

Abstract
The onset of spontaneous seizures in the pilocarpine model of epilepsy causes a hyperpolarized shift in the voltage-dependent activation of hyperpolarization-activated cyclic nucleotide-gated (HCN) channel-mediated current (Ih) in CA1 hippocampal pyramidal neuron dendrites, contributing to neuronal hyperexcitability and possibly to epileptogenesis. However, the specific mechanisms by which spontaneous seizures cause downregulation of HCN channel gating are yet unknown. We asked whether the seizure-dependent downregulation of HCN channel gating was due to altered phosphorylation signaling mediated by the phosphatase calcineurin (CaN) or the kinase p38 mitogen-activated protein kinase (p38 MAPK). We first found that CaN inhibition upregulated HCN channel gating and reduced neuronal excitability under normal conditions, showing that CaN is a strong modulator of HCN channels. We then found that an in vitro model of seizures (1 h in 0 Mg2+ and 50 μm bicuculline at 35–37°C) reproduced the HCN channel gating change seen in vivo. Pharmacological inhibition of CaN or activation of p38 MAPK partially reversed the in vitro seizure-induced hyperpolarized shift in HCN channel gating, and the shift was fully reversed by the combination of CaN inhibition and p38 MAPK activation. We then demonstrated enhanced CaN activity as well as reduced p38 MAPK activity in vivo in the CA1 hippocampal area of chronically epileptic animals. Pharmacological reversal of these phosphorylation changes restored HCN channel gating downregulation and neuronal hyperexcitability in epileptic tissue to control levels. Together, these results suggest that alteration of two different phosphorylation pathways in epilepsy contributes to the downregulation of HCN channel gating, which consequently produces neuronal hyperexcitability and thus may be a target for novel antiepileptic therapies.
Introduction
Inherited and acquired alterations in the properties of various ion channels are associated with epileptogenesis, the development of the epileptic state (Steinlein, 2004; Beck and Yaari, 2008). In hippocampal and neocortical pyramidal neurons, hyperpolarization-activated cyclic nucleotide-gated ion channels (HCN channels) are predominantly localized to the apical dendrites, where they play an important role in integrating synaptic inputs and attenuating neuronal excitability (Magee, 1998). Several studies have shown that loss or downregulation of HCN channels produce neuronal hyperexcitability and contribute to epileptogenesis after either genetic deletion or an acquired insult to the CNS (Ludwig et al., 2003; Shah et al., 2004; Zhang et al., 2006; Huang et al., 2009; Wierschke et al., 2010). We have recently described that loss of HCN channel function after pilocarpine-induced status epilepticus (SE) consists of two separate mechanisms: a loss of HCN channel expression that was independent of spontaneous seizures, and a progressive downregulation of the gating of the remaining HCN channels that was seizure-dependent (Jung et al., 2007). This seizure-dependent change in HCN channel gating produced increased neuronal excitability and was hypothesized to contribute to epileptogenesis in a positive feedback fashion often described as “seizures beget seizures” (Ben-Ari et al., 2008). However, the specific mechanisms by which spontaneous seizures cause downregulation of HCN channel gating are yet unknown.
Phosphorylation signaling modulates HCN channel properties, with prior studies showing that the gating of HCN2 and HCN4 isoforms is sensitive to inhibition of protein tyrosine kinases or the phosphatases PP1 and PP2A (Yu et al., 1995; Yu et al., 2004; Arinsburg et al., 2006). Our previous work showed that HCN channels in hippocampal pyramidal neuron dendrites (largely consisting of the HCN1 isoform) were bidirectionally modulated by p38 mitogen-activated protein kinase (p38 MAPK), with kinase activation upregulating gating, while kinase inhibition downregulated it (Poolos et al., 2006). Calcineurin (CaN, PP2B) is a Ca2+/calmodulin-dependent serine/threonine phosphatase that is highly expressed in the CA1/CA3 regions of the hippocampus and in the dentate gyrus (Yakel, 1997; Zhuo et al., 1999). CaN has been shown to be acutely upregulated after pilocarpine- or hypoxia-induced seizures (Kurz et al., 2001; Sanchez et al., 2005). Thus, we hypothesized that seizure-dependent downregulation of HCN channel gating might be due to CaN activation or loss of p38 MAPK activity.
In this study, we found that CaN inhibition upregulated HCN channel gating and decreased neuronal excitability under normal conditions, showing that CaN is a strong modulator of HCN channels. We then found that an in vitro model of seizures replicated the downregulation of HCN channel gating observed in vivo after pilocarpine-induced SE. Pharmacological inhibition of CaN or activation of p38 MAPK partially reversed the seizure-induced hyperpolarized shift in HCN channel gating both in the in vitro model and in pyramidal neurons from chronically epileptic animals. Finally, we demonstrated that there was enhanced CaN activity as well as reduced p38 MAPK activity in hippocampal area CA1 from chronically epileptic animals, and that pharmacological reversal of this abnormal phosphorylation signaling restored pyramidal neuron excitability to control levels. Together, these results suggest that alteration of two different phosphorylation pathways contribute to downregulation of HCN channel gating in an animal model of epilepsy.
Materials and Methods
Electrophysiology and data analysis.
Male Sprague Dawley rats (Charles River) were housed in a temperature-controlled vivarium on a 12 h light/dark cycle, with food and water ad libitum. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Washington, and conformed to National Institutes of Health guidelines. Hippocampal slices (400 μm) were prepared from 6- to 10-week-old rats in the manner described previously (Jung et al., 2007). In brief, after sectioning, slices were incubated for 10 min in a holding chamber at 34°C and then held at room temperature for 1 h until being moved to a submersion-type chamber at 30–32°C for recordings. The extracellular incubation and recording solutions contained the following (in mm): 125 NaCl, 25 NaHCO3, 10 dextrose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, and 2 MgCl2, pH 7.4 (bubbled with 95% O2 and 5% CO2). The pipette solution for whole-cell current-clamp recordings consisted of the following (in mm): 120 KMeSO4, 20 KCl, 10 HEPES, 4 Na2-ATP, 2 MgCl2, 0.3 Tris-GTP, 0.2 EGTA, pH 7.3 with KOH. The pipette solution for cell-attached voltage-clamp patch recordings contained the following (in mm): 120 KCl, 20 tetraethylammonium-Cl, 10 HEPES, 5 4-aminopyridine, 2 CaCl2, 1 MgCl2, 1 BaCl2, pH 7.4 with KOH. Pyramidal neurons were visualized with differential interference contrast microscopy on a fixed-stage microscope (Zeiss Axioskop). Recordings were accepted for analysis if neurons had resting potentials of at least −56 mV. Whole-cell current-clamp recordings were obtained using a Dagan BVC-700 amplifier, sampled at 10 kHz, and filtered at 2 kHz. Cell-attached patch-clamp recordings in voltage-clamp mode were obtained using an Axopatch 200B amplifier (Molecular Devices), sampled at 2 kHz, and filtered at 500 Hz. Patch pipettes were made from borosilicate glass and pulled with a Sutter P-87 micropipette puller. Pipette resistance was between 5 and 13 MΩ for cell-attached recordings or between 5 and 7 MΩ for whole-cell recordings. For cell-attached recordings, pipettes were coated with Sylgard (Dow Corning) to reduce pipette capacitance. Data from whole-cell recordings were used if the series resistance was <40 MΩ. Voltage-dependent activation of Ih was measured by normalizing the peak tail current amplitudes to a −40 mV voltage command after steady-state activation by various command voltages. This current–voltage relationship was fitted with a Boltzmann function to calculate the half-maximal activation voltage (V1/2). All data collection and analysis were performed with custom software written for the Igor Pro 4.09 analysis environment (Wavemetrics). EPSPs were simulated by the use of current injections in the form of an α function: I = Imax(αt)(1 − e−αt), where α = 1.67 and Imax was set to produce a waveform peak of ∼2 mV. This waveform produces a reasonable approximation of an EPSP using current injection. Temporal summation (TS) was calculated as the ratio of the peak amplitude of the fifth response to that of the first. Membrane potentials were not corrected for calculated liquid junction potentials of 2 mV (cell-attached recordings) or 9 mV (whole-cell recordings). All chemicals were obtained from Sigma or Fisher Scientific unless otherwise stated. ZD7288 (Tocris Bioscience) and bicuculline (Axxora) were prepared from an aqueous stock solution and applied in the bath. For stock solutions, anisomycin, FK506 and A23187 were first dissolved in DMSO and applied in the bath such that the final DMSO concentration was <0.1%.
Pilocarpine model.
The course of pilocarpine-induced SE has been previously described (Arida et al., 1999; Jung et al., 2007). Briefly, male Sprague Dawley rats (Charles River) weighing between 150 and 200 g were given scolpolamine methylnitrate (1 mg/kg, i.p.; Sigma) 30 min before intraperitoneal administration of pilocarpine hydrochloride (385 mg/kg; Sigma) to induce SE. Within the first 15 min after pilocarpine administration, animals exhibited intense salivation, immobility, facial twitching, eye blinking, and head nodding. After 15–60 min, animals showed increased head tremors with forelimb clonus, rearing and falling with generalized convulsions, consistent with SE. After an hour in SE, seizures were terminated with diazepam (DZ; 12 mg/kg, i.p.; Hospira) delivered every 20 min as needed. Animals were observed for 24 h after termination of seizures. To prevent dehydration, Lactated Ringer's solution was subcutaneously injected as needed in the first 24 h.
In vitro seizure model.
Hippocampal slices (400 μm) from naive animals weighing between 150 and 200 g were perfused with modified extracellular recording solution containing 0 Mg2+ and 50 μm bicuculline for 1 h at 35–37°C to provoke seizure-like events (SLEs). During perfusion, extracellular recordings were performed in stratum pyramidale with patch pipettes made from borosilicate glass that were pulled with a Sutter P-87 micropipette puller and filled with a 2 m NaCl solution. Recordings were not DC-filtered, which allowed measurement of slow current sinks or sources. To exclude effects due to temperature, a sham condition was included in which slices were kept in a chamber at 35–37°C for 1 h, but with extracellular solution containing normal Mg2+ and no bicuculline. Following the perfusion, the bath was switched to normal solution and temperature was reduced to 30–32°C. Patch-clamp recordings were made within 3 h of the initial seizure or sham conditions. Each slice was monitored to ensure that SLEs were present in those recordings used for analysis.
Western blotting.
Hippocampal slices prepared from epileptic and age-matched control animals were incubated for 10 min in a holding chamber at 34°C, then held at room temperature for 1 h before incubation for 1 h with pharmacologic agents at 30–32°C. They were then immediately frozen on dry ice and stored at −80°C until processing. The CA1 subregions were then microdissected on dry ice. The tissue samples were homogenized in 300 μl of homogenization buffer containing the following (in mm): 50 Tris-HCl, pH 7.5, 50 NaCl, 10 EGTA, 5 EDTA, 2 sodium pyrophosphate, 4 para-nitrophenylphosphate (pNPP), 1 sodium orthovanadate, 1 phenylmethylsulfonyl fluoride (PMSF), 20 μg/ml leupeptin, and 4 μg/ml aprotinin (Roberson et al., 1999) and normalized for protein content using a Bradford assay. The samples from the experimental and control groups were loaded and run on a 10% acrylamide gel (Bio-Rad Laboratories). Gels were then blotted electrophoretically to nitrocellulose membranes (Bio-Rad) with a transfer tank (transfer buffer: 192 mm glycine, 25 mm Tris at pH 8.3) maintained at 4°C. They were then blocked for an hour at room temperature in blocking buffer (10 mm Tris-HCl at pH 7.5, 150 mm NaCl, 0.05% Tween 20, 5% powdered milk, and 1 mm sodium orthovanadate). The nitrocellulose membrane was incubated with either phospho-DARPP-32 or phospho-p38 MAPK antibodies (Cell Signaling Technology) at a dilution of 1:500, followed by incubation in anti-rabbit secondary antibody (Cell Signaling Technology) at a dilution of 1:500–1000, and visualized by enhanced chemiluminescence (ECL; Fisher Scientific) and film exposure (blue basic autorad film; ISC BioExpress). The membrane was then reprobed using anti-β-tubulin III antibody (1:20,000; Sigma), followed by incubation in anti-mouse secondary antibody (Invitrogen) at a dilution of 1:20,000. To detect levels of total DARPP-32 or p38 MAPK, the nitrocellulose membrane was stripped (Restore PLUS Western Blot Stripping Buffer; Fisher Scientific) and reprobed using anti-DARPP-32 or p38 MAPK antibodies (1:500; Cell Signaling Technology), followed by incubation in anti-rabbit secondary antibody at a dilution of 1:500–1000. Densitometric analysis of visualized bands was conducted with a desktop scanner and National Institutes of Health ImageJ software, and normalized to β-tubulin III immunoreactivity. All densitometric analysis measured bands from three different protein loading amounts and required a linear fit between signal intensity and protein amount (including the origin), so to verify that signal detection was in the linear range of the visualization protocol. Densitometry results are reported as the ratio of the slope of the linear fits under test and control conditions.
All of the values are presented as the means ± SE. Statistical significance was determined by Student's t test (unpaired, two-tailed) or a one-way ANOVA with subsequent Tukey's post hoc analysis.
Results
CaN downregulates HCN channel gating under normal conditions
In this study, we hypothesized that the seizure-dependent downregulation of HCN channel gating in epilepsy might be due either to CaN activation or to loss of p38 MAPK activity. Since there were no prior reports of CaN modulation of HCN channels, we first asked whether CaN affected voltage-dependent activation of HCN channel-mediated current (Ih) and neuronal excitability under normal conditions. We preincubated hippocampal slices from naive animals with FK506 (1 μm), a CaN inhibitor, for a minimum of 30 min, and then performed cell-attached patch-clamp recordings in the apical dendrites of CA1 hippocampal pyramidal neurons. Ih voltage-dependent activation from FK506-treated slices showed a significantly depolarized shift compared with control (half-activation voltage, V1/2 in FK506: −78 ± 2.8 mV, n = 8; control: −89 ± 1.3 mV, n = 22, p < 0.01) (Fig. 1A), implying an increase in activated HCN channels at resting potential through CaN inhibition. Conversely, application of A23187 (5 μm), a nonspecific CaN activator, caused a significant hyperpolarized shift in the voltage-dependent activation of Ih compared with control (V1/2 in A23187: −103 ± 3.0 mV, n = 10; control: −89 ± 1.3 mV, n = 22, p < 0.01) (Fig. 1A), suggesting a downregulation of HCN channel gating through CaN activation. The dendritic recording distances from the soma in FK506- and A23187-treated slices were similar to those in control slices (175 ± 3.7 μm, n = 8; 180 ± 6.9 μm, n = 8; 180 ± 8.7 μm, n = 22, respectively).
Figure 1.

CaN inhibition upregulates Ih activation in dendrites of CA1 hippocampal pyramidal neurons from naive animals. A, Ih voltage-dependent activation after treatment with CaN inhibitor FK506 (open circles) was depolarized compared with control (solid circles), while treatment with a nonspecific CaN activator, A23187, produced a hyperpolarized shift (open triangles). Representative current traces shown are in response to voltage commands of −88, −90, and −92 mV in pyramidal neuron dendrites from control, FK506-treated, and A23187-treated slices, respectively. The dendritic recording distances were 220, 200, and 180 μm, respectively. The approximate dendritic recording location is shown in the representation of a CA1 hippocampal pyramidal neuron. B, Ih amplitudes at maximal activation, obtained after CaN inhibition by FK506 or activation by A23187, were not significantly different from control. Representative current traces shown are in response to voltage commands of −155, −150, and −154 mV in pyramidal neuron dendrites from control, FK506-treated, and A23187-treated slices, respectively. The dendritic recording distances were 180, 200, and 180 μm from the soma, respectively.
Ih amplitudes in pyramidal neuron dendrites, measured at hyperpolarized potentials where voltage-dependent activation was maximal (∼−150 mV), were compared from FK506- and A23187-treated slices, and were not significantly different from control (FK506, 37 ± 8.4 pA, n = 8; A23187, 39 ± 3.5 pA, n = 10; control, 40 ± 6.5 pA, n = 22, p > 0.05 compared by one-way ANOVA with Tukey's post hoc analysis) (Fig. 1B), demonstrating that CaN has an effect on channel gating rather than channel density or conductance. The patch was ruptured to determine the resting potential immediately after cell-attached patch recording. The resting potentials in pyramidal neuron dendrites from FK506- and A23187-treated slices were −60 ± 1.1 mV (n = 6) and −64 ± 1.0 mV (n = 6), respectively, which were similar to control (−65 ± 1.8 mV, n = 11). It is likely that calcineurin affects a variety of ion channels that might influence resting potential, such as KATP channels (Wilson et al., 2000); therefore modulation of CaN activity may have conflicting effects that result in little net change in resting membrane potential. Together, these results showed that CaN is a strong modulator of HCN channel gating, with CaN activation downregulating HCN channel gating in a manner opposite to its upregulation by p38 MAPK (Poolos et al., 2006).
CaN inhibition reduces neuronal excitability via upregulation of HCN channel gating
We then determined how upregulation of HCN channel gating by CaN inhibition affected the excitability of CA1 hippocampal pyramidal neurons. Two parameters sensitive to the amount of Ih active at rest, input resistance (IR) and TS of EPSP-like voltage transients, were examined through current-clamp recordings in pyramidal neuron dendrites in FK506-treated slices from naive animals. For current-clamp recordings, KMeSO4 solution was used for the pipette internal solution because K-gluconate blocks Ih and some K+ currents (Velumian et al., 1997). The steady-state IR was determined in response to hyperpolarizing current pulses (100–300 pA) applied from a holding potential of −65 mV. A depolarizing “sag” in the voltage response to hyperpolarizing current indicated the presence of Ih in pyramidal neuron dendrites (Fig. 2A) (Magee, 1998).
Figure 2.

CaN inhibition decreases neuronal excitability in CA1 hippocampal pyramidal neurons from naive animals. All measurements were performed with resting potential held at −65 mV. A, Current-clamp recording from pyramidal neuron dendrites showed that the mean IR from FK506-treated neurons was significantly reduced compared with control (**p < 0.01). Representative traces in response to 100 pA hyperpolarizing current injection are shown. The dendritic recording distances in control and FK506-treated neurons were 180 and 170 μm, respectively. IR from control and FK506-treated neurons in the presence of HCN channel blocker, ZD7288 (ZD), were increased and similar to each other, implying that the reduction in IR by FK506 was HCN channel dependent. B, TS of five 20 Hz α-function current injections in pyramidal neuron dendrites was also significantly reduced in FK506-treated slices compared with control (**p < 0.01). Representative traces show dendritic current-clamp recordings at 160 μm in both control and FK506-treated neurons. TS from control and FK506-treated neurons in the presence of ZD7288 were increased and similar to each other, implying that the reduction in TS by FK506 was HCN channel dependent. C, AP firing from dendritic depolarizing current injections (500 ms) showed decreased excitability in pyramidal neuron dendrites from FK506-treated neurons compared with control (*p < 0.05). Representative traces show dendritic backpropagating AP firing from dendritic depolarizing current injection (500 pA) at recording distances in control and FK506-treated neurons of 170 and 160 μm, respectively.
The IR in CA1 pyramidal neuron dendrites from FK506-treated slices was significantly lower than under control conditions (FK506, 35 ± 2.8 MΩ, n = 11; control, 51 ± 3.4 MΩ, n = 20, p < 0.01) (Fig. 2A), consistent with upregulation of HCN channel gating (Fig. 1A). To determine whether this change in IR was dependent on HCN channels, ZD7288 (20 μm), a blocker of HCN channels, was applied. ZD7288 caused a similar increase in IR compared with slices treated with both FK506 and ZD7288 (FK506+ZD7288, 135 ± 16 MΩ, n = 9; ZD7288, 143 ± 11 MΩ, n = 10, p > 0.05) (Fig. 2A), demonstrating that FK506 decreases IR in an HCN channel-dependent manner.
We then tested whether TS of EPSP-like voltage transients was affected by CaN inhibition using dendritic current injection of α-function waveforms to approximate EPSPs (as described in Methods). Ih reduces temporal summation of EPSPs, producing its maximal effect on TS at a frequency of ∼20 Hz in CA1 pyramidal neuron dendrites (Magee, 1998; Poolos et al., 2002). Trains of five α-function currents at 20 Hz were injected into the dendrites of CA1 hippocampal pyramidal neurons, and TS was measured as the ratio of the fifth response amplitude to that of the first. TS was significantly reduced in neurons from FK506-treated slices compared with control (FK506, 96 ± 2.0%, n = 11; control, 120 ± 4.5%, n = 20, p < 0.01) (Fig. 2B). ZD7288 (20 μm) applied with FK506 caused a similar increase in TS compared with ZD7288 alone (FK506+ZD7288, 181 ± 10%, n = 9; ZD7288, 173 ± 6.9%, n = 10, p > 0.05) (Fig. 2B), showing that the FK506-induced decrease in TS was specific to HCN channels. Finally, to determine whether inhibition of CaN decreases dendritic excitability in pyramidal neuron dendrites, we investigated the relationship between dendritic current injection and backpropagating dendritic action potential (AP) firing in control and FK506-treated slices. AP firing in CA1 hippocampal pyramidal neurons from FK506-treated slices showed significantly lower frequency compared with control for moderate levels of current injection (Fig. 2C). The dendritic recording distance from the soma in FK506-treated neurons was similar to that in control (163 ± 3.3 μm, n = 11; 174 ± 5.1 μm, n = 18, respectively). These results suggest that CaN inhibition decreases IR and TS via upregulation of HCN channel gating, producing reduced dendritic excitability in pyramidal neurons.
Seizure-dependent downregulation of HCN channel gating in vivo can be replicated with an in vitro model of seizures
We had previously found that HCN channel gating downregulation during epileptogenesis in vivo was dependent on the occurrence of spontaneous seizures, but this led us to ask whether this phenomenon might be reproduced in vitro, so to determine whether HCN channel gating was sensitive to neuronal network hyperactivity per se as opposed to some other factor associated with the epileptic state. Also, an in vitro model of seizures would aid in the pharmacological dissection of the underlying mechanisms of HCN channel modulation. Thus, we combined conditions known from previous work to produce epileptiform activity in hippocampal slices (Bernard, 2006), replacing the usual extracellular bath perfusate with one containing 0 Mg2+ and 50 μm bicuculline, and increasing bath temperature to 35–37°C from the usual 30–32°C. These conditions produced spontaneous epileptiform activity consisting of extracellularly recorded (in stratum pyramidale) interictal-like spikes, which were brief (<100 ms) currents, and SLEs, which were prolonged (>20 s) discharges consisting of multiple spike events superimposed on a current sink (Fig. 3A). These epileptiform events were similar to those seen in other in vitro models using pharmacological means (such as low extracellular Ca2+ or Mg2+ concentration, or elevated extracellular K+ concentration, [K+]o) to produce hippocampal network hyperactivity; in these models, extracellularly recorded “interictal spikes” have correlated with burst firing of pyramidal neurons, while slow extracellular current sinks appear to result from activity-dependent increases in [K+]o (Fisher and Alger, 1984; Konnerth et al., 1986; Poolos et al., 1987; Traynelis and Dingledine, 1988).
Figure 3.

In vitro model replicates seizure-dependent downregulation of HCN channel gating. A, Representative trace shows a spontaneous SLE obtained with an extracellular recording in stratum pyramidale from the CA1 hippocampal area, in the presence of bath solution with 0 Mg2+ and 50 μm bicuculline at 35–37°C. Note prolonged inward current sink and superimposed brief spike events. B, Ih voltage-dependent activation following 1 h of in vitro seizure, control, and sham (1 h at 35–37°C bath temperature only) conditions. The in vitro seizure condition caused a significant hyperpolarization of Ih activation relative to control or sham conditions. C, Ih amplitude at maximal activation was similar across conditions.
We quantified the occurrence of these SLEs in a population of slices exposed to the in vitro seizure protocol for an hour, versus a population subjected only to normal bath perfusate at 35–37°C for an hour (“sham protocol”). We found that 68% of slices (n = 28 in total) subjected to the seizure protocol demonstrated SLEs (on average 3.2 ± 0.3 per hour, n = 19) as well as frequent interictal-like spiking. Slices exposed to the sham protocol of high perfusate temperature only infrequently (9% of total) showed an SLE (these were discarded from further analysis), and no interictal spiking.
We then analyzed HCN channel properties in dendritic cell-attached patch recordings from CA1 pyramidal neurons in slices subjected to the seizure protocol with confirmed SLEs. We compared these values to recordings taken from slices undergoing the sham protocol, in which no SLEs were observed, and to data from control, untreated slices. Similar to what was seen in vivo, dendritic recordings from slices in which SLEs were induced demonstrated a hyperpolarized shift in Ih activation compared with the sham treatment group (V1/2 in seizure group: −101 ± 2.7 mV, n = 20; in sham group: −91 ± 1.1, n = 18, p < 0.01) (Fig. 3B). Under sham conditions, Ih voltage-dependent activation was similar to control (V1/2 in control group: −89 ± 1.3 mV, n = 22, p > 0.05) (Fig. 3B). Ih amplitude was similar under control, seizure and sham conditions (control, 40 ± 6.5 pA, n = 22; seizure, 30 ± 5.3 pA, n = 22; sham, 41 ± 5.8 pA, n = 18, p > 0.05 compared by one-way ANOVA with Tukey's post hoc analysis) (Fig. 3C), thus as in vivo, was not affected by the occurrence of seizures alone (Jung et al., 2007). Further, the resting potentials from neurons exposed to the seizure condition were hyperpolarized relative to control and sham conditions (seizure, −73 ± 2.4 mV, n = 14; control, −65 ± 1.8 mV, n = 11; sham, −67 ± 1.0 mV, n = 15, p < 0.05 for seizure vs control and for seizure vs sham, one-way ANOVA with Tukey's post hoc analysis), consistent with previous in vivo results (Jung et al., 2007). The dendritic recording distances from the soma in seizure and sham conditions were similar to that in control (183 ± 9.4 μm, n = 22; 191 ± 11 μm, n = 18; 180 ± 8.7 μm, n = 22, respectively).
We then determined whether in vitro seizures affected the excitability of CA1 hippocampal pyramidal neurons. Current-clamp recordings in the dendrites of CA1 pyramidal neurons following the seizure-provoking conditions showed increased IR compared with control (seizure, 40 ± 3.6 MΩ, n = 5; control, 26 ± 1.8 MΩ, n = 5, p < 0.01) (Fig. 4A). Interestingly, there was no significant difference in TS between control and seizure conditions (seizure, 102 ± 5.6%, n = 5; control, 92 ± 1.7%, n = 5, p > 0.05) (Fig. 4B). However, this result is similar to previous observations in vivo from chronically epileptic animals (Jung et al., 2007), suggesting that changes in other voltage-gated conductances after SE may oppose the effects of downregulated HCN channels, such as the upregulation of K+ currents or tonic GABAA receptor-mediated currents (Park et al., 2006; Chen et al., 2010). In contrast to TS, AP firing in CA1 hippocampal pyramidal neurons following seizure-provoking conditions showed significantly higher frequency compared with control (Fig. 4C). Dendritic current-clamp recordings were obtained at similar distances (seizure, 204 ± 8.6 μm, n = 5; control, 212 ± 13 μm, n = 5, p > 0.05). Note that IR, TS, and AP firing under control conditions in Figure 4 were lower than those in Figure 2 because the recordings in Figure 4 were obtained at a more distal average distance from the soma (212 μm vs 174 μm) where Ih density is higher. However, comparable average recording sites were sampled between test and control conditions in each experiment.
Figure 4.

Increased neuronal excitability following in vitro SLEs. All measurements were performed with resting potential held at −65 mV. A, Dendritic current-clamp recordings in pyramidal neuron dendrites showed increased IR following in vitro SLEs compared with control. Representative current-clamp recordings in response to 300 pA hyperpolarizing current injection are shown. Dendritic recording distances in control and seizure conditions were 210 and 188 μm, respectively. B, TS of five 20 Hz α-function current injections in pyramidal neuron dendrites following the seizure-provoking conditions (thick trace) was not significantly different from TS in pyramidal neuron dendrites under control conditions (thin trace). Representative traces show dendritic current-clamp recordings at 240 and 188 μm, respectively. C, AP firing with depolarizing current injections (500 ms) showed increased excitability in pyramidal neuron dendrites following in vitro SLEs (**p < 0.01, *p < 0.05). Representative traces show increased dendritic backpropagating AP firing from dendritic depolarizing current injection (500 pA) in seizure conditions compared with control. The dendritic recording distances in control and seizure conditions were 188 and 220 μm, respectively.
These results demonstrate that seizure-dependent modulation of HCN channels can be replicated in an in vitro model of seizures, confirming that HCN channel gating changes are due to seizure discharges per se, and not to conditions specific to the pilocarpine model of epilepsy. Just as in vivo, seizure-like events in vitro produced increased IR, no change in TS, and increased neuronal excitability. And because they can be observed as early as an hour following seizure-provoking conditions, this puts an upper limit on the time course of the development of seizure-dependent HCN channel gating downregulation.
CaN inhibition or p38 MAPK activation reverses the in vitro seizure-induced downregulation of HCN channel gating
To examine the mechanism of seizure-induced downregulation of HCN channel gating, we tested the role of CaN and p38 MAPK on HCN channel gating using the in vitro model of seizures. First, hippocampal slices were incubated with FK506 (1 μm) for a minimum of 30 min following seizure-provoking conditions. Ih activation following FK506 treatment was depolarized relative to the seizure condition (seizures+FK506, V1/2 = −88 ± 3.7 mV, n = 8; seizures alone, −101 ± 2.7 mV, n = 20, p < 0.05), and similar to control (−89 ± 1.3 mV, n = 22, p > 0.05) (Fig. 5A). Thus, FK506 reversed the seizure-induced hyperpolarized shift in Ih activation to control levels, but not to the depolarized levels seen in naive slices treated with FK506 (−78 ± 2.8 mV, n = 8) (Fig. 1A), suggesting that the seizure-induced shift in HCN channel gating was not fully dependent on CaN activity.
Figure 5.
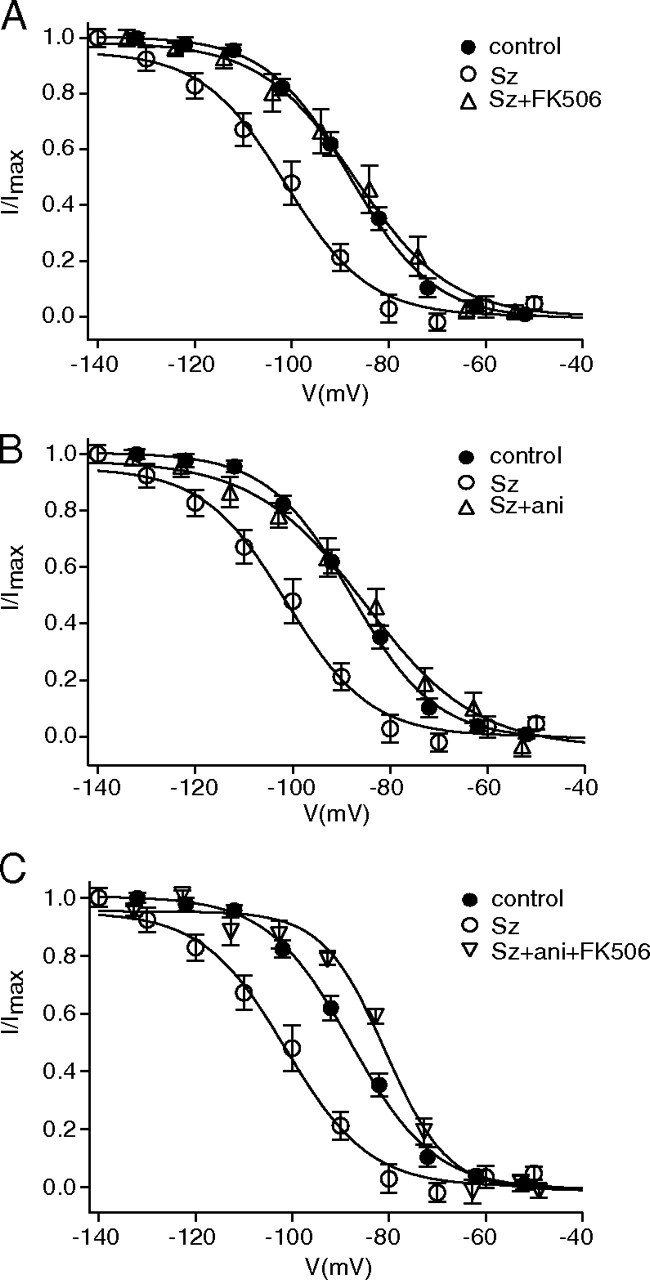
Downregulation of HCN channel gating following in vitro SLEs is partially reversed by p38 MAPK activation or CaN inhibition. A, SLEs (Sz) caused a hyperpolarized shift in Ih voltage-dependent activation. Application of CaN inhibitor FK506 following in vitro SLEs (Sz+FK506) reversed the hyperpolarized shift in Ih voltage-dependent activation to control levels. B, Application of p38 MAPK activator anisomycin following in vitro SLEs (Sz+ani) reversed the hyperpolarized shift in Ih voltage-dependent activation to control levels. C, Coapplication of FK506 and anisomycin following in vitro SLEs (Sz+ani+FK506) depolarized Ih voltage-dependent activation compared with both control and seizure conditions.
Then, we tested the role of p38 MAPK using the in vitro model of seizures. Anisomycin (20 μm) treatment following the seizure-provoking conditions reversed V1/2 to control levels (−85 ± 2.4 mV, n = 8, p < 0.01) (Fig. 5B), but not to the depolarized levels seen in naive slices treated with anisomycin (−78 ± 2.1 mV, n = 7) (Poolos et al., 2006), suggesting that, as with CaN, the seizure-induced hyperpolarized shift in Ih gating was not fully dependent on p38 MAPK activity. However, coapplication of FK506 and anisomycin following the seizure-provoking conditions shifted V1/2 to levels (−80 ± 1.3 mV, n = 7) (Fig. 5C) similar to that seen in naive slices incubated with both FK 506 and anisomycin together (−81 ± 1.6 mV, n = 7, data not shown). These results suggest that downregulation of HCN channel gating following in vitro seizure-like events may be mediated both by reduced p38 MAPK activity and by increased CaN activity as well.
Upregulation of CaN in epileptic animals
We then asked whether the pharmacological evidence for increased CaN activity as a cause of downregulated HCN channel gating following in vitro seizures was supported by biochemical evidence of increased CaN activity in vivo from epileptic animals. To measure CaN activity, we determined the phosphorylation levels of dopamine/cAMP-regulated phospho-protein-32 (DARPP-32), a direct endogenous CaN substrate (Perrino et al., 2002), by immunoblotting with a phospho-specific antibody. To validate our detection methods, we first incubated hippocampal slices from naive animals with FK506 (1 μm) for an hour, and then performed immunoblotting against phosphorylated DARPP-32 (p-DARPP-32) using CA1 hippocampal homogenates. FK506 significantly increased p-DARPP-32 levels compared with control (166 ± 25% of control, n = 7, p < 0.05) (Fig. 6A), consistent with prior results (Kurz et al., 2008), thus validating the sensitivity of our assays to measure the phosphorylation levels of DARPP-32. Then, we determined total DARPP-32 and p-DARPP-32 levels using CA1 hippocampal homogenates from epileptic animals in the chronic period 3 weeks after pilocarpine, when virtually all animals are experiencing spontaneous recurrent seizures (Jung et al., 2007). All levels of total DARPP-32 and p-DARPP-32 were first normalized to β-tubulin III as a marker of neuronal protein content. p-DARPP-32 levels from epileptic animals showed a significant reduction compared with age-matched control animals (69 ± 7.1% of control, n = 5, p < 0.05) (Fig. 6A), while levels of total DARPP-32 were unchanged compared with control (108 ± 5.4% of control, n = 5, p > 0.05). p-DARPP-32 levels normalized to total DARPP-32 levels were also decreased (ratio of p-DARPP-32 to total DARPP-32, 63 ± 5.2% of control, n = 5, p < 0.01) (Fig. 6A). These findings suggest that CaN activity is upregulated in chronically epileptic animals.
Figure 6.
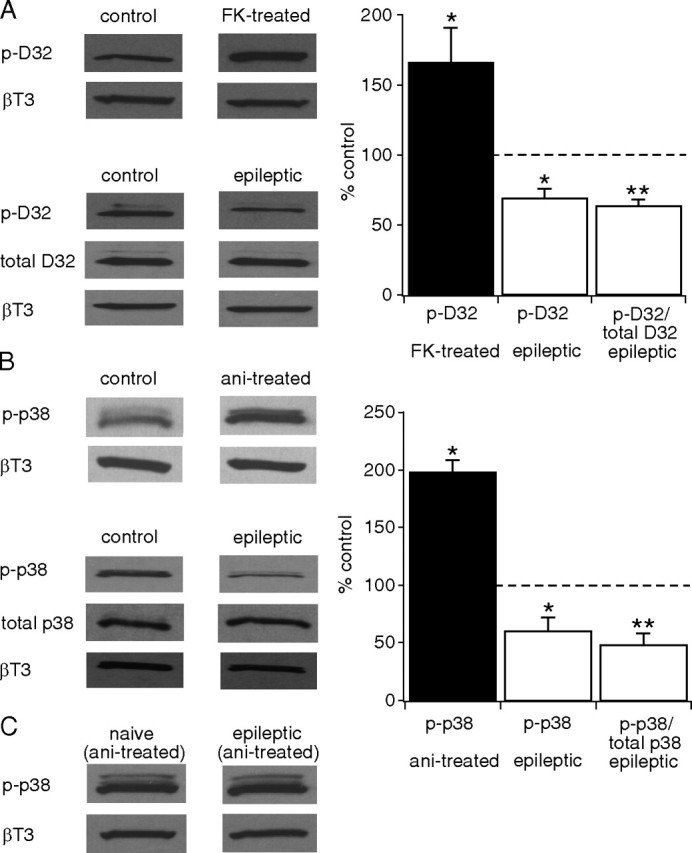
CaN activity is upregulated and p38 MAPK activity is downregulated in the CA1 hippocampal area from epileptic animals. A, Application of FK506 (FK) to hippocampal slices from naive animals increased phosphorylation levels of DARPP-32 (p-D32) compared with untreated slices (control) (*p < 0.05), showing p-DARPP-32 levels are sensitive to dephosphorylation by CaN. p-DARPP-32 levels in CA1 hippocampal tissue were decreased in epileptic animals compared with control, implying increased CaN activity (*p < 0.05). The ratio of p-DARPP-32 to total DARPP-32 was further decreased (**p < 0.01). All p-DARPP-32 and total DARPP-32 levels were normalized to β-tubulin III (βT3) protein levels. Representative Western blots of p-DARPP-32, total DARPP-32 and βT3 protein levels are shown in each condition. Statistical significance compared with control levels. B, Anisomycin (ani) treatment of hippocampal slices from naive animals caused an increase in phosphorylated p38 MAPK (p-p38) levels in the CA1 hippocampal area compared with control (*p < 0.05). p-p38 MAPK levels in CA1 hippocampal tissue were decreased in epileptic animals compared with control (*p < 0.05), implying decreased p38 MAPK activity. The ratio of p-p38 MAPK to total p38 MAPK was further decreased (**p < 0.01). All p-p38 MAPK and total p38 MAPK levels were normalized to βT3 protein levels. Representative Western blots of p-p38 MAPK, total p38 MAPK and βT3 protein levels are shown in each condition. Statistical significance compared with control levels. C, Representative Western blots show that p-p38 MAPK levels following anisomycin treatment of hippocampal slices from both naive and epileptic animals are similar to each other in the CA1 hippocampal area, implying that activation of p38 MAPK cascade by anisomycin is intact in epileptic animals.
Downregulation of p38 MAPK in epileptic animals
We then asked whether similar evidence supported a loss of p38 MAPK activity in epileptic animals. To do so, we determined the levels of phosphorylated p38 MAPK (p-p38 MAPK) from epileptic animals because p38 MAPK activity requires phosphorylation at its Thr180/Tyr182 residues by upstream kinases (Robinson et al., 1996). We first performed immunoblotting against p-p38 MAPK using hippocampal slices prepared from naive animals and preincubated with anisomycin (20 μm) for an hour to validate our detection method for changes in p-p38 MAPK levels. Anisomycin increased p-p38 MAPK levels compared with control (198 ± 11% of control, n = 4, p < 0.01) (Fig. 6B), consistent with prior results (Kang et al., 2006). Further, an analysis of p-p38 MAPK levels using CA1 hippocampal homogenates from epileptic animals at the chronic period 3 weeks after pilocarpine showed a reduction compared with age-matched control animals (60 ± 12% of control, n = 7, p < 0.05) (Fig. 6B), indicating that epilepsy is associated with a loss of p38 MAPK activity. Levels of total p38 MAPK were slightly increased compared with control (127 ± 5.0% of control, n = 5, p < 0.05), and consequently, the p-p38 MAPK level normalized by total p38 MAPK level was further decreased (ratio of p-p38 MAPK to total p38 MAPK, 48 ± 10% of control, n = 7, p < 0.01) (Fig. 6B). As with CaN immunoblotting, all levels of total p38 MAPK and p-p38 MAPK were normalized to β-tubulin III.
We then asked whether decreased p-p38 MAPK levels in epileptic animals were caused by downregulation of the upstream p38 MAPK signaling cascade (Kang et al., 2006). In Figure 6C, representative Western blots show that CA1 hippocampal p-p38 MAPK levels from anisomycin-treated hippocampal slices from both naive and epileptic animals were similar to each other (ratio of epileptic to naive, 104 ± 21%, n = 6, p > 0.05), suggesting that activation of the p38 MAPK cascade by anisomycin is intact in epileptic animals. These in vivo biochemical findings corroborate the in vitro pharmacological results, showing that the seizure-dependent downregulation of HCN channel gating in epilepsy is likely mediated both by reduced p38 MAPK activity and by increased CaN activity as well.
Pharmacological reversal of HCN channel gating downregulation in pyramidal neurons from epileptic animals
The biochemical evidence of altered phosphorylation signaling in chronically epileptic animals prompted us to investigate whether pharmacologically reversing increased CaN and decreased p38 MAPK activation would also reverse the downregulation of HCN channel gating, as occurred in the in vitro model of seizures. We prepared hippocampal slices from chronically epileptic animals, three weeks after pilocarpine, and incubated them for at least 30 min in FK506 (1 μm) before measuring Ih activation in CA1 pyramidal neuron dendrites. As shown in Figure 7A (left), blockade of increased CaN activity with FK506 partially reversed the hyperpolarized shift in Ih activation seen in chronically epileptic animals (V1/2 in epileptic+FK506, −94 ± 2.3 mV, n = 7; in epileptic, −105 ± 2.7 mV, n = 8, p < 0.05), although to a lesser extent than was seen in the in vitro model (Fig. 5A). Preincubation of hippocampal tissue from epileptic animals with the p38 MAPK activator anisomycin (20 μm) also partially reversed the hyperpolarized Ih activation (V1/2 in epileptic+ani, −93 ± 3.1 mV, n = 8; p < 0.05 compared with Ih activation in chronic epilepsy) (Fig. 7A, center). When we preincubated hippocampal tissue in both FK506 and anisomycin, the hyperpolarization of Ih activation in chronic epilepsy was reversed to a greater extent than with either drug alone, nearly reaching control levels (V1/2 in epileptic+ani+FK506, −90 ± 2.1 mV, n = 7; p < 0.01 compared with epileptic conditions) (Fig. 7A, right), but did not reach the depolarized levels seen in the in vitro model (Fig. 5C).
Figure 7.

CaN inhibition and p38 MAPK activation reverse downregulated HCN channel gating and neuronal hyperexcitability in epileptic animals. A, Application of either CaN inhibitor FK506 (epileptic+FK506) or p38 MAPK activator anisomycin (epileptic+ani) partially reversed the hyperpolarized shift in Ih voltage-dependent activation toward control levels in dendrites of CA1 hippocampal pyramidal neurons from chronically epileptic animals. Coapplication of anisomycin and FK506 (epileptic+ani+FK506) restored HCN channel gating nearly to control levels. B, Current-clamp recordings in pyramidal neuron dendrites from chronically epileptic animals showed decreased IR following coapplication of FK506 and anisomycin compared with untreated epileptic tissue. Representative dendritic current-clamp recordings in response to 100 pA hyperpolarizing current injection are shown. All recordings in B–D were performed with resting potential held at −65 mV, and dendritic recording distances in representative traces under drug-treated and untreated epileptic conditions were 180 and 170 μm, respectively. C, TS of five 20 Hz α-EPSP current injections in pyramidal neuron dendrites in epileptic tissue after treatment with anisomycin and FK506 was not significantly different from TS in pyramidal neuron dendrites under untreated epileptic conditions. D, AP firing from depolarizing dendritic current injections (500 ms) in pyramidal neurons from epileptic tissue following coapplication of FK506 and anisomycin showed significantly decreased excitability (*p < 0.05) that was similar to control levels. Representative traces show decreased dendritic backpropagating AP firing from dendritic current injection (700 pA) in drug-treated epileptic conditions compared with untreated epileptic conditions.
Ih amplitudes at maximal activation obtained from chronically epileptic animals (20 ± 4.6 pA, n = 8) were significantly reduced compared with control (40 ± 6.5 pA, n = 22, p < 0.05). Ih amplitudes obtained in epileptic tissue in the presence of FK506, anisomycin, or the two drugs together were not significantly different from those in untreated epileptic tissue (epileptic+FK506, 25 ± 2.5 pA, n = 7; epileptic+ani, 24 ± 3.2 pA, n = 8; epileptic+ani+FK506, 28 ± 7.1 pA, n = 7, p > 0.05 compared by one-way ANOVA with Tukey's post hoc analysis). The dendritic recording distances in epileptic, epileptic+FK506, epileptic+ani and epileptic+ani+FK506 conditions were 168 ± 4.5 μm (n = 8), 169 ± 4.0 μm (n = 7), 174 ± 12 μm (n = 8) and 170 ± 5.2 μm (n = 7), respectively. These results suggest that in hippocampal tissue from epileptic animals, as in the in vitro model of seizures, downregulated HCN channel gating is dependent on both CaN and p38 MAPK signaling; however pharmacological reversal of the abnormal activation of these signaling pathways in epilepsy does not drive HCN channel activation to the depolarized levels seen in neurons from nonepileptic tissue, suggesting the influence of additional signaling mechanisms on HCN channel gating in epilepsy.
Since the combination of CaN inhibition and p38 MAPK activation reversed HCN channel activation nearly to control levels, we then investigated whether these conditions would also reverse the neuronal hyperexcitability seen in epileptic tissue. We preincubated hippocampal slices from epileptic animals in FK506 and anisomycin, and then obtained dendritic whole-cell current-clamp recordings. As expected, the combination of CaN inhibition and p38 MAPK activation significantly reversed the increase in input resistance seen in CA1 pyramidal neurons from chronically epileptic animals (IR in epileptic+ani+FK506, 47 ± 2.5 MΩ, n = 5; IR in epileptic, 67 ± 6.3 MΩ, n = 10; p < 0.05) (Fig. 7B) to control levels (51 ± 3.4 MΩ, n = 20) (Fig. 2A). Consistent with prior results, temporal summation of 20 Hz α-EPSP current injection was not significantly different in chronically epileptic tissue compared with control (TS in epileptic, 116 ± 5.5%, n = 10; TS in control, 120 ± 4.5%, n = 20, p > 0.05), and was not affected by drug application (TS in epileptic+ani+FK506, 121 ± 6.0%, n = 5; p > 0.05) (Fig. 7C). We then measured action potential firing from dendritic current injection and found that the hyperexcitability seen in epileptic tissue was restored to control levels by application of FK506 and anisomycin together (Fig. 7D). The dendritic recording distances in epileptic and epileptic+ani+FK506 conditions were 177 ± 6.1 μm (n = 10) and 162 ± 8.0 μm (n = 5), respectively. The restoration of excitability to control levels was interesting, since in chronic epilepsy, HCN channel function in pyramidal neurons is compromised both by gating downregulation and an ∼60% loss of HCN channel density (Jung et al., 2007). Because drug application reverses HCN channel activation nearly to control levels, but does not alter the current through HCN channels at maximal activation (Fig. 1B) (Poolos et al., 2006), this suggests that combined CaN inhibition and p38 MAPK activation have additional actions on neuronal excitability aside from their effects on HCN channel gating, consistent with previously reported actions of these phosphorylation pathways on various ion channels (Herzig and Neumann, 2000; Misonou et al., 2004; Wittmack et al., 2005; Jin and Gereau, 2006). However, these results do give support to the idea that pharmacological reversal of abnormal phosphorylation signaling can restore neuronal hyperexcitability to normal levels, and thus could potentially exert beneficial antiepileptic effects.
Discussion
Our prior work demonstrated that during epileptogenesis in the pilocarpine model of epilepsy, HCN channels underwent a progressive downregulation of their voltage-dependent gating in concert with the gradual increase in seizure frequency (Jung et al., 2007). This change in HCN channel gating caused neuronal hyperexcitability and possibly contributed to the development or maintenance of epilepsy consistent with the “seizures-beget-seizures” hypothesis (Ben-Ari et al., 2008). Because we and others had previously found that HCN channel gating was modulated under normal conditions by kinase and phosphatase activity, we hypothesized that the seizure-dependent downregulation of HCN channel gating in epilepsy might be mediated by altered phosphorylation signaling. In the present study, we have demonstrated for the first time that the phosphatase calcineurin downregulates HCN channel gating under normal conditions. We then replicated the downregulation of HCN channel gating observed in epileptic animals using an in vitro model of seizures. The seizure-dependent downregulation of HCN channel gating was found to depend both in vitro and in epileptic tissue on CaN activation and loss of p38 MAPK activity, and we observed a concordant increase in hippocampal CaN activity and loss of p38 MAPK activity in chronically epileptic animals. Finally, pharmacological reversal of altered phosphorylation signaling restored pyramidal neuron excitability to control levels in hippocampal tissue from chronically epileptic animals. These results suggest that downregulated HCN channel gating in epilepsy is mediated at least in part by the persistent alteration of two distinct phosphorylation signaling pathways.
CaN is a strong modulator of HCN channel gating
Of the four HCN channel subtypes, HCN1 is the predominantly expressed subtype in neocortex and hippocampus (Santoro and Tibbs, 1999). Previous studies on the role of protein phosphorylation in the modulation of HCN channel properties focused on cardiac HCN2 (Yu et al., 2004) or HCN4 subtypes (Arinsburg et al., 2006), while we showed that p38 MAPK bidirectionally regulated the gating of HCN channels in hippocampal pyramidal neurons (Poolos et al., 2006). However, there are only a few reports of phosphatases modulating HCN channel properties, including the upregulation of cardiac HCN channels by inhibition of PP1 and PP2A (Yu et al., 1995) or the downregulation of HCN2 surface expression by a tyrosine phosphatase (Huang et al., 2008). We have demonstrated here for the first time that HCN channel gating in hippocampal pyramidal neurons is bidirectionally modulated by the serine-threonine phosphatase CaN (PP2B), with phosphatase activation downregulating HCN channel gating, and phosphatase inhibition upregulating it. The actions of p38 MAPK and CaN are consistent in that increased phosphorylation upregulates HCN channel gating while decreased phosphorylation downregulates it. These results show that a kinase and a phosphatase play opposing roles in modulating the properties of HCN channels in CA1 hippocampal pyramidal neurons, broadening the possible signaling mechanisms that may mediate the activity-dependent regulation of HCN channel function.
Seizure-dependent changes in HCN channel gating are replicated by an in vitro model
Using methods to provoke electrographic SLEs in hippocampal slices from naive animals, we were able to reproduce in vitro the downregulation of HCN channel gating and concomitant increase in pyramidal neuron excitability that was observed in an in vivo model of epilepsy as a consequence of spontaneous seizures (Jung et al., 2007). This suggests that the in vivo alteration of HCN channel gating is a result of episodic neuronal network hyperactivity manifested by seizures, and not of some other factor associated with the epileptic state. The modulation of dendritic HCN channel properties by in vitro SLEs occurs rapidly (within a maximum of ∼1 h), suggesting that this process most likely represents a posttranslational modification of existing HCN channels as opposed to the altered transcription of HCN channel subunits and export to the dendrites that occurs in later phases of epileptogenesis (Brewster et al., 2002). This is consistent with other studies that have demonstrated posttranslational modulation of ion channel properties by in vitro seizure-like conditions, such as the upregulation of Kv2.1 gating mediated by CaN-dependent dephosphorylation (Misonou et al., 2004), or changes in the surface expression of Kv4.2 channels (Kim et al., 2007) and GABAA receptors (Goodkin et al., 2008).
Phosphorylation signaling is altered in chronic epilepsy
Because of the results here and in prior work demonstrating the sensitivity of HCN channel gating to phosphorylation signaling, we tested the dependence of seizure-provoked changes in HCN channel gating to modulators of CaN and p38 MAPK. In the in vitro seizure model, the downregulation of HCN channel gating was partially reversed by either CaN inhibition or p38 MAPK activation, and fully reversed by the two together. In hippocampal neurons from chronically epileptic animals, CaN inhibition and p38 MAPK activation produced less pronounced effects than in the in vitro model, suggesting that other signaling pathways may contribute to downregulated HCN channel gating in vivo. Our immunoblotting analysis, however, did show increased CaN activity as well as decreased p38 MAPK activity in the CA1 hippocampus from chronically epileptic animals, corroborating our pharmacological experiments. The decrease in p38 MAPK activation occurred in conjunction with a small increase in total p38 MAPK expression, implying that altered activity of this phosphorylation pathway was due to a loss of activation by upstream signaling processes, not to altered levels of protein expression. Stimulation of the MAPK signaling cascade with anisomycin produced equivalent activation in epileptic tissue compared with tissue from naive animals, suggesting that this signaling pathway is functionally intact, and that a deficit exists either upstream of where anisomycin activates MAPK signaling, or in non-MAPK pathways (Kang et al., 2006).
These results provide novel evidence implicating persistent alteration of the CaN and p38 MAPK pathways in chronic epilepsy. Previous studies have examined only acute upregulation of CaN activity shortly after the induction of seizures by pilocarpine treatment (Kurz et al., 2001), or after hypoxia-induced seizures (Sanchez et al., 2005), while studies of acute changes in p38 MAPK activity after seizures have been more equivocal, showing either downregulation (Mielke et al., 1999) or upregulation (Jeon et al., 2000) of p38 MAPK activity within the first 72 h after kainate treatment. Our results complement those of other recent studies showing alterations of phosphorylation pathways in acquired epilepsy models, such as the mammalian target of rapamycin (mTOR), CaMKII, and ERK MAPK pathways (Churn et al., 2000; Bernard et al., 2004; Buckmaster et al., 2009; Zeng et al., 2009).
Implications for epilepsy and areas of uncertainty
A number of studies have demonstrated that HCN channels act to attenuate the intrinsic excitability of principal neurons in hippocampus and neocortex (Magee, 1999; Nolan et al., 2004; Huang et al., 2009), and that loss of expression and function of these channels occurs in epilepsy (Brewster et al., 2002; Zhang et al., 2006; Jung et al., 2007; Wierschke et al., 2010; but see Dyhrfjeld-Johnsen et al., 2009). The results presented here describe novel alterations of phosphorylation pathways that occur in a model of chronic temporal lobe epilepsy, and that diminish HCN channel function. Pharmacological reversal of altered CaN and p38 MAPK signaling restored the excitability of CA1 hippocampal pyramidal neurons to control levels. This suggests that interventions that would restore HCN channel function might have the potential of antiepileptic actions, and also raises the question of whether altered phosphorylation signaling in epilepsy presents new targets for antiepileptic therapy. In the most straightforward example of this idea, inhibition of mTOR, a pathway dysregulated in tuberous sclerosis, has been found to reduce seizure frequency in an animal model of that disease (Zeng et al., 2008). Whether p38 MAPK activation or CaN inhibition would have beneficial effects on seizures is unclear at present, given the potential number of targets affected by these pathways in addition to HCN channels. However, as additional examples of altered phosphorylation signaling in chronic epilepsy are identified, it may be possible to elucidate multiple signaling pathways that will be amenable to pharmacological intervention.
There are some areas of uncertainty in this study. Phosphorylation sites on the HCN molecule for p38 MAPK and CaN have not been mapped, therefore our results do not demonstrate direct phosphorylation of HCN channels by CaN or p38 MAPK. Thus, although CaN plays a role in dephosphorylation of Kv2.1 channels (Misonou et al., 2004), and p38 MAPK directly phosphorylates voltage-gated sodium channels (Wittmack et al., 2005), we cannot rule out the alternative possibility that CaN or p38 MAPK act through unknown effectors. It is also likely that HCN channel gating is additionally modulated by other kinases and phosphatases, or via phospholipid pathways (Zolles et al., 2006; Fogle et al., 2007). Further experiments will be necessary to evaluate these possibilities and their role in HCN channel gating downregulation in epilepsy.
Footnotes
This work was supported by grants from the National Institutes of Health to N.P.P. (NS046604, NS050229) and by the American Epilepsy Society (S.J.). We appreciate the advice of Dr. Sandy Bajjalieh on the Western blot experiments.
References
- Arida RM, Scorza FA, Peres CA, Cavalheiro EA. The course of untreated seizures in the pilocarpine model of epilepsy. Epilepsy Res. 1999;34:99–107. doi: 10.1016/s0920-1211(98)00092-8. [DOI] [PubMed] [Google Scholar]
- Arinsburg SS, Cohen IS, Yu HG. Constitutively active Src tyrosine kinase changes gating of HCN4 channels through direct binding to the channel proteins. J Cardiovasc Pharmacol. 2006;47:578–586. doi: 10.1097/01.fjc.0000211740.47960.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–369. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Crepel V, Represa A. Seizures beget seizures in temporal lobe epilepsies: the boomerang effects of newly formed aberrant kainatergic synapses. Epilepsy Curr. 2008;8:68–72. doi: 10.1111/j.1535-7511.2008.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C. In: Models of seizures and epilepsy. Pitkanen A, Schwartzkroin PA, Moshe SL, editors. Burlington, MA: Elsevier Academic; 2006. pp. 59–72. [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Brewster A, Bender RA, Chen Y, Dube C, Eghbal-Ahmadi M, Baram TZ. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci. 2002;22:4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci. 2009;29:8259–8269. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shu S, Schwartz LC, Sun C, Kapur J, Bayliss DA. Homeostatic regulation of synaptic excitability: tonic GABA(A) receptor currents replace I(h) in cortical pyramidal neurons of HCN1 knock-out mice. J Neurosci. 2010;30:2611–2622. doi: 10.1523/JNEUROSCI.3771-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churn SB, Kochan LD, DeLorenzo RJ. Chronic inhibition of Ca2+/calmodulin kinase II activity in the pilocarpine model of epilepsy. Brain Res. 2000;875:66–77. doi: 10.1016/s0006-8993(00)02623-8. [DOI] [PubMed] [Google Scholar]
- Dyhrfjeld-Johnsen J, Morgan RJ, Soltesz I. Double trouble? Potential for hyperexcitability following both channelopathic up- and downregulation of I(h) in epilepsy. Front Neurosci. 2009;3:25–33. doi: 10.3389/neuro.01.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS, Alger BE. Electrophysiological mechanisms of kainic acid-induced epileptiform activity in the rat hippocampal slice. J Neurosci 1984. 1984;4:1312–1323. doi: 10.1523/JNEUROSCI.04-05-01312.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogle KJ, Lyashchenko AK, Turbendian HK, Tibbs GR. HCN pacemaker channel activation is controlled by acidic lipids downstream of diacylglycerol kinase and phospholipase A2. J Neurosci. 2007;27:2802–2814. doi: 10.1523/JNEUROSCI.4376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABAA receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80:173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- Huang J, Huang A, Zhang Q, Lin YC, Yu HG. Novel mechanism for suppression of hyperpolarization-activated cyclic nucleotide-gated pacemaker channels by receptor-like tyrosine phosphatase-alpha. J Biol Chem. 2008;283:29912–29919. doi: 10.1074/jbc.M804205200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Walker MC, Shah MM. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci. 2009;29:10979–10988. doi: 10.1523/JNEUROSCI.1531-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SH, Kim YS, Bae CD, Park JB. Activation of JNK and p38 in rat hippocampus after kainic acid induced seizure. Exp Mol Med. 2000;32:227–230. doi: 10.1038/emm.2000.37. [DOI] [PubMed] [Google Scholar]
- Jin X, Gereau RW., 4th Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo JN, Jr, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Seit-Nebi A, Davis RJ, Han J. Multiple activation mechanisms of p38alpha mitogen-activated protein kinase. J Biol Chem. 2006;281:26225–26234. doi: 10.1074/jbc.M606800200. [DOI] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Heinemann U, Yaari Y. Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. I. Development of seizurelike activity in low extracellular calcium. J Neurophysiol. 1986;56:409–423. doi: 10.1152/jn.1986.56.2.409. [DOI] [PubMed] [Google Scholar]
- Kurz JE, Sheets D, Parsons JT, Rana A, Delorenzo RJ, Churn SB. A significant increase in both basal and maximal calcineurin activity in the rat pilocarpine model of status epilepticus. J Neurochem. 2001;78:304–315. doi: 10.1046/j.1471-4159.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- Kurz JE, Moore BJ, Henderson SC, Campbell JN, Churn SB. A cellular mechanism for dendritic spine loss in the pilocarpine model of status epilepticus. Epilepsia. 2008;49:1696–1710. doi: 10.1111/j.1528-1167.2008.01616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:848. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- Mielke K, Brecht S, Dorst A, Herdegen T. Activity and expression of JNK1, p38 and ERK kinases, c-Jun N-terminal phosphorylation, and c-jun promoter binding in the adult rat brain following kainate-induced seizures. Neuroscience. 1999;91:471–483. doi: 10.1016/s0306-4522(98)00667-8. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Perrino BA, Wilson AJ, Ellison P, Clapp LH. Substrate selectivity and sensitivity to inhibition by FK506 and cyclosporin A of calcineurin heterodimers composed of the alpha or beta catalytic subunit. Eur J Biochem. 2002;269:3540–3548. doi: 10.1046/j.1432-1033.2002.03040.x. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Mauk MD, Kocsis JD. Activity-evoked increases in extracellular potassium modulate presynaptic excitability in the CA1 region of the hippocampus. J Neurophysiol. 1987;58:404–416. doi: 10.1152/jn.1987.58.2.404. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Bullis JB, Roth MK. Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogen-activated protein kinase. J Neurosci. 2006;26:7995–8003. doi: 10.1523/JNEUROSCI.2069-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, Cheng M, Khokhlatchev A, Ebert D, Ahn N, Guan KL, Stein B, Goldsmith E, Cobb MH. Contributions of the mitogen-activated protein (MAP) kinase backbone and phosphorylation loop to MEK specificity. J Biol Chem. 1996;271:29734–29739. doi: 10.1074/jbc.271.47.29734. [DOI] [PubMed] [Google Scholar]
- Sanchez RM, Dai W, Levada RE, Lippman JJ, Jensen FE. AMPA/kainate receptor-mediated downregulation of GABAergic synaptic transmission by calcineurin after seizures in the developing rat brain. J Neurosci. 2005;25:3442–3451. doi: 10.1523/JNEUROSCI.0204-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Tibbs GR. The HCN gene family: molecular basis of the hyperpolarization-activated pacemaker channels. Ann N Y Acad Sci. 1999;868:741–764. doi: 10.1111/j.1749-6632.1999.tb11353.x. [DOI] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinlein OK. Genetic mechanisms that underlie epilepsy. Nat Rev Neurosci. 2004;5:400–408. doi: 10.1038/nrn1388. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Velumian AA, Zhang L, Pennefather P, Carlen PL. Reversible inhibition of IK, IAHP, Ih and ICa currents by internally applied gluconate in rat hippocampal pyramidal neurones. Pflugers Arch. 1997;433:343–350. doi: 10.1007/s004240050286. [DOI] [PubMed] [Google Scholar]
- Wierschke S, Lehmann TN, Dehnicke C, Horn P, Nitsch R, Deisz RA. Hyperpolarization-activated cation currents in human epileptogenic neocortex. Epilepsia. 2010;51:404–414. doi: 10.1111/j.1528-1167.2009.02275.x. [DOI] [PubMed] [Google Scholar]
- Wilson AJ, Jabr RI, Clapp LH. Calcium modulation of vascular smooth muscle ATP-sensitive K+ channels—role of protein phosphatase-2B. Circ Res. 2000;87:1019–1025. doi: 10.1161/01.res.87.11.1019. [DOI] [PubMed] [Google Scholar]
- Wittmack EK, Rush AM, Hudmon A, Waxman SG, Dib-Hajj SD. Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J Neurosci. 2005;25:6621–6630. doi: 10.1523/JNEUROSCI.0541-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakel JL. Calcineurin regulation of synaptic function: from ion channels to transmitter release and gene transcription. Trends Pharmacol Sci. 1997;18:124–134. doi: 10.1016/s0165-6147(97)01046-8. [DOI] [PubMed] [Google Scholar]
- Yu H, Chang F, Cohen IS. Pacemaker current i(f) in adult canine cardiac ventricular myocytes. J Physiol. 1995;485:469–483. doi: 10.1113/jphysiol.1995.sp020743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HG, Lu Z, Pan Z, Cohen IS. Tyrosine kinase inhibition differentially regulates heterologously expressed HCN channels. Pflugers Arch. 2004;447:392–400. doi: 10.1007/s00424-003-1204-y. [DOI] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Peng BW, Sanchez RM. Decreased Ih in hippocampal area CA1 pyramidal neurons after perinatal seizure-inducing hypoxia. Epilepsia. 2006;47:1023–1028. doi: 10.1111/j.1528-1167.2006.00574.x. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, Kandel ER. A selective role of calcineurin α in synaptic depotentiation in hippocampus. Proc Natl Acad Sci U S A. 1999;96:4650–4655. doi: 10.1073/pnas.96.8.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolles G, Klöcker N, Wenzel D, Weisser-Thomas J, Fleischmann BK, Roeper J, Fakler B. Pacemaking by HCN channels requires interaction with phosphoinositides. Neuron. 2006;52:1027–1036. doi: 10.1016/j.neuron.2006.12.005. [DOI] [PubMed] [Google Scholar]