

Abstract
Purpose
Ocular albinism type 1 (OA1) is characterized by abnormalities in retinal pigment epithelium (RPE) melanosomes and misrouting of optic axons. The OA1 gene encodes a G-protein–coupled receptor (GPCR) that coimmunoprecipitates with the Gαi-subunit of heterotrimeric G-proteins from human melanocyte extracts. This study was undertaken to test whether one of the Gαi proteins, Gαi3, signals in the same pathway as OA1 to regulate melanosome biogenesis and axonal growth through the optic chiasm.
Methods
Adult Gαi3−/− and Oa1−/− mice were compared with their respective control mice (129Sv and B6/NCrl) to study the effects of the loss of Gαi3 or Oa1 function. Light and electron microscopy were used to analyze the morphology of the retina and the size and density of RPE melanosomes, electroretinograms to study retinal function, and retrograde labeling to investigate the size of the uncrossed optic pathway.
Results
Although Gαi3−/− and Oa1−/− photoreceptors were comparable to those of the corresponding control retinas, the density of their RPE melanosomes was significantly lower than in control RPEs. In addition, the RPE cells of Gαi3−/− and Oa1−/− mice showed abnormal melanosomes that were far larger than the largest 129Sv and B6/NCrl melanosomes, respectively. Although Gαi3−/− and Oa1−/− mice had normal results on electroretinography, retrograde labeling showed a significant reduction from control in the size of their ipsilateral retinofugal projections.
Conclusions
These results indicate that Gαi3, like Oa1, plays an important role in melanosome biogenesis. Furthermore, they suggest a common Oa1-Gαi3 signaling pathway that ultimately affects axonal growth through the optic chiasm.
X-linked OA1 is the most common form of ocular albinism, with an estimated prevalence of 1 in 50,000 live births. 1 Affected males exhibit fundus hypopigmentation, nystagmus, strabismus, foveal hypoplasia, reduced visual acuity, and abnormal crossing of the nerve fibers so that nerve fibers normally destined to project into the ipsilateral optic tract enter instead the contralateral tract.2
The OA1 gene has been isolated from both human and mouse.1,3 The transcript is expressed at high levels in RPE and skin melanocytes and encodes a G-protein–coupled receptor (GPCR) localized to melanosomes. The N-terminal of OA1 is in the melanosomal lumen, whereas its C-terminal is in the cytoplasm.4 OA1 has been suggested to be involved in the trafficking of melanosomal proteins during melanosome biogenesis and to function as a stop signal for melanosome growth; its absence in Oa1 knockout mice may result in the formation of giant melanosomes (macromelanosomes),5 also observed in skin melanocytes and RPE from OA1 patients.6
The Gαi family of proteins comprises three closely related members, Gαi1, Gαi2, and Gαi3, which share 85% to 95% of amino acid sequence identity. These proteins have been localized to intracellular membranes and have been implicated in membrane trafficking and fusion events. In particular, Gαi3 has been localized to Golgi membranes and endosomes in different tissues,7 and it is expressed in fetal and adult human RPE cells8 and in the inner neural retina.9
To determine whether Oa1 activates Gαi3 in the signal transduction cascade resulting in melanosome biogenesis, we studied Gαi3 knockout mice.10 We hypothesized that if Gαi3 and Oa1 proteins signal in the same pathway, the RPE phenotype of Gαi3−/− and Oa1−/− mice should be similar. Therefore, we analyzed the density and morphology of RPE melanosomes in knockout mice. Furthermore, we hypothesized that a defective signaling cascade, starting with the activation of Oa1 by an as yet unidentified ligand but without Gαi3, could also be responsible for the abnormal neural phenotype observed at the optic chiasm of Oa1−/− mice. Consequently, we examined the size of the uncrossed pathway in Gαi3−/− and Oa1−/− mice by counting the number of ipsilaterally projecting ganglion cells. The results presented here demonstrate that loss of Gαi3 indeed produces an RPE phenotype and a reduction of the uncrossed pathway. Our data support the idea that Gαi3 acts in the same signal transduction pathway as Oa1 to regulate melanosome biogenesis and axonal navigation at the optic chiasm.
Methods
Mice
C57BL/6NCrl (B6/NCrl) and congenic Oa1 knockout mice (Oa1−/−) were obtained from The Charles River Laboratories in the United States and Italy, respectively, and were bred at the University of the California, Santa Barbara (UCSB). The latter were confirmed at the Murine Genetic Analysis Laboratory at the University of California, Davis, to be congenic by examination of 1448 SNPs throughout the genome, with the exception of a region surrounding the Oa1 gene on the X chromosome, where 129s1/SvImJ alleles associated with the targeting vector were detected between 139 and 159 Mb. Mice deficient in Gαi3 were generated on the 129Sv background. The genotype of these mice was determined by Southern blot analysis on mouse tail genomic DNA, as described in Jiang et al.10 Wild-type 129Sv (129Sv) and congenic Gαi3 knockout (Gαi3−/−) mice were maintained and bred at the University of California, Los Angeles (UCLA). In addition, C57BL/6J (B6/J) and congenic tyrosinase-negative albino (C57BL/6-Tyrc2J; c2J) mice were obtained from The Jackson Laboratories and bred at UCSB. All experiments involving mice were carried out according to protocols approved by the UCLA and UCSB Animal Research Committees and in accordance with the ARVO Statement for use of Animals in Ophthalmic and Vision Research.
Light and Electron Microscopy
Three-month-old 129Sv, Gαi3−/−, B6/NCrl, and Oa1−/− mice were anesthetized by intraperitoneal injection of 120 mg/kg sodium pentobarbital and were perfused intracardially with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.4. Eyes were enucleated, rinsed in 0.1 M phosphate buffer, postfixed with 1% buffered osmium tetroxide, dehydrated in graded ethanol, and embedded in araldite 502. Sections for light microscopy (1 μm) were photographed with a digital camera (CoolSNAP; Roper Scientific, Duluth, GA) attached to a microscope (210; Zeiss, Thornwood, NY). Images at 100× magnification were merged with the use of software (Image-Pro Plus; Media Cybernetics, Bethesda, MD). For ultrastructural analysis, 60- to 70-nm stained sections (5% uranyl acetate and 0.4% lead citrate) were observed with an electron microscope (EM 910; Zeiss), and RPE fields were photographed with a digital camera (Keenview). The number of melanosomes and their areas in the RPE of 129Sv, Gαi3−/−, B6/NCrl, and Oa1−/− mice were analyzed on 30 random micrographs per line at 16,000× magnification using software (analySIS 2004; Soft Imaging System, Muenster, Germany). Starting at the top left of each section, fields were scanned serially to the right until the whole grid surface was covered. To determine the size and number of melanosomes, first each micrograph was loaded into the software platform (Soft Imaging System); then individual melanosomes were selected with the magic wand tool to measure the area, and the program produced two spreadsheets per micrograph containing the melanosomal count, the individual areas, and their mean size and SD. In total, 1540, 1115, 1550, and 530 melanosomes were analyzed for 129Sv, Gαi3−/−, B6/NCrl, and Oa1−/−, respectively. Images were automatically contrast enhanced with Adobe Photoshop (CS2 9.0.2; Adobe Systems Inc., San Jose, CA). Melanosome densities in the Oa1−/− and Gαi3−/− mice, and their respective control mice, were compared using Student’s t-tests (P < 0.05 was considered significant), and the distribution of melanosomes by size as a function of their frequency in control and knockout mice was displayed in histograms.
Electroretinography
Electroretinograms were performed as previously described.11 Rod-mediated responses were obtained with blue flashes (Wratten 47A; lmax = 470 nm) over an intensity range of 3.5 log units. Cone-mediated responses were obtained with white flashes on a rod-saturating background (32 cd/m2).
Horseradish Peroxidase Injections
Each 129Sv, Gαi3−/−, B6/NCrl, Oa1−/−, B6/J, and c2J mouse between 2 and 6 months of age was anesthetized with an intraperitoneal injection of tribromoethanol and mounted in a stereotaxic headholder. A craniotomy was made overlying the left cerebral cortex, and a 1-μL Hamilton syringe filled with a 40% solution of horseradish peroxidase (HRP) dissolved in 2% dimethyl sulfoxide was positioned sequentially at three rostro-caudal locations to target the left optic tract and visual nuclei. HRP solution (0.1 μL) was pressure injected at each position over a 5- to 7-minute period, and the Hamilton syringe was left in place for another 5 minutes before it was withdrawn. Sterile gel foam was placed in the craniotomy, and the skin was sutured. Mice were injected subcutaneously with sterile physiological saline (0.75 mL) and left to recover on a warm pad.
Two days later, each mouse was anesthetized intraperitoneally with a lethal dose of 120 mg/kg sodium pentobarbital and perfused intracardially with 0.9% saline, followed by 50 mL of 4% paraformaldehyde in sodium phosphate buffer (pH 7.2 at 20°C). Retinas were dissected immediately and postfixed flat in 2.5% glutaraldehyde + 1.25% paraformaldehyde in phosphate buffer for 30 minutes. HRP histochemistry on retinal wholemounts was performed as described previously.12
Retinas were examined for uniformity of labeling throughout the contralateral retina (right eye) and across the temporal crescent of the ipsilateral retina (left eye). The uncrossed pathway emerged almost entirely from this ventrotemporal region of the retina, where the density of cells was conspicuously decreased by comparison with the crossed pathway emerging from this same retinal region in the opposite eye. Occasionally, the heavily labeled uncrossed projection was observed among a sea of faintly labeled though far denser retinal ganglion cells (RGCs), indicative of HRP having spread across the midline to label the crossed projection from the same eye. All retinas showing evidence of midline spread, as well as retinas showing faint labeling or localized absence of labeling in the crossed and uncrossed pathways because of insufficient labeling of the visual targets, were eliminated from analysis.
Each labeled retina ipsilateral to the injected hemisphere was then scanned using a 40× objective on a microscope (BH2; Olympus, Tokyo, Japan). The microscope was mounted with a digital camera (DXC-390; Sony, Tokyo, Japan) and X-Y stage-encoders linked to a computer using specialized software (Bioquant Nova Prime; R&M Biometrics, Nashville, TN). The position of every retrogradely labeled cell in the ganglion cell layer (GCL) and the inner nuclear layer (INL) was plotted. Twenty-seven mice were analyzed (six 129Sv, three Gαi3−/−, four B6/NCrl, five Oa1−/−, three B6/J, and six c2J). Student’s t-tests were conducted to compare the number of uncrossed cells in the c2J, Oa1−/−, and Gαi3−/− mice with their respective control groups. Statistical significance was defined as P < 0.05.
Results
Effects of the Loss of Gαi3 or Oa1 Function on the Size, Shape, and Density of RPE Melanosomes
Light microscopy indicated that the loss of Gαi3 or of the Oa1 protein did not affect retinal morphology. Gαi3−/− and Oa1−/− retinas were structurally identical with those of their corresponding controls, 129Sv and B6/NCrl, respectively, with well-organized photoreceptors (Figs. 1a–d). Electron microscopy showed that RPE melanosomes Gαi3−/− were larger than, fewer than, and comparably shaped (elliptical and round) to 129Sv RPE melanosomes. However, their membranes were blebby (Fig. 1e, white arrows) and not as smooth as control melanosomes (Fig. 1f). The Oa1−/− RPE had also fewer melanosomes than the B6/NCrl RPE, some of them extraordinarily large and round compared with the small and elliptical B6/NCrl melanosomes (Figs. 1g, 1h). Oa1−/− melanosome membranes were smooth (Fig. 1g), unlike those of Gαi3−/− melanosomes (Fig. 1e).
Figure 1.

Appearance of photoreceptors and RPE melanosomes in Gαi3−/−, Oa1−/−, and corresponding control mice. (a–d) Light micrographs of semithin sections show that the photoreceptors of Gαi3−/− and Oa1−/− retinas have normal morphology. Scale bar, 5 μm. (e–h) Electron micrographs of ultrathin sections (scale bar, 0.1 μm) show larger melanosomes in Gαi3−/− (e) than in 129Sv RPE (f). They are oval and have blebby membranes (e, white arrows). Smooth, round, or oval macromelanosomes in Oa1−/− (g) are not present in B6/NCrl RPE (h). OS, outer segment; IS, inner segment; ONL, outer nuclear layer.
Melanosome density (number of melanosomes/RPE μm2) was evaluated in the RPE of Gαi3−/− and Oa1−/− mice and their corresponding control mice. In Gαi3−/− RPE, it was 59.5 ± 3.1 (mean ± SEM), whereas in 129Sv wild-type RPE it was 106.0 ± 4.7 (Fig. 2a). Oa1−/− mice also showed reduced melanosome density, 28.5 ± 1.5, in contrast to 74.0 ± 3.5 in B6/NCrl mice (Fig. 2c). Student’s t-test confirmed a significant effect (P < 0.0001) produced by the loss of either Gαi3 or Oa1.
Figure 2.

Density and size distribution of melanosomes in Gαi3−/− and Oa1−/− and their respective control mice. (a) Melanosome density in Gαi3−/− and 129Sv RPEs. (b) Melanosome size distributions for Gαi3−/− and 129Sv RPEs. (c) Melanosome density in Oa1−/− and B6/NCrl RPEs. (d) Melanosome size distributions for Oa1−/− and B6/NCrl RPEs. *Significant differences.
Many OA1 mutations in ocular albinism result in the formation of macromelanosomes. Similarly, the RPE cells of the Gαi3−/− mice showed very large melanosomes when compared to those of 129Sv mice. A wide range of sizes for individual melanosomes was seen in these animals, the largest reaching 10,000 nm2 (Fig. 2b). Smaller melanosomes (<1000 nm2) were more abundant in control 129Sv RPE, whereas melanosomes larger than 1000 nm2 were more frequent in Gai3−/− RPE. Of the 1115 Gai3−/− and 1540 129Sv melanosomes analyzed, we counted 126 Gai3−/− (11.3%) versus 80 129Sv (5.2%) that measured 3000 to 5000 nm2 and 48 Gai3−/− (4.3%) versus 11 129Sv (0.7%) that measured 5000 to 7000 nm2. Macromelanosomes larger than 7000 nm2 (some of them measuring close to 10,000 nm2) were observed only in the Gai3−/− RPE.
We also observed a remarkable change in the size distribution of melanosomes in Oa1−/− RPE compared with control B6/NCrl RPE (Fig. 2d). Small melanosomes (<2000 nm2) were twice as abundant in wild-type as in Oa1−/− RPE, whereas 2000 to 5000 nm2 melanosomes were comparably frequent in wild-type and Oa1−/− RPEs. In contrast, the percentage of larger melanosomes (>5000 nm2) became outstanding in the Oa1−/− RPE. Of 530 Oa1−/− and 1550 B6/NCrl melanosomes analyzed, we counted 92 Oa1−/− (17.4%) versus 89 B6/NCrl (5.7%) in the 5000 to 10,000 nm2 range and 35 Oa1−/− (6.6%) but only 3 B6/NCrl (0.2%) in the 10,000 to 15,000 nm2 range. Only Oa1−/− RPE contained macromelanosomes (61) with areas greater than 15,000 nm2.
Effects of the Loss of Gαi3 or Oa1 Function on the Electroretinogram
Electroretinographic responses obtained from Gαi3−/− and Oa1−/− mice are summarized in Figures 3a and 3b, respectively. In each panel, mean response amplitudes (±1 SD) are plotted against stimulus intensity (left to right). a-Wave amplitudes are increasingly negative, consistent with changes in signal hyperpolarization with stimulus intensity, and depolarizing b-wave amplitudes are shown as positive values. In each instance, there were no statistically significant differences between the response functions obtained from Gαi3−/− and Oa1−/− mice in comparison to age-matched normal controls for rod- and cone-mediated retinal function. Despite the observed changes within the RPE, the general functioning of the retina, as judged by the electroretinogram, appeared to be normal.
Figure 3.

Standardized electroretinography was performed in all experimental and control mice. Amplitudes and latencies of the photopic a-and b waves, flicker function, scotopic b waves, and dark-adapted maximum stimulation a- and b waves were measured. (a) Responses obtained from Gαi3−/− and control 129Sv mice. (b) Responses obtained from Oa1−/− and control B6/NCrl mice.
Effects of the Loss of Gαi3 or Oa1 Function on the Size of the Uncrossed Optic Pathway
Retrogradely labeled cells in the GCL (Fig. 4a) and the INL (Fig. 4b) were counted across the entire surface of the retina ipsilateral to the HRP injections to quantify the size of the uncrossed pathway. Before analyzing the Oa1−/− and Gαi3−/− mouse retinas, we first sought to validate this technical approach by replicating previous demonstrations of a reduction in the size of the uncrossed pathway in the tyrosinase−/− mouse.13,14
Figure 4.
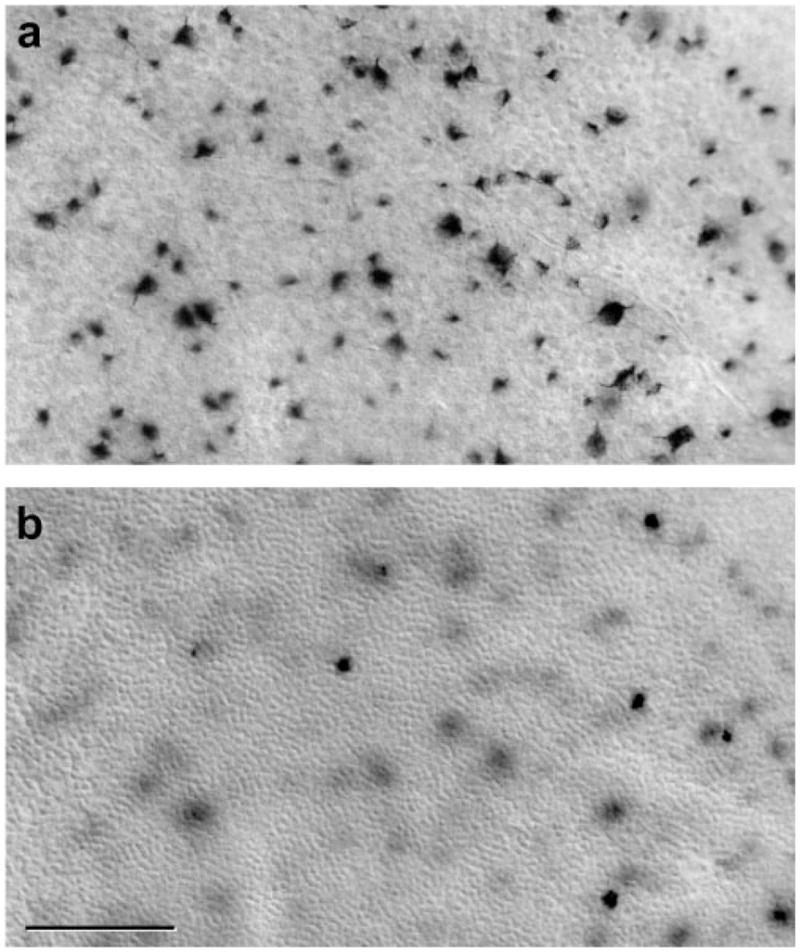
Retrogradely labeled cells in the GCL (a) and INL (b) of the ventrotemporal retina after injection of HRP into the ipsilateral optic pathway. Labeled cells were counted in c2J, B6/J, Oa1−/−, B6/NCrl, Gαi3−/−, and 129Sv mice.
Figure 5a shows two examples of plots of the distribution of labeled cells for c2J and their congenic control, B6/J mice. Labeled cells were found primarily within a crescent-shaped region positioned in the ventrotemporal retina, with some cells displaced to the INL and a small number distributed across the nasal retina. There was no obvious difference in the overall distribution of labeled cells between B6/J and c2J mice. The density of cells in the nasal retina was meager relative to that in the temporal crescent of B6/J retinas (0.9% of the total uncrossed projection), whereas it was slightly, though not significantly, higher (2.2%) in c2J retinas (Fig. 5d).
Figure 5.

(a) Retinal flatmounts showing the distribution of uncrossed RGCs in B6/J and c2J mice. (b) Total number of uncrossed RGCs in retinas of B6/J and c2J mice. (c) Proportion of uncrossed cells in B6/J and c2J displaced to the INL. (d) Proportion of uncrossed cells positioned in the nasal retina of B6/J and c2J mice. (e–h) Retinal distributions and histograms for the 129Sv and Gαi3−/− mice (conventions as in a–d). (i–l) Retinal distributions and histograms for B6/NCrl and Oa1−/− mice (conventions as in a–d). *Significant differences.
B6/J mice had 1350 ± 38.3 uncrossed RGCs, of which roughly 8% were displaced to the INL. The total number of uncrossed cells in c2J mice was significantly reduced (Fig. 5b), to 888 ± 24.8, yet the percentage of displaced cells was no different (Fig. 5c). In general, these results replicated those of Dräger and Olsen,15 but the uncrossed projection we detected was substantially larger in pigmented and albino strains and showed less variability, likely a consequence of a stereotaxic surgical approach to administer the HRP. Thus, we felt this approach should be adequately sensitive to reveal an albinolike neural defect in the Gαi3 and Oa1 knockout mice if one were present.
The six 129Sv mice studied had uncrossed projections that were different from those observed in the B6/J mice. First, the total number of uncrossed RGCs was substantially greater than in B6/J mice, averaging 1761 ± 38.1 cells (compare Figs. 5f and 5b, black bars). Second, the percentage that was displaced to the INL was conspicuously reduced, amounting to less than 3% (compare Figs. 5g and 5c, black bars).
Gαi3 knockout mice had a distribution of uncrossed cells comparable to that of 129Sv control mice (Fig. 5e). However, the total number of uncrossed cells was reduced to 1545 ± 68.2 cells (Fig. 5f), a significant reduction of 12%. The percentage of displaced cells was smaller than in c2J but not significantly different from that in 129Sv mice (Fig. 5g). The percentage of cells found in the nasal retina was also not different (Fig. 5h).
B6/NCrl mice had a consistent though somewhat smaller total number of uncrossed RGCs than B6/J (compare Figs. 5j and 5b, black bars), suggesting the presence of allelic differences through genetic drift, consistent with other demonstrations that these be regarded as substrains of the C57BL6 lineage.16,17 These mice had 1106 ± 32.4 uncrossed RGCs, with a proportion of displaced cells comparable to that observed in B6/J (compare Figs. 5k and 5c, black bars). This difference was not accounted for by any significant difference in the relative frequency of cells in the nasal retina (compare Figs. 5l and 5d, black bars), amounting to less than 1.0% of the total uncrossed projection.
As in Gai3−/− mice, the distribution of uncrossed RGCs in Oa1−/− mice was not obviously different from the distribution in control B6/NCrl mice (Fig. 5i). This included the minuscule proportion of cells in the nasal retina that was slightly higher in Oa1−/− retina (Fig. 5l) than in B6/NCrl mice. The number of uncrossed RGCs in Oa1−/− mice was similar to that found in c2J (871 ± 23.8; compare Figs. 5j and 5b, white bars) but significantly lower than in B6/NCrl mice (Fig. 5j). The proportion of displaced cells was not, however, different (Fig. 5k). Assessed as a percentage of the total uncrossed projection observed in control mice, the Oa1−/− mouse showed a neural defect like that found in the c2J mouse (34% reduction), though smaller in magnitude (21% reduction). In summary, we found reductions in the size of the uncrossed pathway in c2J Oa1−/− and Gαi3−/− mice.
Discussion
GPCRs and G-proteins are important in the regulation of a variety of biological processes, including phototransduction, neurotransmission, olfaction, secretion, chemotaxis, cell proliferation, and cell differentiation.18 Mutations in these genes are known to cause disease. Accordingly, 27 missense, 2 nonsense, 10 frameshift, and 6 splicing mutations, in addition to 18 deletions in the GPCR OA1, have been identified in patients with ocular albinism (http://albinismdb.med.umn.edu).
We investigated the potential involvement of Gαi3 in the transduction pathway controlled by Oa1 and compared in Oa1−/− and Gαi3−/− mice features commonly characterized in patients with ocular albinism: the phenotype of RPE melanosomes, the physiological response of the retina, and the size of the uncrossed retinofugal pathway. Interestingly, we found an unexpected difference in the distribution by size of melanosomes between the two control mice studied, B6/NCrl and 129Sv, stressing the importance of using, for comparison, wild-type mice that are congenic with the individual knockout mice being examined. A reduction in the density of melanosomes and the presence of abnormally large melanosomes were observed in the RPEs of Gαi3−/− and Oa1−/− mice relative to their respective control strains. These macromelanosomes were different in the two knockout strains; they were smaller in the Gαi3−/− than in the Oa1−/− RPE. Whether this difference was simply a consequence of the differences in genetic background between the strains or reflected a real difference in the signaling consequences of defective Oa1 or Gαi3 function remains to be determined. For example, Gαi3 may not be the only endogenous downstream protein that functions as a partner of Oa1 in melanosomal biogenesis. It is possible that other heterotrimeric G-proteins, such as Gαi1 and Gαi2, also interact with Oa1 because they share 85% to 95% homology with Gαi3. Alternatively, Oa1 could be activated by different unknown luminal ligands, possibly stimulating the binding and activation of as yet unidentified heterotrimeric or small G-proteins.
Another notable difference in the macromelanosomes of the Gαi3−/− and Oa1−/− RPEs is their shape—elliptical in Gαi3−/− mice and round in Oa1−/− mice. Furthermore, the Gαi3−/− macromelanosomes display blebby membranes rather than the smooth contour associated with wild-type melanosomes. This feature cannot be a simple consequence associated with the production of an abnormally large melanosome because melanosomes in the Oa1−/− RPE are far larger, yet maintain a smooth membrane. Blebby membranes have been reported in the RPE melanosomes of the pearl mouse, a model for Hermansky-Pudlak syndrome, a disorder of organelle biogenesis affecting three related organelles: melanosomes, lysosomes, and platelets.19 These fully pigmented ellipsoidal melanosomes are similar to Gαi3−/− RPE melanosomes. In the case of the Hermansky-Pudlak syndrome-pearl mutation, the gene product affected is the cytosolic protein β3A subunit of the adaptor protein-3 complex (AP3), which has been shown to function in vesicular trafficking and in docking and fusion along the pathway of melanosome formation and to bind to the trans-Golgi network and endosomal membranes. It is possible that Gαi3, as AP3, plays a role in sorting or delivering components to or from melanosomes and that its absence results in aberrant binding or fusion of vesicles containing melanin or melanosomal proteins important for the biogenesis of these pigment-specialized organelles.
Despite these variations in melanosomal biogenesis, no differences in the electroretinographic (ERG) parameters of Gαi3−/− and Oa1−/− mice, relative to their respective control mice, were observed. The function of the photoreceptors, bipolar cells, and Müller cells, therefore, is not impaired by variations in melanosome biogenesis in the RPE. The absence of an ERG phenotype in the Oa1−/− mice contrasts the findings of an earlier study in which significant reductions in a- and b-wave amplitudes were shown, suggesting that the lack of Oa1 could significantly impact retinal function.20 The reason for this discrepancy is not yet understood but may be the result of differences in ERG methodology or light exposure history for the mice used in the two studies. However, consistent with our observations in mice is the lack of ERG abnormalities in human patients with X-linked ocular albinism.21,22
Our results indicate a novel aspect of the OA1-Gαi3 signaling cascade, namely, its involvement in the correct routing of axons through the optic chiasm. It had been shown previously that oculocutaneous albinos contain a misrouting of axons at the chiasm, such that a number of axons originating in the temporal retina decussate rather than follow their normal uncrossed course.15,23–25 Similar defects in pathway navigation occur in mutants with abnormal melanosomal biogenesis.23,24,26 Abnormalities in neural crest–derived melanin, by contrast, play no role in this function.23,24 Using the sensitive retrograde labeling technique, we confirmed a significant reduction in the size of the uncrossed pathway in tyrosinase-negative mice and showed that this reduction also occurs in Gαi3−/− mice. Because the chiasmatic abnormality in Oa1−/− mice had never been assessed using retrograde labeling, we analyzed it as well, finding again a significant reduction.
Each of these differences could not be ascribed to the “atypical” components of the uncrossed pathway: the significant differences detected were not carried by the uncrossed nasal component (itself a minor proportion of the uncrossed pathway in all strains examined) and nor were they disproportionately manifested in the population displaced to the INL. The former are thought to reflect navigational errors at the chiasm unrelated to the pathway abnormalities in albinism, arising from the earliest differentiating cells near the optic nerve head, the axons of which enter the ipsilateral optic tract; most of these are eliminated during subsequent development.27 The latter are believed to reflect a migratory developmental error, failing to cross the developing inner plexiform layer yet still differentiating an axon that takes a normal uncrossed course.15 Rather, all three strains with altered RPE phenotype (loss of melanin or abnormal melanosome size, density, or morphology) showed a reduction in the number of ganglion cells in the temporal retina projecting ipsilaterally.
Despite recent advances in the understanding of the nature of the pathway signals at the optic chiasm and their reception by axons from the temporal retina,28,29 we still know relatively little about the signaling that arises from the RPE, which, in turn, affects the temporal RGCs. The present study suggests a common pathway initiated through an intramelanosomal signal working through the Oa1 receptor and activating Gαi3 in the RPE cell. Absent melanin, like defective Oa1-Gαi3 signaling, apparently produces comparable alterations within the RPE cells that are ultimately transmitted to RGCs, most likely as the latter are being born at the ventricular surface, possibly through gap junctions.30,31 The three RPE mutations considered here may have consequences that are unique to each, but only those common signals downstream of the Oa1-Gαi3 cascade should be relevant for understanding the nature of the RPE-derived signal that corrupts pathway development at the optic chiasm.
Acknowledgments
The authors thank Karstein Spicher for participating in the generation of the Gαi3−/− mice, Teresita Yang and Marcia Lloyd for their excellent technical assistance, and Novrouz Akhmedov and Clyde Yamashita for helpful discussions.
Supported by National Institutes of Health Grant EY015141 (DBF, BER, MJ), in part by the intramural Research Division of the NIH (LB), and by The Vision of Children Foundation.
Footnotes
Disclosure: A. Young, None; E.B. Powelson, None; I.E. Whitney, None; M.A. Raven, None; S. Nusinowitz, None; M. Jiang, None; L. Birnbaumer, None; B.E. Reese, None; D.B. Farber, None
References
- 1.Schiaffino MV, Bassi MT, Galli L, et al. Analysis of the OA1 gene reveals mutations in only one-third of patients with X-linked ocular albinism. Hum Mol Genet. 1995;4(12):2319–2325. doi: 10.1093/hmg/4.12.2319. [DOI] [PubMed] [Google Scholar]
- 2.King RA, Mentink MM, Oetting WS. Albinism. In: Svriver CR, Beaudet A, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 1995. pp. 4353–4392. [Google Scholar]
- 3.Newton JM, Orlow ST, Barsh GS. Isolation and characterization of the mouse homolog of the X linked ocular albinism (OA1) gene. Genomics. 1996;37(2):219–225. doi: 10.1006/geno.1996.0545. [DOI] [PubMed] [Google Scholar]
- 4.Schiaffino MV, Baschirotto C, Pellegrini G, et al. The ocular albinism type 1 gene product is a membrane glycoprotein localized to melanosomes. Proc Natl Acad Sci U S A. 1996;93(17):9055–9060. doi: 10.1073/pnas.93.17.9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Incerti B, Cortese K, Pizzigoni A, et al. OA1 knockout: new insights on the pathogenesis of ocular albinism type 1. Hum Mol Genet. 2000;9:2781–2788. doi: 10.1093/hmg/9.19.2781. [DOI] [PubMed] [Google Scholar]
- 6.O’Donnell FE, Jr, Hambrick GW, Jr, Green WR, Iliff WJ, Stone DL. X linked ocular albinism: an oculocutaneous macromelanosomal disorder. Arch Ophthalmol. 1976;94:1883–1892. doi: 10.1001/archopht.1976.03910040593001. [DOI] [PubMed] [Google Scholar]
- 7.Gohla A, Klement K, Piekorz RP, et al. An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc Natl Acad Sci U S A. 2007;104(8):3003–3008. doi: 10.1073/pnas.0611434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang M, Pandey S, Tran VT, Fong HK. Guanine nucleotide-binding regulatory proteins in retinal pigment epithelial cells. Proc Natl Acad Sci U S A. 1991;88(9):3907–3911. doi: 10.1073/pnas.88.9.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oguni M, Shinohara H, Asano T, Kato K, Setogawa T. Ontogeny of GTP-binding proteins, Gi and G (o), in rat retina. Histochem Cell Biol. 1996;106(2):235–240. doi: 10.1007/BF02484406. [DOI] [PubMed] [Google Scholar]
- 10.Jiang M, Spicher K, Boulay G, et al. Mouse gene knockout and knockin strategies in application to alpha subunits of Gi/Go family of G proteins. Methods Enzymol. 2002;344:277–298. doi: 10.1016/s0076-6879(02)44721-0. [DOI] [PubMed] [Google Scholar]
- 11.Nusinowitz S, Ridder WH, 3rd, Ramirez J. Temporal response properties of the primary and secondary rod-signaling pathways in normal and Gnat2 mutant mice. Exp Eye Res. 2007;84(6):1104–1114. doi: 10.1016/j.exer.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Reese BE, Cowey A. Large retinal ganglion cells in the rat: their distribution and laterality of projection. Exp Brain Res. 1986;61(2):375–385. doi: 10.1007/BF00239526. [DOI] [PubMed] [Google Scholar]
- 13.Guillery RW, Jeffery G, Cattanach BM. Abnormally high variability in the uncrossed retinofugal pathway of mice with albino mosaicism. Development. 1987;101:857–867. doi: 10.1242/dev.101.4.857. [DOI] [PubMed] [Google Scholar]
- 14.Rice DS, Williams RW, Goldowitz D. Genetic control of retinal projections in inbred strains of albino mice. J Comp Neurol. 1995;354:459–469. doi: 10.1002/cne.903540312. [DOI] [PubMed] [Google Scholar]
- 15.Dräger U, Olsen JF. Origins of crossed and uncrossed retinal projections in pigmented and albino mice. J Comp Neurol. 1980;191:383–412. doi: 10.1002/cne.901910306. [DOI] [PubMed] [Google Scholar]
- 16.Stiedl O, Radulovic J, Lohmann R, et al. Strain and substrain differences in context- and tone-dependent fear conditioning of inbred mice. Behav Brain Res. 1999;104:1–12. doi: 10.1016/s0166-4328(99)00047-9. [DOI] [PubMed] [Google Scholar]
- 17.Sluyter F, Marican CC, Crusio WE. Further phenotypical characterisation of two substrains of C57BL/6J inbred mice differing by a spontaneous single-gene mutation. Behav Brain Res. 1999;98:39–43. doi: 10.1016/s0166-4328(98)00049-7. [DOI] [PubMed] [Google Scholar]
- 18.McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G-protein signaling: back to the future. Cell Mol Life Sci. 2005;62(5):551–577. doi: 10.1007/s00018-004-4462-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen T, Novak EK, Kermani M, et al. Melanosome morphologies in murine models of Hermansky-Pudlak syndrome reflect blocks in organelle development. J Invest Dermatol. 2002;119:1156–1164. doi: 10.1046/j.1523-1747.2002.19535.x. [DOI] [PubMed] [Google Scholar]
- 20.Surace EM, Domenici L, Cortese K, et al. Amelioration of both functional and morphological abnormalities in the retina of a mouse model of ocular albinism following AAV-mediated gene transfer. Mol Ther. 2005;12:652–658. doi: 10.1016/j.ymthe.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Lam BL, Fingert JH, Shutt BC, et al. Clinical and molecular characterization of a family affected with X-linked ocular albinism (OA1) Ophthalmic Genet. 1997;18:175–184. doi: 10.3109/13816819709041432. [DOI] [PubMed] [Google Scholar]
- 22.Rudolph G, Meindl A, Bechmann M, et al. X-linked ocular albinism (Nettleship-Falls): a novel 29-bp deletion in exon 1: carrier detection by ophthalmic examination and DNA analysis. Graefes Arch Clin Exp Ophthalmol. 2001;239:167–172. doi: 10.1007/s004170000234. [DOI] [PubMed] [Google Scholar]
- 23.Witkop CJ, Nance EW, Rawls RF, White JG. Autosomal recessive oculocutaneous albinism in man: evidence for genetic heterogeneity. Am J Hum Genet. 1970;22:55–74. [PMC free article] [PubMed] [Google Scholar]
- 24.Sanderson KJ, Guillery RW, Shackelford RM. Congenitally abnormal visual pathways in mink (Mustela vison) with reduced retinal pigment. J Comp Neurol. 1974;154:225–248. doi: 10.1002/cne.901540302. [DOI] [PubMed] [Google Scholar]
- 25.Jeffery G. The albino retina: an abnormality that provides insight into normal retinal development. Trends Neurosci. 1997;20:165–169. doi: 10.1016/s0166-2236(96)10080-1. [DOI] [PubMed] [Google Scholar]
- 26.Kinear PE, Jay B, Witkop CJ. Albinism. Surv Ophthalmol. 1985;30:75–101. doi: 10.1016/0039-6257(85)90077-3. [DOI] [PubMed] [Google Scholar]
- 27.Godement P, Vanselow J, Thanos S, Bonhoeffer F. A study in developing visual systems with a new method of staining neurones and their processes in fixed tissue. Development. 1987;101:697–713. doi: 10.1242/dev.101.4.697. [DOI] [PubMed] [Google Scholar]
- 28.Williams SE, Mason CA, Herrera E. The optic chiasm as a midline choice point. Curr Opin Neurobiol. 2004;14:51–60. doi: 10.1016/j.conb.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Pak W, Hindges R, Lim YS, Pfaff SL, O’Leary DD. Magnitude of binocular vision controlled by islet-2 repression of a genetic program that specifies laterality of retinal axon pathfinding. Cell. 2004;119(4):567–578. doi: 10.1016/j.cell.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 30.Hayes BP. Intercellular gap junctions in the developing retina and pigment epithelium of the chick. Anat Embryol (Berl) 1977;151:325–333. doi: 10.1007/BF00318934. [DOI] [PubMed] [Google Scholar]
- 31.Tibber MS, Becker D, Jeffery G. Levels of transient gap junctions between the retinal pigment epithelium and the neuroblastic retina re influenced by catecholamines and correlate with patterns of cell production. J Comp Neurol. 2007;503:128–134. doi: 10.1002/cne.21388. [DOI] [PubMed] [Google Scholar]