynthetic pathway for BE-7585A can now be proposed. The combined results set the stage for future biochemical studies of 2-thiosugar biosynthesis and BE-7585A assembly.
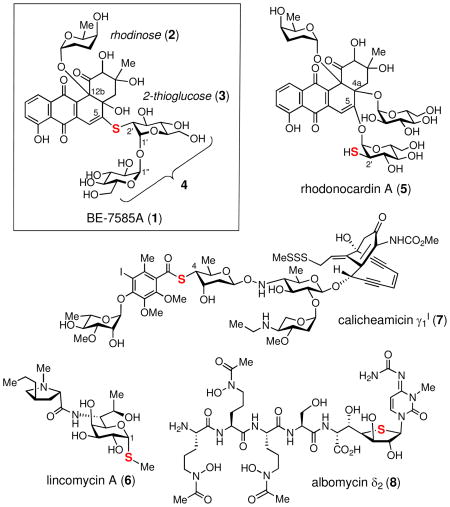
The angucycline class of natural products, which have a characteristic fused four-ring frame assembled in an angular manner, are rich in antibacterial or anticancer activities.1 BE-7585A (1), produced by Amycolatopsis orientalis subsp. vinearia BA-07585, is an angucycline-type natural product that inhibits thymidylate synthase.2 In addition to the benz[a]anthraquinone core, BE-7585A also contains the deoxysugar, rhodinose (2), and a disaccharide appendage (4) containing a highly unusual 2-thioglucose (3). While sulfur is an essential element found ubiquitously in living systems, including amino acids, nucleic acids, metal clusters, enzyme cofactors, and many secondary metabolites, it is seldom found in carbohydrates. The rareness of the thiosugar entity prompted us to explore the biosynthesis of BE-7585A, especially the mode of sulfur incorporation into the 2-thiosugar moiety (3). Mechanistic details of several biological sulfur insertion reactions have recently been unraveled.3 Highly regulated reactive intermediates such as protein persulfide, protein thiocarboxylate or S-adenosylmethionine-dependent radical are often generated in the enzymes carrying out such reactions. Whether the same or different mechanisms are used to form the C–S bond in 2-thiosugar biosynthesis is the focus of this study.
Thus far, only a handful of thiosugar-containing natural products are known. These include BE-7585A (1),2 rhodonocardins (5),4 lincomycins (6),5 celesticetin,6 calicheamicins (7),7 esperamicins,8 namenamicin,9 shishijimicins,10 albomycins (8)11 and SB-21745212 from microorganisms; kotalanol,13 salacinol14 and glucosinolates15 from plants; and 5-thio-D-mannose16 from the marine sponge Clathria pyramida (although the actual producer might be its symbiont). The biosynthetic gene clusters for some of the compounds produced by bacteria have been identified,17–19 but the pathway and mechanisms of the thiosugar biosynthesis in each case remain obscure. Among these compounds, BE-7585A (1) and rhodonocardins (5) are the only two reported natural products containing a 2-thiosugar (3). Interestingly, 1 and 5 are constitutional isomers that share the benz[a]anthraquinone core structure and three sugar components; rhodinose, 2-thioglucose, and glucose, but differ in the attachment of the 2-thioglucose and glucose moieties on the core molecule. In BE-7585A (1), the 2-thioglucose and glucose moieties are linked via their anomeric carbons, and the resulting disaccharide (4) is attached to the C-5 position of the aglycone through a thioether linkage. In contrast, the 2-thioglucose and glucose in 5 are separately attached to C-5 and C-4a of the angucycline core via O-glycosidic bonds, leaving the 2-mercapto-substituent in 3 as a free thiol group. The structure of rhodonocardin (5) is fully documented,4 but that of BE-7585A (1) is not because the structure is reported in a patent.2
Here, we verified the structure of BE-7585A (1) and confirm that the angucycline core of 1 is indeed polyketide-derived. Interestingly, feeding experiments using 13C-labeled acetate suggest that the benz[a]anthraquinone core formation may involve an oxidative ring cleavage followed by rearrangement and recyclization leading to the observed angular-tetracyclic structure. Subsequent cloning and sequencing experiments led to the identification of the bex gene cluster from A. orientalis spanning ~30 kbp. On the basis of the functional prediction of the genes in the cluster, a possible biosynthetic pathway culminating in 1 is proposed. The bexX gene, which encodes a protein homologous to thiazole synthase (ThiG), likely plays a key role in sulfur incorporation during 2-thiosugar formation. The in vitro characterization of BexG2, which catalyzes the coupling of 2-thioglucose-6-phosphate and UDP-glucose to produce the precursor of the disaccharide unit (4) in 1 is also reported. Overall, this work provides significant insights into the biosynthesis of the 2-thiosugar and the unusual ring formation of the benz[a]anthraquinone core in BE-7585A. Elucidation of unusual sugar biosynthesis is important not only for understanding the chemical mechanisms of their formation, but also for future pathway engineering experiments to construct “tailor-made” glycosylated metabolites with new or enhanced biological activities.20
Experimental Procedures
Materials
All chemicals and reagents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA), and were used without further purification unless otherwise specified. Enzymes and molecular weight standards used for the cloning experiments were products of Invitrogen (Carlsbad, CA) or New England Biolabs (Ipswich, MA). Ni-NTA agarose and kits for DNA gel extraction and spin minipreps were obtained from Qiagen (Valencia, CA). PfuUltra DNA polymerase and Gigapack III XL packaging extract were products of Stratagene (La Jolla, CA). Growth medium components were acquired from Becton Dickinson (Sparks, MD). Reagents for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were purchased from Bio-Rad (Hercules, CA), with the exception of the protein molecular weight markers, which were obtained from Invitrogen or New England Biolabs. Sterile syringe filters were products of Fisher Scientific. Amicon YM-10 ultrafiltration membranes were purchased from Millipore (Billerica, MA). The CarboPac PA1 high-performance liquid chromatography (HPLC) column was obtained from Dionex (Sunnyvale, CA). Oligonucleotide primers were prepared by Invitrogen or Integrated DNA Technologies (Coralville, IA).
Bacterial Strains and Plasmids
Amycolatopsis orientalis subsp. vinearia BA-07585 was generously provided by Banyu Pharmaceutical Co. (Tokyo, Japan). Escherichia coli DH5α, acquired from Bethesda Research Laboratories (Gaithersburg, MD), was used for routine cloning experiments. E. coli XL-1 Blue MRF′, purchased from Stratagene, was employed for cosmid manipulations. Protein overexpression host E. coli BL21 star (DE3) was obtained from Invitrogen. Plasmids pGEM-T Easy and pUC119, used for subcloning, were products of Promega (Madison, WI) and Takara Bio (Shiga, Japan), respectively. Vector pET28b(+) for protein overexpression was purchased from Novagen (Madison, WI).
General
NMR spectra were acquired on a Varian Unity 500 MHz spectrometer, and chemical shifts (in ppm) are reported relative to that of the solvent peak (δH = 4.65 for deuterated water, δH = 3.30 and δC = 49.0 for deuterated methanol) or an internal standard (dioxane, δC = 67.4) for spectra taken in D2O solvent. HPLC was performed on a Beckman Coulter System Gold equipped with a UV detector or Corona CAD (charged aerosol detector). DNA sequencing was performed by the core facility of the Institute of Cellular and Molecular Biology at the University of Texas, Austin. Vector NTI Advance 10.1.1 from Invitrogen was used for sequence alignments. Mass spectra were recorded by the Mass Spectrometry core facility in the Department of Chemistry and Biochemistry at the University of Texas, Austin. Standard genetic manipulations of E. coli were performed as described by Sambrook et al.21
Production, Isolation and Identification of BE-7585A (1)
Spores of A. orientalis subsp. vinearia BA-07585 were inoculated into 100 mL of International Streptomyces Project (ISP) medium 2 and grown in a rotary incubator at 29 °C and 250 rpm for 5 days. The resultant seed culture (25 mL) was transferred to 1 L of ISP-2 medium and grown under the same conditions for 10 days. The growth culture was centrifuged at 6000g for 10 min to remove the cells and other insoluble materials. The supernatant was applied to a synthetic polymeric adsorbent Diaion HP20 column (250 mL) and washed with water. The adsorbed compounds were then eluted with 500 mL of methanol. The collected red fractions were pooled and concentrated by rotary evaporation in vacuo and further purified on a Diaion CHP20P column (50 mL) using 5–20% of methanol in water as the eluent. The typical yield was 100–150 mg of BE-7585A (1) from 1-L of culture. The structure of 1 was determined by 1H, 13C, COSY, HSQC, HMBC and NOESY NMR experiments and ESI-MS.
13C-labeling Experiments
Spores of A. orientalis subsp. vinearia BA-07585 were inoculated into 50 mL of ISP-2 medium and grown in a rotary incubator at 30 °C and 250 rpm for 5 days. The resultant seed culture (25 mL) was transferred to 1 L of ISP-2 medium and grown under the same conditions for 2 days. Sodium [1-13C]acetate, in an aqueous solution (4%, 5 mL) sterilized by filtration through a syringe filter (0.2 μm), was added to the culture, which had a light red color. An additional [1-13C]acetate solution was added (4%, 5 mL × 4) to the growth culture every 13–20 h. The final concentration of labeled acetate was 0.1%. After a 10-day incubation period, the culture was centrifuged at 6000g for 10 min. The 13C-labeled BE-7585A (1, 150 mg) was isolated from the supernatant by using Diaion HP20 and CHP20P columns as described above for the purification of the non-labeled 1. The purified compound was analyzed by 13C NMR spectroscopy.
The feeding experiment was also performed with sodium [1,2-13C2]acetate in 1-L ISP-2 medium. A 25 mL aqueous solution containing both sodium [1,2-13C2]acetate (0.68%) and non-labeled sodium acetate (1.3%) was prepared and sterilized by filtration through a syringe filter. The solution (10 mL) was then added to the culture, which had been grown for 2 days and turned to light red. The remaining solution (15 mL) was added 20-h after the first addition. The final concentration of the labeled acetate was 0.017%. After a 10-day incubation period, the culture was centrifuged at 6000g for 10 min to remove cells and any insoluble materials. The 13C-labeled BE-7585A (1, 80 mg) was isolated from the supernatant using Diaion HP20 and CHP20P columns and analyzed by 13C NMR spectroscopy as described above.
Construction of the Cosmid Library
Spores of A. orientalis subsp. vinearia BA-07585 were inoculated into 50 mL of tryptone soya broth (TSB) medium and grown in a rotary incubator at 30 °C and 250 rpm for 3 days. The mycelia were harvested by centrifugation at 5000g for 30 min. The cells were disrupted by lysozyme and SDS, and the cell lysate was treated with proteinase K. The released genomic DNA was isolated by phenol–chloroform extraction followed by sodium acetate–isopropyl alcohol precipitation. The DNA was partially digested with Sau3AI restriction endonuclease in a time dependent manner, and ligated into the BamHI site of the pOJ446 vector.22 The ligated DNA was packaged into phage particles using a Gigapack III XL packaging extract and then introduced into E. coli XL1-Blue MRF′ according to the manufacture’s instructions. Recombinants were selected on Luria-Bertani (LB)-agar plates in the presence of apramycin (50 μg/mL). After incubation for 16 h, the resulting transformants were spotted within grids on new plates. This genomic library contains a total of 454 cosmids.
PCR-based Screening of the Cosmid Library
Polymerase chain reaction (PCR) primers for screening of type II PKS were designed based on multiple sequence alignments of ten known β-ketoacyl synthases α subunit (KSα) of actinomycetes including Streptomyces venezuelae23 and Streptomyces coelicolor A3(2),24 which are available in the National Center for Biotechnology Information (NCBI) database. Similarly, the primers for deoxysugar genes, such as TDP-6-deoxy-4-keto-D-glucose 2,3- dehydratase25 and TDP-2,6-dideoxy-4-keto-D-glucose 3-dehydrase,26 were designed based on the reported gene sequences from eight actinomycete strains including Streptomyces fradiae27 and Streptomyces cyanogenus.28 The sequences of the primers used for the PCR-based screening are listed in Table 1. PCR was performed using PfuUltra DNA polymerase with these primers and the cosmid DNA from the genomic library. Amplified DNA fragments were ligated into the pGEM-T Easy vector for cloning and sequencing.
Table 1.
Primers used for cosmid library screening.
| Primer name | Sequence |
|---|---|
| KSα -forward | 5′-CGACGCVCCSATCDCVCCSATC-3′ |
| KSα -reverse | 5′-GGAANCCDCCGAABCCGCTGCC-3′ |
| 2,3-dehydratase-forward | 5′-AGCTSTCSCCSACVGTBCAGC-3′ |
| 2,3-dehydratase-reverse | 5′-WRGAAVCGRCCSCCYTCYTCSG-3′ |
| 3-dehydrase-forward | 5′-TSAACCCGMTCVTSCAGACGG-3′ |
| 3-dehydrase-reverse | 5′-CSGGRTGSCKGGTSAKGTTSCC-3′ |
M = AC, R =AG, W = AT, S = CG, Y = CT, K = GT, V = ACG, H = ACT, D = AGT, B = CGT, N = ACGT
Sequencing and Gene Cluster Identification
Cosmids A108 and C006, which together span the entire bex cluster, were digested with either KpnI, NcoI or PstI. The resulting 2.5–12 kb DNA fragments were subcloned into a modified pUC119 vector, whose multiple cloning site harbors extra restriction sites.29 Both strands of the subclones were sequenced using M13 universal primers or by primer walking. Sequencing data were obtained using a capillary-based AB 3700 DNA analyzer and assembled using Vector NTI Suite program. Open reading frame assignments were made with the assistance of FramePlot 2.3.2.30 Homologous protein sequences were identified in the NCBI database using the basic local alignment search tool (BLAST).
Cloning of bexG2
The bexG2 gene was PCR-amplified from cosmid C006 using primers with engineered NdeI and HindIII restriction sites. The sequences of the primers are 5′-GGTTAGGCAT-ATGAGGATTCTGACCTGCAGC-3′ (forward) and 5′-GTAGAAGCTTAGGTCAGAGCCCGAAAA-CCCC-3′ (reverse). The engineered restriction sites are shown in bold, the start codon in bold and also underlined, and the stop codon underlined. The PCR-amplified gene fragments were purified, digested with NdeI and HindIII, and ligated into pET28b(+) vector digested with the same enzymes. The resulting plasmid, bexG2/pET28b(+), was used to transform E. coli BL21 star (DE3) strain for protein overexpression. The BexG2 enzyme was expressed as an N-terminal-His6-tagged protein.
Expression and Purification of BexG2
An overnight culture of E. coli BL21 star (DE3)- bexG2/pET28b(+), grown in the LB medium (10 mL) containing 50 μg/mL of kanamycin at 37 °C, was used to inoculate 1 L of the same growth medium. The culture was incubated at 37 °C with shaking (230 rpm) until the OD600 reached ~0.5. Protein expression was then induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM, and the cells were allowed to grow at 18 °C and 125 rpm for an additional 24 h. The cells were harvested by centrifugation at 4500g for 15 min and stored at −80 °C until lysis. All purification steps were carried out at 4 °C using Ni-NTA resin according to the manufacturer’s protocol with minor modifications. Specifically, the thawed cells (3 g) were re-suspended in the lysis buffer (15 mL) containing 10% (v/v) glycerol and 10 mM imidazole. After incubation with lysozyme (15 mg) for 30 min, the cells were disrupted by sonication using 10 × 10-s pulses with a 30-s cooling pause between each pulse. The resulting lysate was centrifuged at 10000g for 20 min, and the supernatant was subjected to Ni-NTA chromatography. Bound protein was eluted using buffer containing 10% glycerol and 250 mM imidazole. The collected protein solution was dialyzed against 3 × 1-L of 50 mM Tris•HCl buffer (pH 8) containing 300 mM NaCl and 15% glycerol. The protein solution was then flash-frozen in liquid nitrogen and stored at −80 °C until use. Protein concentration was determined by the Bradford assay31 using bovine serum albumin as the standard. The yield of BexG2 was 40 mg from 1 L culture. The molecular mass and purity of BexG2 were estimated by SDS-PAGE analysis (Figure 1).
Figure 1.

SDS-PAGE gel of the purified N-His6 tagged BexG2 (482 aa, 53.6 kDa). The molecular weight marks are 220, 160, 120, 100, 90, 80, 70, 60, 50, 40, 30, 25, 20, 15, and 10 kDa (top to bottom).
Synthesis of 2-Thio-D-glucose (3)
1,3,4,6-Tetra-O-acetyl-β-D-mannopyranose (11)
As shown in Scheme 1, compound 11 was synthesized from D-mannose (9, 16.5 g, 92 mmol) following a previously reported procedure with slight modifications.32 Accordingly, to a small portion of 9 (~30 mg) in acetic anhydride (63 mL) was added 70% perchloric acid (5 drops). The remaining 9 was slowly added to the reaction mixture over a 20-min period, keeping the reaction temperature at 40–45 °C. The resulting mixture was stirred at room temperature for 1 h and then cooled to 15 °C. Phosphorus tribromide (13 mL) was added dropwise to the reaction with the reaction temperature kept at 20–25 °C. Cold water (8.3 mL) was slowly added to the resulting mixture, and the reaction was kept at room temperature for 1.5 h. After cooling to 10 °C, a chilled solution of sodium acetate trihydrate (50 g, 370 mmol) in water was slowly added with the temperature kept at 25–30 °C. The reaction mixture was stirred at room temperature for 20 min and poured onto ice. The resulting mixture was extracted with methylene chloride (100 mL × 2) and washed sequentially with cold saturated sodium bicarbonate and cold water. The solution was dried over anhydrous Na2SO4 and evaporated in vacuo. The obtained crude mixture, which contained 2,3,4,6-tetra-O-acetyl-α-D-mannopyranose (10) byproduct, was thoroughly washed with anhydrous diethyl ether to give the desired product 11 as a white powder (6.7 g, 19 mmol, 21%). The 1H NMR spectral data of 11 is consistent with those previously reported.32b
Scheme 1.

1,3,4,6-Tetra-O-acetyl-2-O-trifluoromethylsulfonyl-β-D-mannopyranose (12)
Compound 12 was synthesized from 11 using a previously reported procedure with minor modifications.33 Specifically, to a solution of 11 (3.5 g, 10 mmol) in anhydrous methylene chloride (100 mL) was added anhydrous pyridine (1.8 mL, 22 mmol). The solution was cooled to −20 °C and treated with trifluoromethane- sulfonic anhydride (3.6 mL, 22 mmol), which was added dropwise in 1 h under N2. The resulting suspension was stirred at room temperature for 1 h. The mixture was washed sequentially with cold water, saturated sodium bicarbonate and water, dried over anhydrous Na2SO4, and concentrated in vacuo. The product 12 was obtained as a pale yellow solid (4.6 g, 9.6 mmol, 96%). The 1H NMR spectral data of 12 matches those reported previously.33b
1,2,3,4,6-Penta-O,S,O,O,O-acetyl-2-thio-β-D-glucopyranose (13)
Compound 13 was prepared from 12 following a reported procedure with minor modifications.34 Accordingly, to a solution of 12 (4.1 g, 8.5 mmol) in anhydrous dimethylformamide (170 mL) was added potassium thioacetate (9.7 g, 85 mmol). The resulting mixture was stirred at room temperature for 1 h under N2. After evaporation of the solvent in vacuo, the residue was mixed with methylene chloride and water. The organic layer was separated and washed sequentially with water and saturated sodium chloride, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes–EtOAc: 2:1) and recrystallized from hexanes–EtOAc. The product 13 was obtained as a white powder (3.0 g, 7.4 mmol, 87%). The 1H NMR spectrum of 13 is consistent with the literature report.34a
2-Thio-D-glucose (3)
To a solution of 13 (510 mg, 1.25 mmol) in anhydrous methanol (15 mL) was added Dowex 50W X8 (~200 mg). The reaction was gently refluxed for 15 h. The resulting solution was filtered and the filtrate was concentrated under reduced pressure to give an α:β mixture of 1-O-methyl-2- thio-D-glucopyranose (α:β = 1:3, determined by 1H NMR spectroscopy). The mixture was dissolved in water (20 mL) and stirred at 80 °C in the presence of Dowex 50W X8 for 20 h under N2. The resulting solution was filtered and lyophilized (260 mg). A part of the residue (100 mg) was used for further purification by silica gel column chromatography (water–acetonitrile: 1:50). The product 3 was obtained as a white powder (30 mg, 0.15 mmol, 32%). In solution, 3 exists as a mixture of α and β isomers (α:β = 9:10, under the 1H NMR conditions). 1H NMR (500 MHz, D2O) δ 5.13 (d, J = 3.3 Hz, 1H, 1α-H), 4.55 (d, J = 8.6 Hz, 1H, 1β-H), 3.75 (dd, J = 12.3, 2.3 Hz, 1H, 6β1-H), 3.74 (m, 1H, 5α-H), 3.69 (dd, J = 12.3, 2.4 Hz, 1H, 6α1-H), 3.62 (dd, J = 12.3, 5.1 Hz, 1H, 6α2-H), 3.58 (dd, J = 12.3, 5.9 Hz, 1H, 6β2-H), 3.48 (dd, J = 10.7, 9.0 Hz, 1H, 3α-H), 3.34 (ddd, J = 9.7, 5.9, 2.3 Hz, 1H, 5β2-H), 3.27 (m, 1H, 3β-H), 3.26 (m, 1H, 4α-H), 3.23 (m, 1H, 4β-H), 2.75 (dd, J = 10.7, 3.3 Hz, 1H, 2α-H), 2.57 (dd, J = 10.2, 8.6 Hz, 1H, 2β-H); 13C NMR (125 MHz, D2O) δ 98.2 (1β-C), 94.5, (1α-C), 77.2 (3β-C), 76.7 (5β-C), 74.6 (3α-C), 72.8 (5α-C), 71.5 (4α-C), 71.2 (4β-C), 61.6 (6β-C), 61.4 (6α-C), 48.5 (2β-C), 45.8 (2α-C); HRMS (ESI+) calcd for C6H12O5NaS+ [M + Na]+ 219.0298, found 219.0303. (ESI−) calcd for C6H11O5S− [M − H]− 195.0333, found 195.0330.
BexG2 in vitro Activity Assay
As depicted in Scheme 2, a typical BexG2 assay mixture (0.1 mL) contained 2-thio-D-glucose (3, 1 mM), ATP (1 mM), UDP-D-glucose (15, 1 mM), MgCl2 (10 mM), hexokinase (5 μg) and BexG2 (16 μM) in 50 mM Tris•HCl buffer, pH 8.0. The substrate for BexG2, 2-thio-D-glucose 6-phosphate (14), was produced in situ from 3 and ATP by hexokinase. The reaction mixture was incubated at 30 °C for 1.5 h and filtered through an Amicon ultrafiltration unit equipped with a YM-10 membrane to remove the proteins. HPLC analysis was performed using a Dionex CarboPac PA1 analytical column (4 × 250 mm). The sample was eluted with a gradient of water (solvent A) and 1 M NH4OAc (solvent B). The gradient was ran from 30 to 50% B over 10 min, 50 to 80% B over 5 min, with a 5 min wash at 80% B, and 80 to 30% B over 5 min, followed by re-equilibration at 30% B for 10 min. The flow rate was 1 mL/min, and the detector was set at 276 nm.
Scheme 2.

Isolation and Characterization of BexG2 Product (16)
A large scale BexG2 reaction (1 mL) containing 2-thio-D-glucose (3, 10 mM), ATP (12 mM), UDP-D-glucose (15, 12 mM), MgCl2 (3 mM), hexokinase (20 μg) and BexG2 (30 μM) in 100 mM Tris•HCl buffer, pH 8.0, was carried out. The reaction mixture was incubated at 30 °C for 6.5 h and filtered through an Amicon ultrafiltration unit equipped with a YM-10 membrane to remove the proteins. HPLC analysis and purification were performed using a Dionex CarboPac PA1 semi-preparative column (9 × 250 mm). The sample (330 μL × 3) was eluted with 0.1 M NH4OAc at 5 mL/min. The resulting flow through was split to a collection fraction (4.85 mL/min) and a detection fraction (0.15 mL/min) by a splitter. The elution was monitored by a Corona CAD. The retention time of 16 was 10.4 min under the HPLC conditions. The fraction containing 16 was collected and lyophilized. 1H NMR (500 MHz, D2O) δ 5.11 (d, J = 3.5 Hz, 1H, 1-H), 5.06 (d, J = 3.8 Hz, 1H, 1′−H), 3.96 (m, 2H, 61,2-H), 3.93 (m, 1H, 5′−H), 3.85 (m, 1H, 5-H), 3.76 (dd, J = 12.5, 2.2 Hz, 1H, 6′1-H), 3.68 (m, 1H, 3′−H), 3.66 (m, 1H, 3-H), 3.65 (m, 1H, 6′2-H), 3.53 (dd, J = 9.9, 3.8 Hz, 1H, 2′−H), 3.44 (dd, J = 9.9, 9.2 Hz, 1H, 4-H), 3.34 (dd, J = 10.1, 9.2 Hz, 1H, 4′−H), 2.90 (dd, J = 10.8, 3.5 Hz, 1H, 2-H); 13C NMR (125 MHz, D2O) δ 94.8 (1-C), 93.4 (1′−C), 73.8 (3-C), 72.9 (5′−C), 72.7 (3′−C), 71.9 (5-C), 71.1 (2′−C), 70.4 (4-C), 69.8 (4′−C), 63.8 (6-C), 60.8 (6′−C), 44.5 (2-C); HRMS (ESI−) calcd for C12H22O13SP− [M − H]− 437.0524, found 437.0520.
Results and Discussion
Production, Isolation and Identification of BE-7585A
To determine that the A. orientalis subsp. vinearia BA-07585 strain can produce BE-7585A (1) in our hand and to verify the assigned structure, the bacterial strain was grown in ISP-2 medium. The major product, which is red, was purified and subjected to spectroscopic analyses. The HRMS results (HRMS (ESI+) calcd for C37H46O20NaS [M+Na]+ 865.2201, found 865.2208; HRMS (ESI−) calcd for C37H45O20S [M–H]−, 841.2225; found, 841.2222) are consistent with the reported structure. Further verification of the structure of 1 relied on a series of NMR experiments (see Figures S1-6 for 1H and 13C NMR spectra).35 The 13C chemical shift of C-2′ at δ 52.4 is further upfield than that typically expected for an O-methine carbon and thus argues for the presence of a thiol substituent at this position rather than a hydroxyl group. The connectivity of the three sugar moieties in the structure is established by HMBC analysis. The results (listed in Table S1)35 clearly reveal the presence of a 1′,1″-O-glycosyl bond between the 2-thioglucose and the glucose moieties (see Figure 2). The 3JH-H values (3.1–3.5 Hz) of the anomeric protons (H-1′, H-1″ and H-1‴) and the NOESY experiment established an α,α-linkage between the disaccharide and an anomeric α-configuration of the rhodinose moiety (2) to C-12b of the aglycone can also be deduced. The observed HMBC correlation between H-2′ and C-5 implicates a thioether linkage between the 2-thiosugar and the aglycone. These results clearly distinguish the structure of BE-7585A (1) from its constitutional isomer, rhodonocardin A (5), and thus exclude the possibility that 1 is identical to rhodonocardin A.
Figure 2.

Selected HMBC results for BE-7585A (1) and selected NOESY results for 16.
13C-labeling Experiments
The angucycline core structures are generally derived from the assembly of ten acetate units catalyzed by type II polyketide synthases (PKSs).36 To confirm the biosynthetic origin of the ring carbons of BE-7585A (1), [1-13C]acetate was added into a growing A. orientalis culture. The 13C-labeled product was purified and analyzed by 13C NMR spectroscopy. Enhanced signals for C-1, C-3, C-4a, C-6, C-7, C-8, C-10, C-11a and C-12a of the ring structure of 1 are observed, indicating these carbons are derived from C-1 of acetate (Scheme 3, and see Figure S7).35 To determine the orientation of these acetate units during assembly, [1,2-13C2]acetate was also utilized in the feeding experiment. The 13C–13C spin couplings were observed for C-1/C-2, C-3/C-13, C-4a/C-12b, C-5/C-6, C- 6a/C-7, C-7a/C-8, C-9/C-10, C-11/C-11a and C-12/C-12a (see Figure S8),35 indicating that these pairs of carbons are derived from intact acetate units. This assignment was confirmed by the INADEQUATE spectrum (see Figure S9).35 Thus, the position and orientation of acetate units in the angucycline core structure of 1 were unambiguously determined.
Scheme 3.

Interestingly, the acetate incorporation pattern observed here is unusual for angucycline class natural products (see Scheme 3). As exemplified by the biosynthesis of urdamycin A (17),37 vineomycin A1,38 PD116740,39 kinamycin D40 and gilvocarcin V (30),41 in the typical ring closure reaction the linear decaketide (18) formed through the action of PKS is cyclized into the benz[a]anthracene backbone (19) in a manner in which the head (C-20) and the tail (C-2) of the decaketide chain coincide with C-13 and C-2, respectively, in the cyclic product (18 → 19). A decarboxylation of the last acetate unit (at C-2 to release C-1) is a necessary step in this pathway to account for the incorporation pattern. While such a mode of skeletal assembly involving simple folding and condensation is found for most augucycline-type natural products studied thus far, an alternative pathway has been found in the biosynthesis of PD116198 (32, see Scheme 4) from Streptomyces phaeochromogenes WP 3688.42 In this case, chain elongation and ring condensation leads to a linearly fused tetracyclic anthracyclinone intermediate (20). Subsequent oxidative bond cleavage between C-10a and C-11 followed by rearrangement and recylcization are proposed to give the angular angucyclinone core (20 → 21, Scheme 3). Since BE- 7585A displays the same acetate incorporation pattern as PD116198 (32), an analogous ring reconstruction via an enzyme-catalyzed Baeyer-Villiger oxidation may also be operative in the biosynthesis of 1. Similar oxidative C–C bond cleavages of polyketide-derived tetracyclic substrates have also been proposed for the biosynthesis of gilvocarcin (30) and jadomycin (31) based on gene disruption and gene complementation experiments.43,44 Enzymes responsible for this type of reactions are flavin-dependent monooxygenases. Notably, a flavin monooxygenase (MtmOIV) catalyzing the Baeyer-Villiger ring opening in the biosynthesis of mithramycin (33) was recently characterized and its crystal structure determined.45,46
Scheme 4.

Identification of the Gene Cluster
As mentioned above, urdamycin A (17)47 is an angucycline class antibiotic having a similar structure as BE-7585A (1). It is composed of a benz[a]anthraquinone core, two L-rhodinose (2) and two D-olivose moieties, with one of the rhodinosyl groups attached at the C-12b position, as seen in the structure of 1. The gene cluster of 17 has been identified and sequenced, and the biosynthetic pathway of 17 has been studied.48 In view of the close resemblance between 17 and 1, homologous genes encoding the type II polyketide synthase (PKS), the aglycone tailoring enzymes, TDP-rhodinose biosynthetic enzymes, and a rhodinosyltransferase found in the urdamycin gene cluster are expected to exist in the gene cluster of 1 as well. To locate the BE-7585A biosynthetic gene cluster, a cosmid library was constructed from the genomic DNA of A. orientalis. This library was screened using PCR probes designed based on multiple sequence alignment of several KSαs, hexose 2,3- dehydratases and hexose 3-dehydrases found in the gene clusters of 17 and several other related compounds. Five cosmids were found to harbor these genes. Sequencing of these cosmids led to a contiguous DNA sequence of 48.4 kb, from which 42-open reading frames (ORFs) (orf1–42) were identified (Figure 3). Among them, orf9–36 are believed to be involved in BE-7585A biosynthesis based on BLAST search (Table 2). As predicted, the cluster includes type II PKSs, post-PKS aglycone tailoring genes, and the deoxysugar biosynthetic genes. The remaining ORFs (orf1–8 and orf37–42) are likely boundary ORFs, since they appear to be the biosynthetic genes for primary metabolites (See Table S2).35 Overall, the BE-7585A biosynthetic gene cluster (bex cluster) spans a region of 28.9 kb and contains 28 ORFs. The nucleotide sequences reported herein have been deposited into the GenBank database under the accession number HM055942.
Figure 3.

Organization of the BE-7585A biosynthetic gene cluster (cluster). The gene cluster spans 28.9 kbp and contains 28 open reading frames.
Table 2.
Proposed functions of ORFs in the BE-7585A biosynthetic cluster (bex cluster).
| gene | proposed function | protein homologue and origin | identity/similarity (%) | protein accession number |
|---|---|---|---|---|
| bexR1 | regulator | repressor-response regulator [Streptomyces sp. AM-7161] | 56/68 | BAC79018 |
| bexE | oxygenase | putative oxygenase (Aur1A) [Streptomyces aureofaciens] | 75/83 | AAX57188 |
| bexF | cyclase | putative cyclase (UrdF) [Streptomyces fradiae] | 78/85 | CAA60568 |
| bexA | ketosynthase α subunit | putative ketoacyl synthase (UrdA) [Streptomyces fradiae] | 85/92 | CAA60569 |
| bexB | ketosynthase β subunit | keto-acyl synthase beta [Streptomyces antibioticus] | 74/83 | CAG14966 |
| bexC | acyl carrier protein | putative acyl carrier protein (SimA3) antibioticus] [Streptomyces | 60/74 | AAK06786 |
| bexD | ketoreductase | ketoreductase [Streptomyces antibioticus] | 81/89 | CAG14968 |
| bexL | cyclase | putative aromatase [Streptomyces sp. SCC 2136] | 70/78 | CAH10113 |
| bexM | oxygenase-reductase | oxygenase-reductase (UrdM) [Streptomyces fradiae] | 62/71 | AAF00206 |
| bexJ1 | transporter | Transporter (UrdJ2) [Streptomyces fradiae] | 47/61 | AAF00207 |
| bexN | carboxyl transferase | decarboxylase (JadN) [Streptomyces sviceus ATCC 29083] | 43/54 | ZP_05020741 |
| bexH | sugar epimerase/reductase | predicted nucleoside-diphosphate sugar epimerase [Streptosporangium roseum DSM 43021] | 48/65 | ZP_04471118 |
| bexI | oxygenase | putative anthrone monooxygenase (GilOII) [Streptomyces griseoflavus] | 27/38 | AAP69583 |
| bexK | hydroxylase | cytochrome P-450-like enzyme [Saccharopolyspora erythraea NRRL 2338] | 41/54 | YP_001107923 |
| bexO | ferredoxin | ferredoxin [Streptomyces coelicolor A3(2)] | 40/59 | NP_625075 |
| bexP | methyltransferase | putative methyltransferase [Saccharopolyspora erythraea NRRL 2338] | 37/52 | YP_001105373 |
| bexG1 | glycosyltransferase | glycosyl transferase (UrdGT1a) [Streptomyces fradiae] | 49/62 | AAF00214 |
| bexQ | sugar 3-ketoreductase | sugar 3-ketoreductase (KijD10) [Actinomadura kijaniata] | 52/65 | ACB46498 |
| bexS | sugar 4,6-dehydratase | putative dNDP-glucose 4,6-dehydratase [Streptomyces aureofaciens] | 72/80 | ACK77743 |
| bexT | sugar 3-dehydrase | NDP-hexose 3,4-dehydratase homolog [Streptomyces cyanogenus] | 78/86 | AAD13547 |
| bexU | TDP-glucose synthase | putative TDP-glucose synthase [Streptomyces nogalater] | 69/82 | AAF01820 |
| bexV | sugar 2,3-dehydratase | NDP-hexose 2,3-dehydratase [Frankia sp. CcI3] | 58/72 | YP_483211 |
| bexR2 | regulator | TetR family transcriptional regulator [Saccharopolyspora erythraea NRRL 2338] | 50/61 | YP_001103502 |
| bexW | reductase | putative reductase [Streptomyces griseoflavus] | 67/74 | AAP69587 |
| bexJ2 | transporter | EmrB/QacA family drug resistance transporter [Frankia sp. EAN1pec] | 36/53 | YP_001507963 |
| bexX | thiosugar synthase | Thiazole biosynthesis protein (ThiG) [Stigmatella aurantiaca DW4/3-1] | 58/75 | ZP_01459245 |
| bexJ3 | transporter | major facilitator superfamily transporter [Mycobacterium vanbaalenii PYR-1] | 36/50 | YP_956695 |
| bexG2 | glycosyltransferase | α,α-trehalose-phosphate synthase [Methanothermobacter thermautotrophicus str. Delta H] | 30/47 | NP_276863 |
Functional Predication of Genes Involved in Angucycline Core Formation
The deduced gene products of bexA, bexB, and bexC display high sequence similarities to the type II minimal PKS, KSα, β-ketoacyl synthase β subunit (KSβ), and acyl carrier protein (ACP), respectively. The minimal PKS (BexA, BexB and BexC) assembles 10 malonyl-CoAs from 10 acetate units into a linear decaketide chain (18). As shown in Scheme 4, to accommodate the unusual labeling pattern observed in the 13C-labeling experiments (see Scheme 3), 18 should be cyclized to a linear structure 23 rather than an angular structure (i.e., 19). On basis of the established biosynthesis of other angucycline type compounds, a ketoreductase homologue, BexD, and two cyclase homologues, BexL and BexF, are predicted to act together with PKS to construct the ring structure.36 Specifically, the reduction of the 9- keto group by BexD likely initiates the ring formation reaction catalyzed by BexL. Subsequent decarboxylation and cyclization by BexF yields 23 as a key intermediate.
The resulting ring structure 23 would then be tailored by a set of oxygenases to the angular angucyclinone core (29). Recent studies indicated that multi-oxygenase complexes are required to perform the oxidative C–C bond cleavage and ring rearrangement in both gilvocarcin (30) and jadomycin (31) biosynthesis.44,49 Since a similar oxidative ring opening process (23 → 29) likely occurs in the formation of BE-7585A, a complex of oxygenases may also be involved. A few possible candidates include a flavin-dependent oxygenase homologue, BexE, a bifunctional oxygenase-reductase homologue, BexM, and the anthrone monooxygenase homologue BexI, which displays sequence similarity to GilOII and JadG from the gilvocarcin and jadomycin pathways, respectively.44,50 Although it is difficult to define the cascade of the oxidation events solely based on sequence analysis, the identification of genes encoding these oxygenases (BexE, BexI and BexM) in the bex cluster provides important leads for future biochemical experiments to address these tailoring oxidation reactions in the biosynthesis of BE-7585A (1).
Other genes likely involved in aglycone biosynthesis are bexK, bexN and bexW. BexK is a cytochrome P450 hydroxylase homologue, which may be responsible for hydroxylation at C-2 of 28 to produce 29. This assignment is supported by an early observation that a cytochrome P450 inhibitor blocked C-2 hydroxylation in PD116198 (32) biosynthesis, implicating C-2 hydroxylation in both pathways as a P450-dependent reaction.51 BexN is an acetyl-CoA carboxylase homologue, which may be involved in supplying malonyl-CoA in PKS-catalyzed reactions. Precedence is found in the jadomycin pathway where a BexN homologue, JadN, is believed to play a similar role.52 BexW is a NADPH-dependent FMN reductase homologue, which may serve as the reductase for the flavin-dependent oxygenases involved in the aglycone biosynthesis.
Functional Predication of Genes Involved in Rhodinose Formation and Rhodinosyltransfer
Rhodinose and rhodinose-derivatives are found in several natural products whose biosynthetic gene clusters have been isolated and sequenced.27,28,53–55 Sequence comparison of the putative sugar biosynthetic genes in these clusters and those in the bex cluster allowed the identification of the rhodinose biosynthetic genes. Scheme 5 shows the correlation of their proposed functions and the chemical transformation steps required to convert glucose 1-phosphate (34), which is the biosynthetic precursor for most deoxyhexoses, to TDP-L-rhodinose (40).20b,d The pathway is initiated by the activation of 34 to TDP-glucose (35), catalyzed by a TDP-glucose synthase homologue, BexU. The second step is the conversion of 35 to 36, catalyzed by a TDP-glucose 4,6-dehydratase homologue, BexS. Subsequent C-2 deoxygnation (36 → 37) is catalyzed by a 2,3-dehydratase homologue, BexV, and a 3-ketoreductase homologue, BexQ. The resulting product 37 then undergoes C-3 deoxygenation to 38 catalyzed by a [2Fe-2S]-containing and PMP-dependent BexT, which displays good sequence homology to hexose 3-dehydrase enzymes (78% identity to LanQ from S. cyanogenus; 73% identity to SpnQ from Saccharopolyspora spinosa).26,56 While this reaction typically requires an iron-sulfur containing flavoprotein reductase for conezyme regeneration,57 no gene encoding such a specific reductase can be located in the bex cluster. It is possible that a general cellular reductase or BexO, which is a ferredoxin homologue, assumes this role.26,58
Scheme 5.

The bex cluster also contains the gene bexH that is believed to encode a sugar epimerase. Thus, a 5- epimerization reaction (38 → 39) followed by 4-ketoreduction (39 → 40) is proposed to complete the biosynthesis of TDP-L-rhodinose (40). However, no hexose 4-ketoreductase gene can be located in the bex cluster. Whether the 4-ketoreduction of 39 is mediated by a non-specific reductase in the cellular pool or BexH functions as a bifunctional epimerase-ketoreductase59 will be determined in future biochemical studies. Further support for the assignment of 40 as an L-sugar comes from another gene product in the bex cluster, BexG1. This glycosyltransferase homologue demonstrates high sequence similarity to the TDP-L-rhodinosyltransferase, UrdGT1a, from the urdamycin pathway having 49% identity and 62% similarity.60 Therefore, BexG1 is predicted to have a similar function catalyzing the transfer of the L-rhodinosyl group of 40 to the C-12b position of the angucycline core (29) to generate 41. Since the UrdGT1a reaction proceeds with inversion of stereochemistry at the anomeric center of rhodinose and both BexG1 and UrdGT1a are members of the GT-1 glycosyltransferase family of enzymes,61 BexG1 is expected to be an inverting enzyme as well. This implies that the TDP leaving group of the sugar donor should be β-oriented as shown in 40, because the glycosidic linkage of the rhodinose moiety in 41 is in the α-configuration. An epimerization at C-5 of 38 is thus necessary to fix the anomeric configuration as β in 39 and 40.
Biosynthesis of the Thiosugar Moiety
Among all orfs in the bex cluster, the bexX gene, which displays good sequence similarity to thiazole synthases, ThiGs, (58% identity to ThiG from Stigmatella aurantiaca DW4/3-1; 38% identity to ThiG from Bacillus subtilis subsp. subtilis str 168) is clearly important for the biosynthesis of the thiosugar (3). ThiG is a key enzyme involved in the formation of thiazole of thiamine. Studies of thiamine biosynthesis in B. subtilis showed that the ThiG reaction is initiated by the Schiff base formation between one of lysine residues (Lys96) of ThiG and the 2-keto group of its substrate, 1-deoxy-D-xylulose-5-phosphate (DXP, 42). As illustrated in Scheme 6, the sulfur atom is transferred from the thiocarboxylate (43) at the C-terminus of a sulfur carrier protein, ThiS, and incorporated into the ThiG-DXP adduct (44).62,63 Because of the presence of the bexX gene in the cluster, an analogous mechanism of sulfur incorporation can be proposed for the biosynthesis of the 2-thioglucose (Scheme 7). Accordingly, the putative substrate, glucose 6-phosphate (45), may form an imine adduct (46) with the active site lysine of BexX at C-1 position. Deprotonation of C-2 of 46 followed by tautomerization via an enamine 47 generates a 2-keto intermediate 48, which can then react with a nucleophilic sulfur group to incorporate a sulfur atom at C-2 in 49. The reactive sulfur donor is likely a ThiS-thiocarboxylate or a protein persulfide equivalent. Dehydration of 49 to 50 followed by C-1 deprotonation gives an enamine intermediate 51, which can tautomerize to C-1 imine 52. Upon hydrolysis, the BexX-bound product 52 is released to give 2-thioglucose 6-phosphate (53/14).
Scheme 6.

Scheme 7.

The sulfur atom of ThiS–thiocarboxylate has been shown to be derived from cysteine mediated by a cysteine desulfurase.62 In B. subtilis (and E. coli) this sulfur loading reaction also requires ThiF, which plays a role in ThiS activation.64 Unfortunately, neither ThiS homologue nor a cysteine desulfurase homologue is found in the bex cluster. However, since the substrate and the function of ThiG and BexX are analogous, it is possible that the endogenous ThiS used in thiamin biosynthesis may be recruited to serve as the sulfur donor for thiosugar biosynthesis in Amycolatopsis orientalis. Further investigation of the sulfur delivery process is in progress.
Pathway of the Disaccharide Formation and Its Transfer to the Aglycone
Once formed, the BexX product, 2-thioglucose 6-phosphate (14), is expected to accept a glucose moiety from UDP-glucose (15) to form 2-thiotrehalose 6-phosphate (16). This disaccharide moiety of BE-7585A (1) is structurally related to trehalose. The latter is commonly formed by linking the glucose moiety of UDP-glucose (15) to glucose 6-phosphate (45) and the reaction is catalyzed by trehalose 6-phosphate synthases (TPSs).65 Since the bexG2-encoded protein exhibits sequence similarity to TPSs (30% identity and 47% similarity to TPS from Methanothermobacter thermautotrophicus str. Delta H), BexG2 is likely the 2-thiotrehalose 6-phosphate synthase catalyzing the coupling of 14 and 15 to form 16, whose 6-phosphate group is removed at a later stage. In archaea, bacteria, fungi, plants and animals, the dephosphorylation of trehalose 6-phosphate to trehalose is catalyzed by trehalose 6-phosphate phosphatase (TPP).65 In fact, the TPS- and TPP-encoding genes are clustered in S. avermitilis and Saccharopolyspora erythraea genomes.66,67 Although no TPP homologous gene is found in the bex cluster or in the boundary region, the phosphate group of 16 may be hydrolyzed by an endogenous phosphatase encoded elsewhere in the genome. The hydrolysis may occur on the disaccharide 16 or after the disaccharide has been attached to the aglycone. Interestingly, a new trehalose biosynthetic route (catalyzed by TreTs), which utilizes UDP-glucose (15) and glucose instead of glucose 6-phosphate (45), was recently found.68 However, the poor sequence alignment results between BexG2 and these trehalose glycosyltransferring synthases (TreTs) renders this route less likely for the BexG2 reaction.
How the thiodisaccharide (16 or its hydrolyzed product without 6-phosphate group) is attached to C-5 of the benz[a]anthraquinone core is not clear. It is conceivable that the thioether bond formation may simply be a result of a non-enzymatic attack by the nucleophilic 2-thiol group of the disaccharide at the C-5 position of benz[a]anthraquinone (29 or 41). The C-5 site is expected to be electrophilic and thus can function as a Michael acceptor. Subsequent deprotonation at C-5 leads to a hydroquinone intermediate (54), which can be readily oxidized to the quinone form (55) by molecular oxygen (see Scheme 8). A similar path has been demonstrated in urdamycin E (56) biosynthesis.69
Scheme 8.

Regulatory and Resistant Genes
The remaining genes in the BE-7585A gene cluster are mainly regulatory and resistance genes. Among them, BexR1 and BexR2 display sequence similarity to transcriptional regulators. BexJ1, BexJ2 and BexJ3 are transporter homologues and are possibly involved in self-resistance mechanisms.70 A methyltransferase homologue, BexP, may also be involved in the self-resistance mechanism by catalyzing methylation of ribosomal RNA.
BexG2 Activity and Substrate Specificity Assay
To verify the proposed pathway for disaccharide formation, the putative glycosyltransferase, BexG2, was expressed and purified to near homogeneity (see Figure 1). The predicted substrate, 2-thioglucose 6-phosphate (14) was produced in situ from chemically synthesized 2-thioglucose (3) using hexokinase (Scheme 2). The standard reaction mixture contained 14, ATP, UDP-glucose (15), MgCl2, hexokinase, and BexG2 in Tris•HCl buffer. HPLC analysis of the reaction mixture showed the consumption of ATP and 15 and the production of ADP and UDP (Figure 4A, trace c). This observation indicated that the tandem reactions carried out by hexokinase and BexG2 were successful. In the absence of BexG2, only consumption of ATP and the production of ADP were observed (Figure 4A, trace b). Therefore, the conversion of 15 to UDP must be BexG2-dependent. When both enzymes were omitted in a control, no reaction occurred (Figure 4A, trace a). To confirm the identity of the reaction product, a Corona CAD was used to detect product formation. As shown in Figure 4B, a new peak was observed in the hexokinase-BexG2 reaction. The corresponding fraction was collected, and the isolated product was subjected to NMR and MS analyses. All data are consistent with the assigned structure, 16, which is linked by an α,α-1′,1″-glycosidic bond (see Figure 2).
Figure 4.

BexG2 activity and substrate specificity assay. (A) In situ production of 2-thioglucose 6-phosphate (14) from 2-thioglucose (3) using hexokinase was monitored by the consumption of ATP (21.4 min) and the production of ADP (16.5 min) at 276 nm. The conversion of 14 to 16 catalyzed by BexG2 was detected by the consumption of UDP-glucose (15) (6.1 min) and the production of UDP (10.5 min) at 276 nm. (a) Control reaction, no enzymes; (b) Control reaction, no BexG2; (c) The complete reaction containing 3 (1 mM), ATP (1 mM), 15 (1 mM), MgCl2 (10 mM), hexokinase (5 μg) and BexG2 (16 μM) in 50 mM Tris•HCl buffer, pH 8.0 (0.1 mL); (d) Control reaction to investigate the substrate specificity, no ATP and hexokinase. (B) Isolation of BexG2 product 16. The large scale reaction contained 3 (10 mM), ATP (12 mM), 15 (12 mM), MgCl2 (3 mM), hexokinase (20 μg) and BexG2 (30 μM) in 100 mM Tris•HCl buffer, pH 8.0 (1 mL). The resulting product 16 (10.4 min, monitored by the Corona CAD) was collected and characterized.
We then tested if 2-thioglucose (3) can be processed by BexG2. The reaction conditions were the same as described above except for the omission of hexokinase and ATP. HPLC analysis of the reaction showed that no UDP was produced (Figure 4A, trace d). Thus, it appears that BexG2 cannot process 3 as a substrate, and phosphorylation at C-6 position of 2-thioglucose is necessary for the BexG2 reaction. This is consistent with the sequence analysis of BexG2, which exhibits higher sequence similarity to trehalose phosphate synthases than to trehalose glycosyltransferring synthases (TreTs)68 as discussed above.
Conclusions
BE-7585A (1), which is an angucycline-type natural product produced by Amycolatopsis orientalis subsp. vinearia BA-07585, contains a rare 2-thioglucose (3) attached to the polyketide core. In this work, the BE-7585A biosynthetic cluster (bex cluster) containing 28 ORFs was identified by PCR-based cosmid screening of the genomic DNA of the producing strain. The cluster harbors genes typical for type II polyketide synthesis. Also contained in the cluster is a thiazole synthase (ThiG) homologue, BexX, which may catalyze the key reaction producing 2-thioglucose 6-phosphate (14) from glucose 6-phoshate (45). On the basis of sequence analysis, a biosynthetic pathway is proposed for BE-7585A including the benz[a]anthraquinone core formation, rhodinose formation, and 2-thiosugar-containing disaccharide formation. Further isotopic tracer experiments using [1-13C] and [1,2-13C2]acetate revealed that the construction of the angucycline skeleton involves an unusual oxidative ring opening and rearrangement. To obtain support for the proposed biosynthetic pathways, the disaccharide formation was demonstrated in vitro using purified enzyme BexG2 and the chemoenzymatically synthesized 2-thioglucose 6-phosphate. We have also confirmed that 2-thioglucose 6-phosphate instead of 2-thioglucose is the substrate for the BexG2 enzyme. Although the direct sulfur donor has not yet been identified, an endogenous sulfur carrier protein, such as ThiS used in thiamin biosynthesis, may be recruited for the thiosugar production. This work provided the first insights into the biosynthesis of a 2-thiosugar. Studies of the molecular mechanism of the sulfur incorporation and unusual angucycline core construction are in progress.
Supplementary Material
Acknowledgments
We are grateful to Banyu Pharmaceutical Co. (Tokyo, Japan) for providing the Amycolatopsis orientalis subsp. vinearia BA-07585 strain. We also thank David Kim for his assistance with protein purification. This work was supported in part by the National Institutes of Health Grants (GM035906).
Abbreviations
- ACP
acyl carrier protein
- BLAST
basic local alignment search tool
- ATP
adenosine 5′-triphosphate
- ADP
adenosine 5′-diphosphate
- CAD
charged aerosol detector
- COSY
correlated spectroscopy
- DXP
1-deoxy-D-xylulose-5-phosphate
- ESI
electrospray ionization
- FMN
flavin mononucleotide
- HMBC
heteronuclear multiple bond correlation
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectrometry
- HSQC
heteronuclear single quantum coherence
- INADEQUATE
incredible natural abundance double quantum transfer experiment
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- ISP
International Streptomyces Project
- KSα
β-ketoacyl synthase α subunit
- KSβ
β-ketoacyl synthase β subunit
- LB
Luria-Bertani
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced form)
- NCBI
National Center for Biotechnology Information
- NOESY
nuclear Overhauser enhancement (effect) spectroscopy
- NTA
nitrilotriacetic acid
- PCR
polymerase chain reaction
- PLP
pyridoxal-5′-phosphate
- SAM
S-adenosyl-L-methionine
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TDP
thymidine 5′-diphosphate
- TPP
trehalose 6-phosphate phosphatase
- TPS
trehalose 6-phosphate synthases
- TSB
tryptone soya broth
- UDP
uridine 5′-diphosphate
References
- 1.Rohr J, Thiericke R. Nat Prod Rep. 1992;9:103–137. doi: 10.1039/np9920900103. [DOI] [PubMed] [Google Scholar]
- 2.Okabe T, Suda H, Sato F, Okanishi M. Banyu Pharmaceutical Co., Ltd; Japan: 02–16894 A. Jpn Kokai Tokkyo Koho JP. 1990
- 3.(a) Begley TP, Xi J, Kinsland C, Taylor S, McLafferty F. Curr Opin Chem Biol. 1999;3:623–629. doi: 10.1016/s1367-5931(99)00018-6. [DOI] [PubMed] [Google Scholar]; (b) Fontecave M, Ollagnier-de-Choudens S, Mulliez E. Chem Rev. 2003;103:2149–2166. doi: 10.1021/cr020427j. [DOI] [PubMed] [Google Scholar]; (c) Mueller EG. Nat Chem Biol. 2006;2:185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]; (d) Kessler D. FEMS Microbiol Rev. 2006;30:825–840. doi: 10.1111/j.1574-6976.2006.00036.x. [DOI] [PubMed] [Google Scholar]; (e) Booker SJ, Cicchillo RM, Grove TL. Curr Opin Chem Biol. 2007;11:543–552. doi: 10.1016/j.cbpa.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Jurgenson CT, Begley TP, Ealick SE. Annu Rev Biochem. 2009;78:569–603. doi: 10.1146/annurev.biochem.78.072407.102340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Etoh H, Iguchi M, Nagasawa T, Tani Y, Yamada H, Fukami H. Agric Biol Chem. 1987;51:1819–1824. [Google Scholar]
- 5.Hoeksema H, Bannister B, Birkenmeyer RD, Kagan F, Magerlein BJ, Mackellar FA, Schroeder W, Slomp G, Herr RR. J Am Chem Soc. 1964;86:4223–4224. [Google Scholar]
- 6.Hoeksema H. J Am Chem Soc. 1964;86:4224–4225. [Google Scholar]
- 7.Lee MD, Dunne TS, Siegel MM, Chang CC, Morton GO, Borders DB. J Am Chem Soc. 1987;109:3464–3466. [Google Scholar]
- 8.Golik J, Clardy J, Dubay G, Groenewold G, Kawaguchi H, Konishi M, Krishnan B, Ohkuma H, Saitoh K, Doyle TW. J Am Chem Soc. 1987;109:3461–3462. [Google Scholar]
- 9.McDonald LA, Capson TL, Krishnamurthy G, Ding WD, Ellestad GA, Bernan VS, Maiese WM, Lassota P, Discafani C, Kramer RA, Ireland CM. J Am Chem Soc. 1996;118:10898–10899. [Google Scholar]
- 10.Oku N, Matsunaga S, Fusetani N. J Am Chem Soc. 2003;125:2044–2045. doi: 10.1021/ja0296780. [DOI] [PubMed] [Google Scholar]
- 11.Benz G, Schröder T, Kurz J, Wünsche C, Karl W, Steffens G, Pfitzner J, Schmidt D. Angew Chem Int Ed. 1982;21:527–528. [Google Scholar]
- 12.Stefanska AL, Fulston M, Houge-Frydrych CSV, Jones JJ, Warr SR. J Antibiot. 2000;53:1346–1353. doi: 10.7164/antibiotics.53.1346. [DOI] [PubMed] [Google Scholar]
- 13.Yoshikawa M, Murakami T, Yashiro K, Matsuda H. Chem Pharm Bull. 1998;46:1339–1340. doi: 10.1248/cpb.46.1339. [DOI] [PubMed] [Google Scholar]
- 14.Yoshikawa M, Morikawa T, Matsuda H, Tanabe G, Muraoka O. Bioorg Med Chem. 2002;10:1547–1554. doi: 10.1016/s0968-0896(01)00422-9. [DOI] [PubMed] [Google Scholar]
- 15.Gershenzon J, Halkier BA. Annu Rev Plant Biol. 2006;57:303–333. doi: 10.1146/annurev.arplant.57.032905.105228. [DOI] [PubMed] [Google Scholar]
- 16.Capaon RJ, MacLeod JK. J Chem Soc Chem Commun. 1987;15:1200–1201. [Google Scholar]
- 17.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. Science. 2002;297:1173–1174. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 18.Peschke U, Schmidt H, Zhang H-Z, Piepersberg W. Mol Microbiol. 1995;16:1137–1156. doi: 10.1111/j.1365-2958.1995.tb02338.x. [DOI] [PubMed] [Google Scholar]
- 19.Čermák L, Novotná J, Ságová-Marečková M, Kopecký J, Najmanová L, Janata J. Folia Microbiol. 2007;52:457–462. doi: 10.1007/BF02932104. [DOI] [PubMed] [Google Scholar]
- 20.(a) He XM, Liu H-w. Annu Rev Biochem. 2002;71:701–754. doi: 10.1146/annurev.biochem.71.110601.135339. [DOI] [PubMed] [Google Scholar]; (b) Thibodeaux CJ, Melancon CE, III, Liu H-w. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]; (c) Mendez C, Luzhetskyy A, Bechthold A, Salas JA. Curr Top Med Chem. 2008;8:710–724. doi: 10.2174/156802608784221532. [DOI] [PubMed] [Google Scholar]; (d) Thibodeaux CJ, Melancon CE, III, Liu H-w. Angew Chem Int Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Mannual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 22.Bierman M, Logan R, O’Brien K, Seno ET, Nagaraja Rao R, Schoner BE. Gene. 1992;116:43–49. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]
- 23.Han L, Yang K, Ramalingam E, Mosher RH, Vining LC. Microbiology. 1994;140:3379–3389. doi: 10.1099/13500872-140-12-3379. [DOI] [PubMed] [Google Scholar]
- 24.(a) Malpartida F, Hopwood DA. Nature. 1984;309:462–464. doi: 10.1038/309462a0. [DOI] [PubMed] [Google Scholar]; (b) Malpartida F, Hallam SE, Kieser HM, Motamedi H, Hutchinson CR, Butler MJ, Sugden DA, Warren M, McKillop C, Bailey CR, Humphreys GO, Hopwood DA. Nature. 1987;325:818–821. doi: 10.1038/325818a0. [DOI] [PubMed] [Google Scholar]
- 25.(a) Chen H, Agnihotri G, Guo Z, Que NLS, Chen X, Liu H-w. J Am Chem Soc. 1999;121:8124–8125. [Google Scholar]; (b) Draeger G, Park SH, Floss HG. J Am Chem Soc. 1999;121:2611–2612. [Google Scholar]; (c) Zhang H, White-Phillip JA, Melancon CE, III, Kwon H-j, Yu W-l, Liu H-w. J Am Chem Soc. 2007;129:14670–14683. doi: 10.1021/ja0744854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong L, Zhao Z, Liu H-w. J Am Chem Soc. 2006;128:14262–14263. doi: 10.1021/ja0649670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmeister D, Ichinose K, Domann S, Faust B, Trefzer A, Dräger, Kirschning A, Fischer C, Künzel E, Bearden DW, Rohr J, Bechthold A. Chem Biol. 2000;7:821–831. doi: 10.1016/s1074-5521(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 28.Westrich L, Domann S, Faust B, Bedford D, Hopwood DA, Bechthold A. FEMS Microbiol Lett. 1999:381–387. doi: 10.1111/j.1574-6968.1999.tb13398.x. [DOI] [PubMed] [Google Scholar]
- 29.Melancon CE, III, Liu H-w. J Am Chem Soc. 2007;129:4896–4897. doi: 10.1021/ja068254t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishikawa J, Hotta K. FEMS Microbiol Lett. 1999;174:251–253. doi: 10.1111/j.1574-6968.1999.tb13576.x. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 32.(a) Deferrari JO, Gros EG, Mastronardi IO. Carbohydr Res. 1967;4:432–434. [Google Scholar]; (b) Kováč P. Carbohydr Res. 1986;153:168–170. doi: 10.1016/s0008-6215(00)90209-x. [DOI] [PubMed] [Google Scholar]
- 33.(a) Hamacher K. Carbohydr Res. 1984;128:291–295. [Google Scholar]; (b) Pavliak V, Kováč P. Carbohydr Res. 1991;210:333–337. doi: 10.1016/0008-6215(91)80134-9. [DOI] [PubMed] [Google Scholar]
- 34.(a) Kirk BA, Knapp S. Tetrahedron Lett. 2003;44:7601–7605. [Google Scholar]; (b) Knapp S, Naughton ABJ, Jaramillo C, Pipik B. J Org Chem. 1992;57:7328–7334. [Google Scholar]
- 35.See supporting information.
- 36.Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Nat Prod Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- 37.Rohr J. J Antibiot. 1989;42:1151–1157. doi: 10.7164/antibiotics.42.1151. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka H, Omura S. J Antibiot. 1982;35:602–608. doi: 10.7164/antibiotics.35.602. [DOI] [PubMed] [Google Scholar]
- 39.Gould SJ, Cheng XC, Halley KA. J Am Chem Soc. 1992;114:10066–10068. [Google Scholar]
- 40.(a) Sato Y, Geckle M, Gould SJ. Tetrahedron Lett. 1985;26:4023–4026. [Google Scholar]; (b) Sato Y, Gould SJ. J Am Chem Soc. 1986;108:4625–4631. [Google Scholar]; (c) Seaton PJ, Gould SJ. J Am Chem Soc. 1987;109:5282–5284. [Google Scholar]
- 41.(a) Takahashi K, Tomita F. J Antibiot. 1983;36:1531–1535. doi: 10.7164/antibiotics.36.1531. [DOI] [PubMed] [Google Scholar]; (b) Carter GT, Fantini AA, James JC, Borders DB, White RJ. J Antibiot. 1985;38:242–248. doi: 10.7164/antibiotics.38.242. [DOI] [PubMed] [Google Scholar]
- 42.Gould SJ, Halley KA. J Am Chem Soc. 1991;113:5092–5093. [Google Scholar]
- 43.Liu T, Fischer C, Beninga C, Rohr J. J Am Chem Soc. 2004;126:12262–12263. doi: 10.1021/ja0467521. [DOI] [PubMed] [Google Scholar]
- 44.Rix U, Wang C, Chen Y, Lipata FM, Rix LLR, Greenwell LM, Vining LC, Yang K, Rohr J. Chembiochem. 2005;6:838–845. doi: 10.1002/cbic.200400395. [DOI] [PubMed] [Google Scholar]
- 45.Gibson M, Nur-e-alam M, Lipata F, Oliveira MA, Rohr J. J Am Chem Soc. 2005;127:17594–17595. doi: 10.1021/ja055750t. [DOI] [PubMed] [Google Scholar]
- 46.Beam MP, Bosserman MA, Noinaj N, Wehenkel M, Rohr J. Biochemistry. 2009;48:4476–4487. doi: 10.1021/bi8023509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drautz H, Zähner H, Rohr J, Zeeck A. J Antibiot. 1986;12:1657–1669. doi: 10.7164/antibiotics.39.1657. [DOI] [PubMed] [Google Scholar]
- 48.(a) Decker H, Haag S. J Bacteriol. 1995;177:6126–6136. doi: 10.1128/jb.177.21.6126-6136.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Faust B, Hoffmeister D, Weitnauer G, Westrich L, Haag S, Schneider P, Decker H, Künzel E, Rohr J, Bechthold A. Microbiology. 2000;146:147–154. doi: 10.1099/00221287-146-1-147. [DOI] [PubMed] [Google Scholar]
- 49.Fischer C, Lipata F, Rohr J. J Am Chem Soc. 2003;125:7818–7819. doi: 10.1021/ja034781q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kharel MK, Zhu L, Liu T, Rohr J. J Am Chem Soc. 2007;129:3780–3781. doi: 10.1021/ja0680515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gould S, Cheng XC. J Org Chem. 1994;59:400–405. [Google Scholar]
- 52.Wang L, McVey J, Vining LC. Microbiology. 2001;147:1535–1545. doi: 10.1099/00221287-147-6-1535. [DOI] [PubMed] [Google Scholar]
- 53.Räty K, Kunnari T, Hakala J, Mäntsälä P, Ylihonko K. Mol Gen Genet. 2000;264:164–172. doi: 10.1007/s004380000306. [DOI] [PubMed] [Google Scholar]
- 54.Ichinose K, Bedford DJ, Tornus D, Bechthold A, Bibb MJ, Revill WP, Floss HG, Hopwood DA. Chem Biol. 1998;5:647–659. doi: 10.1016/s1074-5521(98)90292-7. [DOI] [PubMed] [Google Scholar]
- 55.Olano C, Gómez C, Pérez M, Palomino M, Pineda-Lucena A, Carbajo RJ, Braña AF, Méndez C, Salas JA. Chem Biol. 2009;16:1031–1044. doi: 10.1016/j.chembiol.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 56.Thorson JS, Liu H-w. J Am Chem Soc. 1993;115:7539–7540. [Google Scholar]
- 57.(a) Hallis TM, Liu H-w. Acc Chem Res. 1999;32:579–588. [Google Scholar]; (b) He X, Agnihotri G, Liu H-w. Chem Rev. 2000;100:4615–4661. doi: 10.1021/cr9902998. [DOI] [PubMed] [Google Scholar]
- 58.Hong L, Zhao Z, Melancon CE, III, Zhang H, Liu H-w. J Am Chem Soc. 2008;130:4954–4967. doi: 10.1021/ja0771383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.(a) Chang S, Duerr B, Serif G. J Biol Chem. 1988;263:1693–1697. [PubMed] [Google Scholar]; (b) Alam J, Beyer N, Liu H-w. Biochemistry. 2004;43:16450–16460. doi: 10.1021/bi0483763. [DOI] [PubMed] [Google Scholar]
- 60.Trefzer A, Hoffmeister D, Künzel E, Stockert S, Weitnauer G, Westrich L, Rix U, Fuchser J, Bindseil KU, Rohr J, Bechthold A. Chem Biol. 2000;7:133–142. doi: 10.1016/s1074-5521(00)00079-x. [DOI] [PubMed] [Google Scholar]
- 61.Schuman B, Alfaro JA, Evans SV. Top Curr Chem. 2007;272:217–257. [Google Scholar]
- 62.Begley TP. Nat Prod Rep. 2006;23:15–25. doi: 10.1039/b207131m. [DOI] [PubMed] [Google Scholar]
- 63.Settembre EC, Dorrestein PC, Zhai H, Chatterjee A, McLafferty FW, Begley TP, Ealick SE. Biochemistry. 2004;43:11647–11657. doi: 10.1021/bi0488911. [DOI] [PubMed] [Google Scholar]
- 64.(a) Xi J, Ge Y, Kinsland C, McLafferty FW, Begley TP. Proc Natl Acad Sci USA. 2001;98:8513–8518. doi: 10.1073/pnas.141226698. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lehmann C, Begley TP, Ealick SE. Biochemistry. 2006;45:11–19. doi: 10.1021/bi051502y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Avonce N, Mendoza-Vargas A, Morett E, Iturriaga G. BMC Evol Biol. 2006;6:109. doi: 10.1186/1471-2148-6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S. Nat Biotech. 2003;21:526–531. doi: 10.1038/nbt820. [DOI] [PubMed] [Google Scholar]
- 67.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF. Nat Biotech. 2007;25:447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
- 68.(a) Qu Q, Lee SJ, Boos W. J Biol Chem. 2004;279:47890–47897. doi: 10.1074/jbc.M404955200. [DOI] [PubMed] [Google Scholar]; (b) Ryu SI, Park CS, Cha J, Woo EJ, Lee SB. Biochem Biophys Res Commun. 2005;329:429–436. doi: 10.1016/j.bbrc.2005.01.149. [DOI] [PubMed] [Google Scholar]; (c) Kouril T, Zaparty M, Marrero J, Brinkmann H, Siebers B. Arch Microbiol. 2008;190:355–369. doi: 10.1007/s00203-008-0377-3. [DOI] [PubMed] [Google Scholar]; (d) Nobre A, Alarico S, Fernandes C, Empadinhas N, da Costa MS. J Bacteriol. 2008;190:7939–7946. doi: 10.1128/JB.01055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rohr J. J Chem Soc Chem Commun. 1989;8:492–493. [Google Scholar]
- 70.Walsh C. Nature. 2000;406:775–781. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.