

Abstract
Deposition of islet amyloid polypeptide (IAPP) as islet amyloid in type 2 diabetes contributes to loss of β-cell function and mass, yet the mechanism for its occurrence is unclear. Neprilysin is a metallopeptidase known to degrade amyloid in Alzheimer disease. We previously demonstrated neprilysin to be present in pancreatic islets and now sought to determine whether it plays a role in degrading islet amyloid. We used an in vitro model where cultured human IAPP (hIAPP) transgenic mouse islets develop amyloid and thereby have increased β-cell apoptosis. Islet neprilysin activity was inhibited or up-regulated using a specific inhibitor or adenovirus encoding neprilysin, respectively. Following neprilysin inhibition, islet amyloid deposition and β-cell apoptosis increased by 54 and 75%, respectively, whereas when neprilysin was up-regulated islet amyloid deposition and β-cell apoptosis both decreased by 79%. To determine if neprilysin modulated amyloid deposition by cleaving hIAPP, analysis of hIAPP incubated with neprilysin was performed by mass spectrometry, which failed to demonstrate neprilysin-induced cleavage. Rather, neprilysin may act by reducing hIAPP fibrillogenesis, which we showed to be the case by fluorescence-based thioflavin T binding studies and electron microscopy. In summary, neprilysin decreases islet amyloid deposition by inhibiting hIAPP fibril formation, rather than degrading hIAPP. These findings suggest that targeting the role of neprilysin in IAPP fibril assembly, in addition to IAPP cleavage by other peptidases, may provide a novel approach to reduce and/or prevent islet amyloid deposition in type 2 diabetes.
Keywords: Amyloid, Insulin Secretion, Pancreas, Pancreatic Islet, Peptidases, Islet Amyloid, Islet Amyloid Polypeptide, Neprilysin, Peptide Degradation, Type 2 Diabetes
Introduction
Type 2 diabetes is a progressive disease in which the formation of islet amyloid deposits occurs in over 90% of subjects (1). Islet amyloid deposition involves aggregation of the normally soluble β-cell peptide, islet amyloid polypeptide (IAPP4 or amylin), and contributes to the deterioration of β-cell function and reduced β-cell mass observed in the disease (1). Given that IAPP is co-secreted with insulin (2), factors such as insulin resistance that increase insulin secretion also result in elevated IAPP secretion (3–5) and, when occurring in conjunction with β-cell dysfunction, can enhance IAPP aggregation (1). Likewise, a decrease in the clearance of IAPP can potentially accelerate amyloidogenesis, as has been suggested by studies of insulin degrading enzyme (IDE), where inhibition of IDE activity reduced degradation of synthetic IAPP peptide and increased its aggregation as amyloid (6, 7).
Recently we demonstrated that another amyloid-degrading enzyme, neprilysin, is present and active in islets and may play a role in islet amyloid accumulation (8). Neprilysin is a 90- to 110-kDa zinc metallopeptidase with a short cytoplasmic NH2 terminus, a transmembrane hydrophobic region, and a large extracellular domain containing the active site. It has broad substrate specificity, with its physiological role depending on its tissue localization. In the brain, neprilysin has been shown to be involved in degradation of amyloid; specifically, it can degrade amyloid-β (Aβ(1–42)), the peptide component of Alzheimer's amyloid, in vitro and in vivo (9–12).
Given that amyloidosis in type 2 diabetes and Alzheimer disease shares some similarities (13), and our previous observation of a relationship between islet amyloid deposition and neprilysin levels in vivo (8), we sought to determine whether neprilysin can indeed inhibit islet amyloid formation and its associated β-cell toxicity in vitro and, if so, by what mechanism. We used isolated islets from our human IAPP (hIAPP) transgenic mice, an amyloidogenic model that we developed (14) because mouse IAPP (mIAPP) does not form amyloid (15–17). A major advantage of our in vitro model is that it is particularly relevant to islet amyloid formation in humans, because the amyloid deposits form from endogenous hIAPP and are histologically comparable to the classic light microscopy-visible deposits observed in human type 2 diabetes (1, 18). Further, it enables the study of a direct effect of neprilysin on hIAPP fibrillogenesis in the islet, with non-amyloidogenic non-transgenic control islets from littermates for comparison.
EXPERIMENTAL PROCEDURES
Transgenic Mice
Hemizygous transgenic mice with β-cell production of hIAPP (19) on an F1 C57BL/6 × DBA/2J background were used with non-transgenic littermates as controls. Transgenic status was determined by PCR as previously described (20). The study was approved by the Institutional Animal Care and Use Committee at the VA Puget Sound Health Care System.
Isolation and Culture of Pancreatic Islets
Islets were isolated from the pancreases of 10-week-old female and male mice by collagenase digestion using methods previously described (21). Islets were cultured overnight in RPMI 1640 medium (containing 11.1 mm glucose), in a 37 °C humidified atmosphere of 95% air:5% CO2 to allow them to recover from the isolation procedure. They were then transferred to media containing 16.7 mm glucose (an amyloidogenic condition) and cultured for 144 h in the absence or presence of the specific neprilysin inhibitor dl-[N-(3-mercapto-2-benzylpropanoyl)]glycine (dl-thiorphan, 50 μm) (22). For up-regulation of neprilysin activity, freshly isolated islets were infected with an adenovirus containing neprilysin cDNA (AdV-NEP, gift from Dr. Jeffrey Robbins, Cincinnati, OH) (23) for 20 h, then transferred to media containing 16.7 mm glucose and cultured for 144 h. Varying titers of AdV-NEP ranging from 2.1 × 102 to 2.1 × 108 pfu/ml were tested for their ability to increase neprilysin activity without causing toxicity. A titer of 2.1 × 106 pfu/ml was considered optimal and thus used in all experiments. In a subset of experiments, control infections were performed with an adenovirus containing green fluorescent protein (AdV-GFP, gift from Dr. Christopher Rhodes, Chicago, IL). AdV-GFP did not affect islet amyloid formation in hIAPP transgenic islets.
Real-time Quantitative Reverse Transcription-PCR
Gene expression of neprilysin, hIAPP, mIAPP, and insulin in isolated islets was determined with real-time quantitative reverse transcription-PCR performed using the TaqMan system (ABI Prism 7000, Applied Biosystems, Foster City, CA) as previously described (8). TaqMan Assays on Demand neprilysin (Mm00485028_m1), hIAPP (Hs00169095_m1), mIAPP (Mm00439403_m1), and insulin II (Mm00731595_gH) gene expression mixes were from Applied Biosystems. TaqMan eukaryotic 18 S rRNA (Hs99999901_s1, Applied Biosystems) was used as endogenous control. Each sample was run in triplicate.
Neprilysin Enzymatic Activity Assay
Islet neprilysin enzyme activity was determined fluorometrically as previously described (8).
Histological Assessment of Islet Amyloid and β-Cell Apoptosis
Islets were fixed in 4% (w/v) phosphate-buffered paraformaldehyde and embedded in paraffin (14). Sections (10 μm) were cut and stained with thioflavin S to visualize amyloid deposits. For quantification of β-cell apoptosis, sections were stained with propidium iodide and anti-insulin antibody as previously described (24, 25). Histological assessments of amyloid and apoptosis were made in a blinded manner on an average of 16 islets per culture condition.
Mass Spectrometry
Stock solutions of hIAPP and Aβ(1–42) peptides (10 μg/μl in DMSO, Bachem, Torrance, CA) were diluted to 10 μm in phosphate-buffered saline with 1 mm CaCl2 and 0.5 mm MgCl2. Recombinant human neprilysin (R&D Systems, Minneapolis, MN) was added for a final concentration of 0.4 μm, and the mixture was incubated at 37 °C for 16 h. Peptide samples without neprilysin were analyzed immediately. Samples were desalted using C4 ZipTips (Millipore, Billerica, MA) and analyzed on a matrix-assisted laser desorption ionization-time of flight mass spectrometer (Voyager DE-STR, Applied Biosystems) with recrystallized α-cyano-4-hydroxycinnamic acid as the matrix. Spectra were collected from >100 shots from 500 to 5000 m/z. External calibration was performed using melitten (Sigma) as a calibration standard. Mass spectrometry was performed in triplicate on three independent occasions.
Thioflavin T Assay
A stock solution of hIAPP (2.5 mm in DMSO) was diluted in a 25 mm Tris-HCl plus 100 mm NaCl buffer at pH 8.0 for a final concentration of 50 μm hIAPP and 2% DMSO. A 1 μm stock of recombinant human neprilysin (R&D Systems) was prepared in the same buffer. Reactions were started by adding equal amounts of 50 μm hIAPP stock and 1 μm neprilysin stock to tubes for final reaction conditions of 25 μm hIAPP, 0.5 μm neprilysin, and 1% DMSO. In a subset of experiments, 1 μm neprilysin was incubated with 50 μm thiorphan for 30 min prior to addition of hIAPP. Control reactions contained no neprilysin and/or thiorphan. Also, to control for the possibility that the mere presence of any protein may affect fibrillogenesis, hen egg lysozyme was substituted for neprilysin and added to hIAPP for final reaction conditions of 25 μm hIAPP and 5 μm lysozyme. For all reactions, an emission wavelength scan was performed for thioflavin T after mixing (t = 0), and the reactions were placed in a 37 °C incubator for 16 h after which an emission wavelength scan was again taken. Thioflavin T fluorescence was measured using 450 nm excitation and 480 nm emission wavelengths. The assay was performed in triplicate on six independent occasions.
Transmission Electron Microscopy
A 15-μl sample from the end of the thioflavin T experiments (at t = 16 h) was placed on a carbon-coated Formvar 300-mesh copper grid and negatively stained with saturated uranyl acetate as previously described (26).
Statistical Analyses
Data are presented as mean ± S.E. for the number of experiments indicated. Statistical significance was determined using analysis of variance with post hoc analysis or with Mann-Whitney U test if data were not normally distributed. A p value of <0.05 was considered statistically significant.
RESULTS
Effect of Neprilysin Inhibition on Islet Amyloid Formation
Isolated islets were cultured in amyloidogenic conditions for 144 h in the absence or presence of the neprilysin inhibitor thiorphan, after which neprilysin mRNA and activity levels were measured. Although thiorphan did not alter neprilysin mRNA levels (Fig. 1A), it significantly reduced neprilysin activity levels (Fig. 1B) in hIAPP transgenic islets. Inhibition of neprilysin activity levels by thiorphan was comparable in non-transgenic islets (control 46.4 ± 4.8 versus inhibitor 8.7 ± 2.1 pmol of methoxy-2-naphthylamine/h/μg of protein; p < 0.0001, n = 6). Thioflavin S staining demonstrated that culture with thiorphan increased amyloid deposition in hIAPP transgenic islets by 54% (Fig. 1C); as expected, no amyloid was detected in non-transgenic islets.
FIGURE 1.

Neprilysin mRNA (A) and activity (B) levels and amyloid severity (C) in hIAPP transgenic islets following 144-h culture in the absence (open bars) or presence (closed bars) of the specific neprilysin inhibitor thiorphan. *, p < 0.05 versus control; n = 6.
Effect of Neprilysin Up-regulation on Islet Amyloid Formation
Isolated islets were infected with AdV-NEP, then cultured in amyloidogenic conditions. After 144-h culture of infected hIAPP transgenic islets, neprilysin mRNA (Fig. 2A) and activity (Fig. 2B) levels remained significantly elevated compared with control non-infected hIAPP transgenic islets. Neprilysin activity levels were similarly elevated in non-transgenic islets infected with AdV-NEP and cultured for 144 h (control 43.0 ± 4.3 versus AdV-NEP 500.3 ± 30.4 pmol of methoxy-2-naphthylamine/h/μg of protein; p < 0.001, n = 3). Quantitation of thioflavin S staining in hIAPP transgenic islets infected with AdV-NEP revealed a 79% decrease in amyloid deposition (Fig. 2C) compared with control islets. In a subset of experiments, islets were infected with AdV-GFP. AdV-GFP did not affect islet amyloid formation in hIAPP transgenic islets (data not shown).
FIGURE 2.

Neprilysin mRNA (A) and activity (B) levels and amyloid severity (C) in control non-infected (open bars) or neprilysin adenovirus (AdV-NEP)-infected (closed bars) hIAPP transgenic islets following 144-h culture. *, p < 0.05 versus control; n = 6.
Effect of Neprilysin Inhibition or Up-regulation on β-Cell Apoptosis
To determine whether the changes in islet amyloid formation due to neprilysin inhibition or up-regulation were paralleled by changes in β-cell apoptosis, condensed or fragmented nuclei were quantified in insulin-positive islet cells (Fig. 3). As expected, hIAPP transgenic islets cultured in amyloidogenic conditions exhibited increased β-cell apoptosis compared with non-transgenic islets. When amyloid formation was increased by inhibiting neprilysin with thiorphan, the rate of β-cell apoptosis in hIAPP transgenic islets increased by 75% compared with control hIAPP transgenic islets, while remaining unchanged in non-transgenic islets. When neprilysin activity was up-regulated in hIAPP transgenic islets with AdV-NEP infection and amyloid formation was reduced, β-cell apoptosis decreased by 79% compared with control hIAPP transgenic islets, while again remaining unchanged in non-transgenic islets.
FIGURE 3.
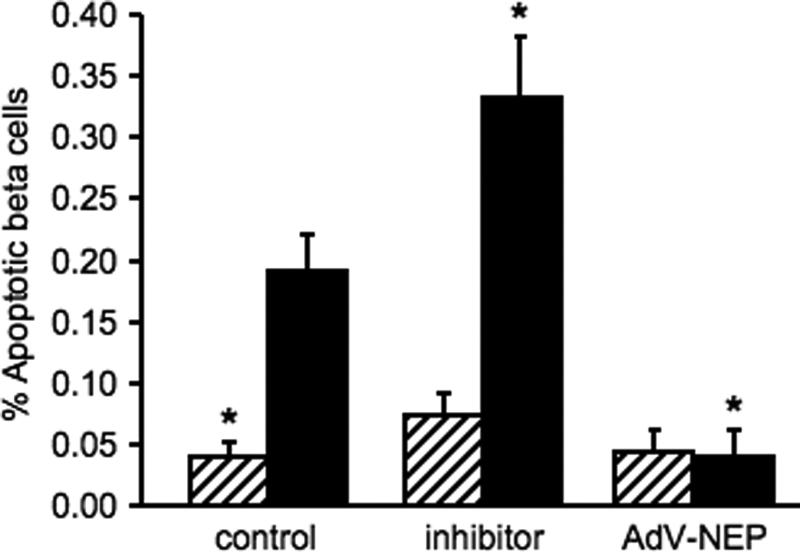
β-Cell apoptosis in non-transgenic (hatched bars) or hIAPP transgenic (closed bars) islets following treatment with the neprilysin inhibitor thiorphan or infection with a neprilysin-containing adenovirus (AdV-NEP). *, p < 0.05 versus hIAPP transgenic control; n = 6.
hIAPP, mIAPP, and Insulin mRNA Levels in Cultured Islets
mRNA levels of IAPP and insulin were determined in hIAPP transgenic islets after 144-h culture to assess the effect of inhibition or up-regulation of neprilysin. Neither hIAPP nor mouse IAPP mRNA levels were significantly altered in inhibitor or AdV-NEP-treated islets compared with controls (Table 1). Similarly, insulin mRNA levels remained unchanged in inhibitor and AdV-NEP-treated islets compared with controls.
TABLE 1.
hIAPP, mIAPP, and insulin mRNA levels in hIAPP transgenic islets following 144-h culture
Neprilysin was inhibited by co-culture of islets with the neprilysin inhibitor thiorphan, or up-regulated by infection with a neprilysin-containing adenovirus (AdV-NEP). Levels are expressed relative to that in control islets (n = 5/group).
| Control | Inhibitor | AdV-NEP | |
|---|---|---|---|
| hIAPP mRNA | 1.00 | 0.64 ± 0.16 | 1.15 ± 0.12 |
| mIAPP mRNA | 1.00 | 0.99 ± 0.17 | 1.16 ± 0.15 |
| Insulin mRNA | 1.00 | 0.91 ± 0.09 | 1.34 ± 0.23 |
hIAPP Cleavage and Fibril Formation in the Presence of Neprilysin
Mass spectrometry was performed to determine whether neprilysin mediates its effects on islet amyloid formation by cleaving hIAPP. In the absence of neprilysin, the mass spectrum for hIAPP indicated only one peak corresponding to the molecular mass of full-length hIAPP (Fig. 4A). After incubation of hIAPP with neprilysin for 16 h, hIAPP was not cleaved, as indicated by the single peak once again corresponding to full-length hIAPP (Fig. 4B). To ensure neprilysin was still active under the conditions used, Aβ(1–42) was incubated with neprilysin for 16 h, and the resultant mass spectrum (Fig. 4D) was compared with that obtained when Aβ(1–42) was analyzed in the absence of neprilysin (Fig. 4C). As previously reported (27, 28), in the presence of neprilysin multiple peaks were observed indicative of Aβ(1–42) cleavage and confirming that the enzyme was active.
FIGURE 4.

Representative mass spectra of hIAPP alone (A), hIAPP incubated with neprilysin (B), Aβ(1–42) alone (C) or Aβ(1–42) incubated with neprilysin (D). In both A and B, a single peak is observed with an m/z value of 3904.3 corresponding to the molecular mass of full-length hIAPP. In C, a single peak is observed with an m/z value of 4514.3 corresponding to the molecular mass of full-length Aβ(1–42), whereas in D multiple peaks with smaller molecular masses are observed, representing neprilysin-mediated Aβ(1–42) cleavage products.
Given that hIAPP was not cleaved by neprilysin, the thioflavin T assay was utilized to determine whether neprilysin mediates its effects on islet amyloid formation via inhibition of hIAPP fibril formation. Thioflavin T is a fluorescent dye that binds to amyloid fibrils but not to soluble proteins. Thus, fibril formation can be followed by monitoring increases in fluorescence intensity. At t = 0, thioflavin T fluorescence was negligible both in the presence and absence of neprilysin, indicating that fibrils had not yet formed (Fig. 5A). At 16 h, there was a large increase in thioflavin T fluorescence in the absence of neprilysin indicative of fibril formation, but no or very little increase in fluorescence in the presence of neprilysin (Fig. 5A). TEM images indicated that by 16 h fibril formation had occurred in the absence of neprilysin (Fig. 5B). In the presence of neprilysin, there was little to no presence of fibrils, and the majority of the structures seen throughout the TEM grids appeared to be amorphous globular aggregates. Control TEM images of neprilysin alone at t = 0 and t = 16 h revealed only large dark debris with no evidence of amorphous globular aggregates.
FIGURE 5.

A, representative thioflavin T fluorescence profile of hIAPP in the absence (closed symbols) or presence (open symbols) of neprilysin at t = 0 (circles) and t = 16 (triangles) h. At t = 0, fluorescence was negligible both in the presence and absence of neprilysin, indicating that fibrils had not yet formed. At t = 16 h, there was a significant increase in fluorescence in the absence of neprilysin indicative of fibril formation, but almost no increase in fluorescence in the presence of neprilysin. B, representative TEM images of hIAPP in the absence or presence of neprilysin after incubation for 16 h. Scale bar, 500 nm. C, thioflavin T fluorescence of hIAPP in the absence or presence of neprilysin, neprilysin pre-inhibited with thiorphan (inhibitor), or hen egg lysozyme at t = 16 h. *, p < 0.05 versus hIAPP alone; n = 3.
In a subset of experiments, neprilysin was pre-inhibited with the specific inhibitor, thiorphan, prior to analysis of hIAPP fibril formation by thioflavin T fluorescence and TEM. Pre-inhibited neprilysin was unable to prevent fibril formation, as evidenced by increased thioflavin T fluorescence (Fig. 5C) and the presence of fibrils by TEM. Thiorphan in the absence of neprilysin did not affect thioflavin T fluorescence (Fig. 5C) or the presence of fibrils by TEM, as compared with reactions in which either neprilysin was pre-inhibited with thiorphan or neprilysin was not present.
To determine if the mere presence of any other protein can inhibit hIAPP fibril formation, hen egg lysozyme was substituted for neprilysin, and thioflavin T fluorescence was monitored for up to 16 h. At 16 h, an increase in thioflavin T fluorescence was observed in the presence of hen egg lysozyme that was comparable to that seen in the absence of hen egg lysozyme (Fig. 5C), indicating that the presence of lysozyme did not alter hIAPP fibril formation. TEM images confirmed the presence of fibrils under these conditions (data not shown).
DISCUSSION
Islet amyloidogenesis in type 2 diabetes contributes to loss of β-cell function and mass (1). The amyloidogenic process involves aggregation of the β-cell peptide hIAPP from a monomeric state to oligomers, fibrils, and ultimately mature amyloid deposits. It is unclear what exactly triggers aggregation, although reduced degradation of IAPP and/or amyloid may play a role in the process. In the present study, we sought to determine whether the enzyme neprilysin, which has been shown to degrade the peptide constituent of Alzheimer's amyloid (Aβ) (27, 28), is also capable of degrading hIAPP. Because islet and Alzheimer's amyloid are in many ways similar (13), it could be hypothesized that neprilysin should degrade hIAPP. Interestingly, we demonstrate that neprilysin is able to decrease islet amyloid deposition but that its mechanism of action is not via degradation of hIAPP, but rather by inhibition of hIAPP fibril formation. Thus, whereas in Alzheimer disease neprilysin is capable of decreasing amyloid formation by degrading the amyloidogenic peptide constituent, in the islet the mechanism is distinctly different.
Using our in vitro model of islet amyloid formation and amyloid-induced β-cell apoptosis (25), we show that inhibition of islet neprilysin activity with thiorphan increased amyloid formation and thereby β-cell apoptosis. To confirm that the effect of thiorphan to increase amyloid formation and β-cell apoptosis was due to altered neprilysin activity, we also increased neprilysin expression using an adenovirus encoding neprilysin and showed that this intervention decreased islet amyloid formation and also β-cell apoptosis. That the adenoviral manipulation per se may have decreased amyloid formation was ruled out in experiments using a control adenovirus encoding GFP. Similarly, adenovirus infection and/or increased neprilysin activity did not decrease β-cell apoptosis independent of islet amyloid, because non-transgenic islets infected with AdV-NEP had the same rate of β-cell apoptosis as non-infected islets. Furthermore, given that IAPP regulation closely mirrors that of insulin and IAPP is co-secreted with insulin (2, 29), any manipulation that modulates one of these peptides can potentially affect the other. This said, we found that neither inhibition nor up-regulation of neprilysin altered mRNA levels of hIAPP, mIAPP, or insulin. Together, these data indicate that neprilysin is capable of modulating islet amyloid formation and β-cell apoptosis independent of any potential effects on IAPP production.
Neprilysin has been extensively studied in relation to amyloid formation in the brain. It has been shown to cleave Aβ both in vitro and in vivo (reviewed in Ref. 30). Further, it has been shown to hydrolyze a number of other peptides, some of which are produced in pancreatic islets like glucagon (31) and angiotensin II (32). Interestingly, we did not observe neprilysin-mediated hIAPP cleavage. This suggests that other enzymes may be largely responsible for clearing hIAPP. One such candidate is IDE, which has previously been shown to degrade synthetic hIAPP peptide. Studies of IDE showed that its inhibition in RIN-m5F insulinoma cells increased amyloid formation and hIAPP-induced cytotoxicity when hIAPP was applied to cultured cells (6, 7). Similarly, we have recently observed that matrix metalloproteinase-9 is capable of degrading hIAPP (33).
Given that neprilysin does not cleave hIAPP, we next examined the effect of neprilysin on hIAPP fibril formation. Thioflavin T fluorescence was used to monitor the kinetics of fibril formation, wherein the thioflavin T fluorescence significantly increases upon binding to fibrils. In the presence of neprilysin, little to no fibrils formed (i.e. lack of fluorescence) compared with hIAPP in the absence of neprilysin (i.e. strong fluorescence). Alone, these data may be interpreted a number of ways, because use of the thioflavin T assay as the sole means to investigate fibril formation is not definitive. Importantly, thioflavin T assays can yield false negatives in inhibitor studies in which the inhibitors displace bound thioflavin T or quench its fluorescence (26). Thus, we also performed TEM to visually determine whether or not neprilysin does in fact inhibit fibril formation. TEM images confirmed that very few fibrils were formed in the presence of neprilysin. The small, isolated regions of the TEM grid that did contain fibrils also appeared to contain amorphous aggregates and globular objects. Others have speculated that, in the presence of inhibitors of fibril formation, the observation of such globular objects suggests fibril disaggregation (34, 35).
The exact mechanism by which neprilysin inhibits IAPP fibril formation is currently unknown. Studies of neprilysin have shown that it can directly bind other proteins to mediate cellular events like cell adhesion, independent of protein hydrolysis (36–39). Such protein-protein interaction could explain the ability of neprilysin to reduce islet amyloid formation; neprilysin could bind hIAPP, thereby preventing its aggregation by locking it in a fixed conformation or by occupying binding sites that would normally bind additional hIAPP molecules and/or other amyloid components. The fact that the neprilysin inhibitor increased amyloid formation in islets may suggest that the inhibitor binds to or somehow blocks the site at which hIAPP binds neprilysin and thus the hIAPP peptide is free to aggregate. This hypothesis is corroborated by our thioflavin T and TEM data in which we found pre-inhibited neprilysin (i.e. active site occupied by thiorphan) was unable to inhibit fibril formation in vitro. Conversely, in islets overexpressing neprilysin, amyloid formation decreased perhaps because more neprilysin was available to bind hIAPP. Some support for the protein-protein interaction hypothesis comes from a study utilizing a soluble non-amyloidogenic IAPP molecular mimic that binds hIAPP and completely blocks its self-assembly and fibrillogenesis (40).
Knowledge regarding the subcellular localization and conformational changes of neprilysin may provide clues about its interaction with hIAPP. At this time, the specific localization of neprilysin within the islet has not been clearly defined. Previously, using an immunohistochemical approach, we showed that islet neprilysin localized to both β-cells and non-β-cells and is largely membranous with some cytoplasmic localization (8). Reports describing neprilysin in non-islet tissues also document that it is predominantly plasma membrane-bound; however, there is evidence in neurons that neprilysin is also localized in secretory vesicles (41). Furthermore, the neprilysin homologue neprilysin 2, which shares many of the same properties as neprilysin (42), exists in the endoplasmic reticulum (43). Thus, when considering where in the islet neprilysin may be acting to inhibit hIAPP fibril formation, any of these sites are possible. In line with its ability to trap peptides via conformational changes at the plasma membrane (44), neprilysin could prevent aggregation of hIAPP secreted into the extracellular space, a site where amyloid deposits commonly occur (18). Neprilysin in secretory vesicles or the endoplasmic reticulum could also act to sequester excess hIAPP prior to its release from the β-cell and thus impede fibril formation. Clearly, more extensive studies are required to understand any potential neprilysin-hIAPP interactions that may impact islet amyloid formation.
In summary, we have demonstrated that the enzyme neprilysin impacts islet amyloid formation in vitro. Neprilysin does not cleave the hIAPP peptide, and therefore reduces fibril formation via another mechanism. Although enzymatic activity may not be required, the active site of neprilysin does appear to be involved and hIAPP fibril formation may be impeded via a protein-protein interaction with neprilysin. Importantly, our studies highlight that amyloidogenesis in the islet can differ markedly from that in the brain, where neprilysin has been shown to reduce amyloid plaque formation primarily by cleaving the peptide constituent of Alzheimer's amyloid. Our findings also have implications for approaches taken in developing amyloid inhibitors, which could focus on inhibition of IAPP fibril assembly in addition to IAPP cleavage.
Acknowledgments
We thank B. Barrow, C. Braddock, M. Cone, M. Peters, M. Watts, and J. Willard for excellent technical support.
This work was supported, in whole or in part, by National Institutes of Health Grants DK-080945 (to S. Z.), DK-074404 (to R. L. H.), and GM-078114 (to D. P. R.). This work was also supported by the Dept. of Veterans Affairs, Virginia Affairs Puget Sound Health Care System, Seattle, WA. Mass spectrometry work was performed at the University of Washington's Diabetes Endocrinology Research Center Mass Spectrometry Core, which is supported by NIH Grant DK-017047.
- IAPP
- islet amyloid polypeptide
- AdV-NEP
- neprilysin-containing adenovirus
- GFP
- green fluorescent protein
- hIAPP
- human islet amyloid polypeptide
- IDE
- insulin-degrading enzyme
- mIAPP
- mouse islet amyloid polypeptide
- TEM
- transmission electron microscopy
- Aβ(1–42)
- amyloid-β
- thiorphan
- dl-[N-(3-mercapto-2-benzylpropanoyl)]glycine.
REFERENCES
- 1.Hull R. L., Westermark G. T., Westermark P., Kahn S. E. (2004) J. Clin. Endocrinol. Metab. 89, 3629–3643 [DOI] [PubMed] [Google Scholar]
- 2.Kahn S. E., D'Alessio D. A., Schwartz M. W., Fujimoto W. Y., Ensinck J. W., Taborsky G. J., Jr., Porte D., Jr. (1990) Diabetes 39, 634–638 [DOI] [PubMed] [Google Scholar]
- 3.Clark A., Saad M. F., Nezzer T., Uren C., Knowler W. C., Bennett P. H., Turner R. C. (1990) Diabetologia 33, 285–289 [DOI] [PubMed] [Google Scholar]
- 4.Enoki S., Mitsukawa T., Takemura J., Nakazato M., Aburaya J., Toshimori H., Matsukara S. (1992) Diabetes Res. Clin. Pract. 15, 97–102 [DOI] [PubMed] [Google Scholar]
- 5.Kautzky-Willer A., Thomaseth K., Pacini G., Clodi M., Ludvik B., Streli C., Waldhäusl W., Prager R. (1994) Diabetologia 37, 188–194 [DOI] [PubMed] [Google Scholar]
- 6.Bennett R. G., Duckworth W. C., Hamel F. G. (2000) J. Biol. Chem. 275, 36621–36625 [DOI] [PubMed] [Google Scholar]
- 7.Bennett R. G., Hamel F. G., Duckworth W. C. (2003) Diabetes 52, 2315–2320 [DOI] [PubMed] [Google Scholar]
- 8.Zraika S., Hull R. L., Udayasankar J., Clark A., Utzschneider K. M., Tong J., Gerchman F., Kahn S. E. (2007) Diabetes 56, 304–310 [DOI] [PubMed] [Google Scholar]
- 9.Iwata N., Tsubuki S., Takaki Y., Shirotani K., Lu B., Gerard N. P., Gerard C., Hama E., Lee H. J., Saido T. C. (2001) Science 292, 1550–1552 [DOI] [PubMed] [Google Scholar]
- 10.Iwata N., Tsubuki S., Takaki Y., Watanabe K., Sekiguchi M., Hosoki E., Kawashima-Morishima M., Lee H. J., Hama E., Sekine-Aizawa Y., Saido T. C. (2000) Nat. Med. 6, 143–150 [DOI] [PubMed] [Google Scholar]
- 11.Marr R. A., Rockenstein E., Mukherjee A., Kindy M. S., Hersh L. B., Gage F. H., Verma I. M., Masliah E. (2003) J. Neurosci. 23, 1992–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckman E. A., Adams S. K., Troendle F. J., Stodola B. A., Kahn M. A., Fauq A. H., Xiao H. D., Bernstein K. E., Eckman C. B. (2006) J. Biol. Chem. 281, 30471–30478 [DOI] [PubMed] [Google Scholar]
- 13.Götz J., Ittner L. M., Lim Y. A. (2009) Cell. Mol. Life Sci. 66, 1321–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zraika S., Hull R. L., Udayasankar J., Utzschneider K. M., Tong J., Gerchman F., Kahn S. E. (2007) Biochem. Biophys. Res. Commun. 354, 234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Betsholtz C., Christmansson L., Engström U., Rorsman F., Svensson V., Johnson K. H., Westermark P. (1989) FEBS Lett. 251, 261–264 [DOI] [PubMed] [Google Scholar]
- 16.Betsholtz C., Svensson V., Rorsman F., Engström U., Westermark G. T., Wilander E., Johnson K., Westermark P. (1989) Exp. Cell Res. 183, 484–493 [DOI] [PubMed] [Google Scholar]
- 17.Westermark P., Engström U., Johnson K. H., Westermark G. T., Betsholtz C. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 5036–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verchere C. B., D'Alessio D. A., Palmiter R. D., Weir G. C., Bonner-Weir S., Baskin D. G., Kahn S. E. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 3492–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D'Alessio D. A., Verchere C. B., Kahn S. E., Hoagland V., Baskin D. G., Palmiter R. D., Ensinck J. W. (1994) Diabetes 43, 1457–1461 [DOI] [PubMed] [Google Scholar]
- 20.Andrikopoulos S., Verchere C. B., Terauchi Y., Kadowaki T., Kahn S. E. (2000) Diabetes 49, 2056–2062 [DOI] [PubMed] [Google Scholar]
- 21.Zraika S., Dunlop M., Proietto J., Andrikopoulos S. (2002) Arch. Biochem. Biophys. 405, 275–279 [DOI] [PubMed] [Google Scholar]
- 22.Roques B. P., Noble F., Crine P., Fournié-Zaluski M. C. (1995) Methods Enzymol. 248, 263–283 [DOI] [PubMed] [Google Scholar]
- 23.Maloyan A., Gulick J., Glabe C. G., Kayed R., Robbins J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 5995–6000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scaglia L., Cahill C. J., Finegood D. T., Bonner-Weir S. (1997) Endocrinology 138, 1736–1741 [DOI] [PubMed] [Google Scholar]
- 25.Zraika S., Hull R. L., Udayasankar J., Aston-Mourney K., Subramanian S. L., Kisilevsky R., Szarek W. A., Kahn S. E. (2009) Diabetologia 52, 626–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng F., Marek P., Potter K. J., Verchere C. B., Raleigh D. P. (2008) Biochemistry 47, 6016–6024 [DOI] [PubMed] [Google Scholar]
- 27.Howell S., Nalbantoglu J., Crine P. (1995) Peptides 16, 647–652 [DOI] [PubMed] [Google Scholar]
- 28.Leissring M. A., Lu A., Condron M. M., Teplow D. B., Stein R. L., Farris W., Selkoe D. J. (2003) J. Biol. Chem. 278, 37314–37320 [DOI] [PubMed] [Google Scholar]
- 29.Kahn S. E., Fujimoto W. Y., D'Alessio D. A., Ensinck J. W., Porte D., Jr. (1991) Horm. Metab. Res. 23, 577–580 [DOI] [PubMed] [Google Scholar]
- 30.Hersh L. B., Rodgers D. W. (2008) Curr. Alzheimer Res. 5, 225–231 [DOI] [PubMed] [Google Scholar]
- 31.Trebbien R., Klarskov L., Olesen M., Holst J. J., Carr R. D., Deacon C. F. (2004) Am. J. Physiol. Endocrinol. Metab. 287, E431–E438 [DOI] [PubMed] [Google Scholar]
- 32.Lau T., Carlsson P. O., Leung P. S. (2004) Diabetologia 47, 240–248 [DOI] [PubMed] [Google Scholar]
- 33.Aston-Mourney K., Zraika S., Udayasankar J., Subramanian S. L., Kahn S. E., Hull R. L. (2008) in Diabetes Vol. 57, pp. A457, American Diabetes Association 68th Scientific Sessions, San Francisco, CA [Google Scholar]
- 34.Yan P., Hu X., Song H., Yin K., Bateman R. J., Cirrito J. R., Xiao Q., Hsu F. F., Turk J. W., Xu J., Hsu C. Y., Holtzman D. M., Lee J. M. (2006) J. Biol. Chem. 281, 24566–24574 [DOI] [PubMed] [Google Scholar]
- 35.Yang F., Lim G. P., Begum A. N., Ubeda O. J., Simmons M. R., Ambegaokar S. S., Chen P. P., Kayed R., Glabe C. G., Frautschy S. A., Cole G. M. (2005) J. Biol. Chem. 280, 5892–5901 [DOI] [PubMed] [Google Scholar]
- 36.Ganju R. K., Shpektor R. G., Brenner D. G., Shipp M. A. (1996) Blood 88, 4159–4165 [PubMed] [Google Scholar]
- 37.Iwase A., Shen R., Navarro D., Nanus D. M. (2004) J. Biol. Chem. 279, 11898–11905 [DOI] [PubMed] [Google Scholar]
- 38.Shen R., Milowsky M. I., Ozaki N., Navarro D., Sumitomo M., Xu Y., Nanus D. M. (2002) Anticancer Res. 22, 2533–2538 [PubMed] [Google Scholar]
- 39.Sumitomo M., Iwase A., Zheng R., Navarro D., Kaminetzky D., Shen R., Georgescu M. M., Nanus D. M. (2004) Cancer Cell 5, 67–78 [DOI] [PubMed] [Google Scholar]
- 40.Yan L. M., Tatarek-Nossol M., Velkova A., Kazantzis A., Kapurniotu A. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 2046–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukami S., Watanabe K., Iwata N., Haraoka J., Lu B., Gerard N. P., Gerard C., Fraser P., Westaway D., St George-Hyslop P., Saido T. C. (2002) Neurosci. Res. 43, 39–56 [DOI] [PubMed] [Google Scholar]
- 42.Rose C., Voisin S., Gros C., Schwartz J. C., Ouimet T. (2002) Biochem. J. 363, 697–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raharjo S. B., Emoto N., Ikeda K., Sato R., Yokoyama M., Matsuo M. (2001) J. Biol. Chem. 276, 25612–25620 [DOI] [PubMed] [Google Scholar]
- 44.Malito E., Hulse R. E., Tang W. J. (2008) Cell Mol. Life Sci. 65, 2574–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]