

Abstract
Growth plate abnormalities, associated with impaired hypertrophic chondrocyte apoptosis, are observed in humans and animals with abnormalities of vitamin D action and renal phosphate reabsorption. Low circulating phosphate levels impair hypertrophic chondrocyte apoptosis, whereas treatment of these cells with phosphate activates the mitochondrial apoptotic pathway. Because phosphate-mediated apoptosis of chondrocytes is differentiation-dependent, studies were performed to identify factors that contribute to hypertrophic chondrocyte apoptosis. An increase in the percentage of cells with low mitochondrial membrane potential, evaluated by JC-1 fluorescence, was observed during hypertrophic differentiation of primary murine chondrocytes in culture. This percentage was further increased by treatment of hypertrophic, but not proliferative, chondrocytes with phosphate. Phosphate-mediated apoptosis was observed as early as 30 min post-treatment and was dependent upon Erk1/2 phosphorylation. Inhibition of Erk1/2 phosphorylation in vivo confirmed an important role for this signaling pathway in regulating hypertrophic chondrocyte apoptosis in growing mice. Murine embryonic metatarsals cultured under phosphate-restricted conditions demonstrated a 2.5-fold increase in parathyroid hormone-related protein mRNA expression accompanied by a marked attenuation in phospho-Erk immunoreactivity in hypertrophic chondrocytes. Thus, these investigations point to an important role for phosphate in regulating mitochondrial membrane potential in hypertrophic chondrocytes and growth plate maturation by the parathyroid hormone-related protein signaling pathway.
Keywords: Apoptosis, Bone, Differentiation, ERK, Mitochondrial Apoptosis, Apoptosis, Chondrocyte, Growth Plate, Phosphate, Rickets
Introduction
Maturation of the growth of long bones is dependent upon the differentiation of proliferative chondrocytes into prehypertrophic and subsequently hypertrophic chondrocytes. Terminal differentiation of hypertrophic chondrocytes is characterized by the expression of signaling molecules that promote vascular invasion, apoptosis, and replacement of hypertrophic chondrocytes by osteoblasts that lay down the primary spongiosa of bone. Aberrant regulation of this developmental process results in growth plate disorders. Although calcium has been shown to play an important role in regulating chondrocyte maturation (1), apoptosis of terminally differentiated hypertrophic chondrocytes is dependent upon normal levels of circulating phosphate (2).
Rickets is a growth plate anomaly observed in growing animals and humans with abnormalities of vitamin D action and renal phosphate reabsorption (3–6). In vivo investigations in genetically modified and dietary-manipulated mouse models demonstrate that hypophosphatemia is the underlying metabolic abnormality that impairs growth plate maturation in these disorders: low circulating phosphate levels result in impaired apoptosis of terminally differentiated hypertrophic chondrocytes in the growth plate, leading to rickets (2). The observation that inhibition of phosphate transport prevents phosphate-mediated apoptosis in hypertrophic chondrocytes (7–9) further reinforces that circulating phosphate, rather than the presence of local mineralized matrix, is a key determinant of hypertrophic chondrocyte apoptosis. In vitro investigations demonstrate that the mitochondrial apoptotic pathway is activated by phosphate, resulting in caspase-9 cleavage and induction of hypertrophic chondrocyte apoptosis. Treatment of wild-type mice with caspase-9 inhibitors confirmed a critical role for the mitochondrial apoptotic pathway in hypertrophic chondrocyte apoptosis and growth plate maturation in vivo (2).
Chondrocyte susceptibility to phosphate-induced apoptosis is differentiation-dependent (2). Because proliferative chondrocytes are not susceptible to phosphate-mediated apoptosis, studies were performed to determine whether primary proliferating chondrocytes acquire an increased susceptibility to phosphate-induced apoptosis during differentiation and to identify pathways that contribute to activation of the mitochondrial apoptotic pathway by phosphate.
EXPERIMENTAL PROCEDURES
Cell Culture
Primary chondrocytes were isolated from ventral rib cages of newborn mice by sequential collagenase II digestions and plated onto gelatin-coated plates at a density of 3 × 105/cm2 as described previously (2, 10). Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 25 μg/ml ascorbic acid at 37 °C and 5% CO2. To evaluate activation of signaling and apoptotic pathways, cells were incubated with sodium phosphate or control anion (sodium chloride or sodium sulfate) in Dulbecco's modified Eagle's medium with 0.5% fetal bovine serum. To evaluate the contribution of Erk2 phosphorylation to caspase-9 activation, cells were pretreated for 1 h with vehicle or with the MEK inhibitor U0126.
Flow Cytometry
Mitochondrial membrane potential was assessed using the APO LOGIX JC-1 mitochondrial potential detection kit (Bachem). The cationic dye JC-1 accumulates and aggregates in intact mitochondria, emitting a bright red fluorescence. With disruption of the mitochondrial membrane potential, mitochondrial aggregates do not form, but rather the dye remains in monomeric form in the cytoplasm, emitting green fluorescence. Chondrocytes were incubated with JC-1 for 15 min in medium at 37 °C according to the manufacturer's instructions. Cells were then placed on ice until being subjected to flow cytometry on a FACSCalibur flow cytometer. Data analyses were performed using FlowJo v8.7.1 software. Annexin V binds to phosphatidylserine, which is externalized to the outer cell membrane in early apoptosis. Chondrocytes were treated overnight with control anion or phosphate prior to evaluation of annexin V binding using a Guava PCA system and Guava Nexin reagents.
Western Analyses
Chondrocytes were lysed in Tris-buffered saline containing 2% SDS, 2% Triton X-100, 1 mm EDTA, 1 mm sodium fluoride, 20 mm β-glycerol phosphate, 2 mm sodium orthovanadate, and protease inhibitor mixture (Roche Applied Science). Lysates were subjected to two freeze/thaw cycles. Protein concentration was calculated using the BCA protein assay, following which 10 μg of protein was subjected to SDS-PAGE. Caspase-9 cleavage, Akt phosphorylation, and Erk1/2 phosphorylation were visualized using primary polyclonal antibodies against caspase-9, phospho-Akt, Erk1/2, and phospho-Erk1/2 (Cell Signaling, Danvers, MA) and horseradish peroxidase-conjugated IgG secondary antibody and visualized using ECL PlusTM (Amersham Biosciences) according to the manufacturer's instructions.
Reverse Transcription (RT)-PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, CA). RNA was subjected to reverse transcription and quantitative real-time PCR using the QuantiTect SYBR Green RT-PCR kit (Qiagen) on an Opticon DNA engine (MJ Research, Waltham, MA). The expression of the genes of interest was normalized to that of actin in each sample using the formula of Livak and Schmittgen (11).
Animal Studies
All mice were in the C57BL/6J background. Animals were maintained in a virus- and parasite-free barrier facility and exposed to a 12-h light/dark cycle. To evaluate the effect of Erk inhibition on growth plate maturation and apoptosis, wild-type mice were treated twice daily from 18 to 21 days with 100 mg/kg U0126 or vehicle (12) and killed on day 21 or 31.
Histology
Immunohistochemical detection of phospho-Erk was performed on fixed frozen sections as described previously (13). Apoptosis was evaluated using the TUNEL-based in situ cell death detection kit (Roche Diagnostics) and an antibody to cleaved caspase-3 (Cell Signaling) (2). In situ hybridization was performed on fixed frozen or paraffin sections using 35S-UTP-labeled antisense RNA probes as described previously (4). The presence of endothelial cells was evaluated using an anti-CD31 antibody (BD Biosciences).
Metatarsal Cultures
Metatarsals were isolated from day 15.5 post-coitum C57BL/6J embryos and cultured in phosphate-free Dulbecco's modified Eagle's medium supplemented with 0.25% defined fetal bovine serum, 50 μg/ml ascorbic acid, antibiotic/antimycotic, sodium pyruvate, and sodium phosphate at a final concentration of 1.25 mm (control) or 0.05 mm. After a 24-h incubation at 37 °C and 5% CO2, RNA was isolated from six pooled metatarsals for each condition (RNeasy mini kit) (14).
Statistics
Student's t test was used to identify significant differences. p < 0.05 was considered to be significant.
RESULTS
Previous investigations have demonstrated that neither 3T3 fibroblastic cells nor primary chondrocytes in the proliferative phase of differentiation are susceptible to caspase-9 activation by phosphate in vitro (2). To determine whether the susceptibility of hypertrophic versus proliferative chondrocytes to phosphate-mediated apoptosis was associated with a differentiation-dependent decrease in mitochondrial membrane potential (ΔΨ) and/or a decrease in ΔΨ in response to phosphate, the percentage of cells with low ΔΨ was examined over the course of chondrocyte differentiation in vitro. Mitochondrial membrane integrity was evaluated using the cationic dye JC-1, a highly specific probe for detecting changes in mitochondrial ΔΨ. JC-1 forms red aggregates in intact mitochondria. However, permeabilization of the mitochondrial membrane leads to a decrease in the electrochemical gradient across the membrane, resulting in the release of green fluorescent JC-1 monomers into the cytosol. A 3.2-fold increase in the percentage of cells with low ΔΨ was observed in chondrocytes cultured in differentiation medium for 5 versus 14 days (Fig. 1A, white bars). In addition, sodium phosphate treatment (Fig. 1A, black bars) significantly increased the percentage of cells with low ΔΨ cultured for 14 days, but not at earlier time points. To confirm that this was associated with apoptosis, annexin V binding was evaluated. Annexin binds to phosphatidylserine, which is exteriorized during apoptosis. The increase in the number of cells with low ΔΨ was associated with a 1.8-fold increase in annexin V-labeled cells in response to phosphate treatment at 14 days in culture (4.7 ± 0.3% to 8.6 ± 0.2%). Phosphate had no effect on annexin V binding by proliferative chondrocytes at 5 days in culture. To substantiate that these changes in JC-1 fluorescence and annexin V binding in response to phosphate treatment correlated with hypertrophic differentiation, RT-PCR analyses were performed to evaluate the expression of osteopontin, a marker of late hypertrophic chondrocytes. A significant increase in osteopontin expression was observed between 7 and 14 days in culture (Fig. 1B), correlating with an increase in the number of cells with low ΔΨ, their susceptibility to reduction in ΔΨ by phosphate, and the ability of phosphate to induce cleavage of caspase-9 (Fig. 1C). Chondrocytes cultured for 2 weeks under conditions that promote hypertrophic differentiation were then treated with control anion or sodium phosphate for a period of 30 min to 2 h to examine the time course of caspase-9 activation. As demonstrated in Fig. 2A, cleavage of caspase-9 was observed as early as 30 min post-exposure to 7 mm phosphate. Initial investigations examining phosphate-induced caspase-9 activation were performed with 7 mm phosphate, a level analogous to that in the circulation of the late gestational fetus (2). To examine whether lower levels of phosphate were also able to activate the mitochondrial apoptotic pathway in hypertrophic chondrocytes, dose-response studies were performed. These investigations demonstrated that caspase-9 was cleaved within 1 h of exposure to 5 mm phosphate and, to a lesser extent, to 3 mm phosphate (Fig. 2B).
FIGURE 1.

Hypertrophic chondrocyte differentiation is associated with a decrease in ΔΨ and susceptibility to phosphate-mediated apoptosis. A, JC-1 fluorescence was evaluated after a 4-h exposure of chondrocytes to 7 mm sodium phosphate (black bars) or to control anion (sodium chloride or sodium sulfate; white bars) from 5 to 14 days in culture. *, p < 0.01 versus control at 2 weeks and control and phosphate values from 5 to 10 days in culture; **, p < 0.001 versus control and phosphate values from 5 to 10 days in culture. B, expression of osteopontin was determined by quantitative real-time RT-PCR and normalized to that of actin in the same sample. Data are presented as expression relative to day 5 in culture. C, chondrocyte cell lysates were subjected to Western analyses to evaluate caspase-9 cleavage in response to 7 mm control anion (C) or phosphate (Pi) at 5 and 14 days in culture. Upper bands represent total caspase-9, and the arrow points to the band representing cleaved caspase-9. Data are representative of those obtained from at least four independent primary chondrocyte cell preparations.
FIGURE 2.
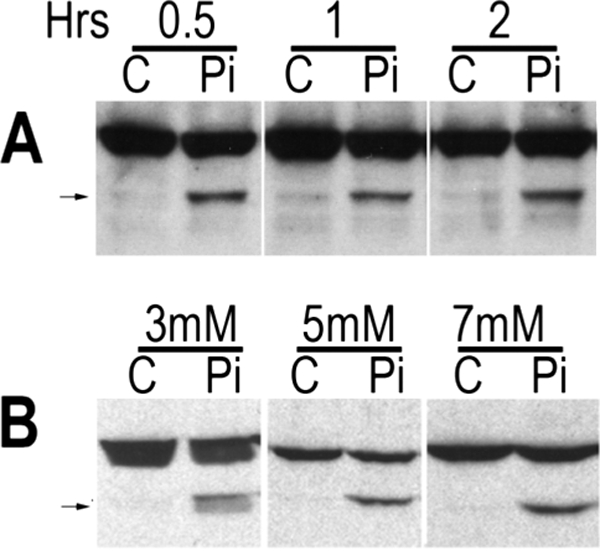
Time course and dose response of phosphate-mediated hypertrophic chondrocyte apoptosis. Hypertrophic chondrocytes were exposed to control anion (C; sodium chloride or sodium sulfate) or phosphate (Pi; sodium phosphate) prior to evaluation of caspase-9 activation by Western analyses. A, cells were treated with control anion or phosphate at a concentration of 7 mm for 30 min to 2 h. B, cells were treated for 1 h with 3, 5, or 7 mm control anion or phosphate. Upper bands represent total caspase-9, and the arrows to the bands representing cleaved caspase-9. Data are representative of those obtained from three independent chondrocyte preparations.
Based on the rapid activation of caspase-9 following treatment of hypertrophic chondrocytes with phosphate, studies were performed to determine whether a specific signal transduction pathway could be implicated in this process. Studies by other investigators have demonstrated activation of Erk1/2 upon exposure of clonal and primary murine chondrocytes to 10 mm phosphate (15). To investigate whether the doses of phosphate capable of activating caspase-9 could reproduce this effect or activate alternative signaling pathways, hypertrophic chondrocytes were treated with 7 mm sodium phosphate or control anion for 60 min. Although phosphate treatment did not alter Akt phosphorylation, Erk phosphorylation was markedly induced by phosphate (Fig. 3A). Phosphate treatment of proliferative chondrocytes did not induce Erk1/2 phosphorylation (Fig. 3B). Time course studies demonstrated that Erk phosphorylation was induced in hypertrophic chondrocytes as early as 5 min post-exposure to phosphate, peaked at 15 min, and was sustained for 2 h (Fig. 3C). To determine whether Erk activation could be implicated in the apoptotic process, cells were pretreated with the MEK1/2 inhibitor U0126 prior to exposure to phosphate. As shown in Fig. 3D, a 1-h pretreatment of chondrocytes with 30 μm U0126, a dose previously shown to inhibit Erk1/2 phosphorylation and phosphate-induced matrix Gla protein mRNA expression (15), prevented caspase-9 cleavage in response to 3 and 5 mm phosphate and markedly impaired caspase-9 activation in response to 7 mm phosphate.
FIGURE 3.

Phosphate induction of Erk phosphorylation. A, Erk and Akt phosphorylation was examined by Western analyses after a 1-h exposure of hypertrophic chondrocytes to 7 mm sodium sulfate (Pi−) or 7 mm sodium phosphate (Pi+). B, Erk phosphorylation (pErk1/2) was examined after a 1-h exposure of proliferative chondrocytes to 7 mm sodium sulfate or 7 mm sodium phosphate. Total Erk immunoreactivity is shown below. C, Erk phosphorylation was examined 5–120 min after exposure of hypertrophic chondrocytes to 7 mm sodium sulfate or 7 mm sodium phosphate. D, cells were pretreated with Me2SO (U0−) or 30 μm U0126 (U0+) for 1 h before the addition of sodium phosphate (Pi+) or sodium sulfate (Pi−) at the concentrations indicated. One hour later, caspase-9 activation was evaluated by Western analyses of cell lysates. The arrow points to the band representing cleaved caspase-9. Data are representative of those obtained from three independent chondrocyte preparations.
To investigate the relevance of these data to in vivo maturation of the growth plate, Erk phosphorylation was inhibited by treating wild-type mice twice daily for 4 days with U0126 or vehicle. This resulted in a decrease in phospho-Erk1/2 immunoreactivity in the growth plate associated with expansion of the hypertrophic chondrocyte layer, characterized by an increase in the region of collagen type X- and vascular endothelial growth factor-expressing chondrocytes. Impairment of Erk1/2 phosphorylation did not alter blood vessel invasion based on immunohistochemistry for the endothelial cell marker CD31 (Fig. 4B). However, a decrease in apoptosis of hypertrophic chondrocytes as assessed by TUNEL labeling (Fig. 4B, middle panels, arrowheads) and cleaved caspase-3 immunoreactivity was observed. Thus, these data confirm that Erk phosphorylation is required for normal growth plate maturation, including hypertrophic chondrocyte apoptosis. Characterization of the growth plate of mice killed 10 days post-U0126 treatment demonstrated reversal of this phenotype.
FIGURE 4.

Inhibition of Erk activation results in expansion of the growth plate and impaired hypertrophic chondrocyte apoptosis. Wild-type mice were treated for 4 days with vehicle or U0126. A, histological sections of the tibial growth plate were used to evaluate the presence of phospho-Erk (pERK) by immunohistochemistry of collagen type X (ColX), vascular endothelial growth factor (VEGF), and MMP-9 by in situ hybridization. B, immunohistochemistry was performed to evaluate the presence of blood vessels at the chondro-osseous junction (CD31), apoptotic hypertrophic chondrocytes by TUNEL labeling (with arrowheads pointing to apoptotic nuclei), and cleaved caspase-3. Brackets indicate the chondro-osseous junction. Data are representative of those obtained from three mice for each treatment condition.
To address whether impaired Erk1/2 phosphorylation could be implicated in the rachitic phenotype observed in hypophosphatemic states, studies were performed to address the effect of phosphate restriction on Erk1/2 phosphorylation. Cultured metatarsals from day 15.5 mouse embryos have been used by numerous investigators to characterize factors that regulate endochondral bone formation. Metatarsal elements were therefore isolated from wild-type embryos and cultured for 24 h under control (1.25 mm) or phosphate-restricted (0.05 mm) conditions. As shown in Fig. 5A, metatarsals cultured under control conditions demonstrated significant phospho-Erk1/2 immunoreactivity in the region of hypertrophic chondrocytes (bracketed region, shown externally in high power). However, phospho-Erk1/2 immunoreactivity was markedly attenuated under phosphate-restricted conditions.
FIGURE 5.

Phosphate restriction increases PTHrP expression and impairs Erk1/2 phosphorylation. Metatarsals isolated from day 15.5 post-coitum mouse embryos were cultured for 24 h under control (1.25 mm) and phosphate-restricted (0.05 mm) conditions. A, Erk phosphorylation was evaluated by immunohistochemistry. High power images of the hypertrophic chondrocyte region (bracketed) are shown adjacent to the metatarsals. Data are representative of immunohistochemical analyses performed in three independent metatarsal elements. Pi, phosphate. B, expression of PTHrP mRNA was evaluated by quantitative real-time RT-PCR and normalized to that of actin in the same sample. *, p < 0.05. Data represent the mean ± S.E. of three independent RNA preparations subjected to quantitative RT-PCR. C, phospho-Erk immunoreactivity was examined in the growth plates of wild-type mice (WT) and hyp mice, the murine homologue of X-linked hypophosphatemia, at 8 days of age. D, in situ hybridization analyses were performed to evaluate the expression of PTHrP in the periarticular proliferating chondrocytes (arrows) at 8 days of age. Data are representative of those obtained from three mice of each genotype.
A number of signaling molecules have been shown to regulate the maturation and differentiation of growth plate chondrocytes. Although Erk phosphorylation associated with activation of FGFR3 impairs chondrocyte differentiation, lack of parathyroid hormone-related protein (PTHrP) leads to increased Erk phosphorylation and apoptosis (16). Therefore, studies were undertaken to address whether alterations in PTHrP expression could be implicated in the chondrocyte phenotype observed with phosphate restriction. RNA was isolated from three independent pools of six metatarsals for each condition and subjected to quantitative real-time RT-PCR. As shown in Fig. 5B, phosphate restriction led to a 2.5-fold increase in PTHrP mRNA levels but did not alter expression of the mRNAs encoding BMP2 or FGF18 (data not shown).
To address whether impaired Erk1/2 phosphorylation in hypertrophic chondrocytes is observed under hypophosphatemic conditions in vivo, the growth plates of 1-week-old hyp and wild-type mice were examined. hyp mice are the murine model of the human disorder X-linked hypophosphatemia. These mice exhibit hypophosphatemia by 1 week of age and rickets associated with impaired hypertrophic chondrocyte apoptosis (2). In contrast to the prominent phospho-Erk1/2 immunoreactivity observed in the growth plates of the wild-type mice, a marked reduction in phospho-Erk1/2 immunoreactivity was observed in the hyp growth plates, consistent with the role of phosphate in activating Erk phosphorylation (Fig. 5C). This was associated with an increase in PTHrP mRNA expression in the periarticular region of the growth plate of the hyp mouse (Fig. 5D).
DISCUSSION
The underlying basis for the susceptibility of hypertrophic chondrocytes to phosphate-mediated apoptosis has led to numerous investigations in cellular models. Chondrocyte hypertrophy in chicken growth plates is accompanied by a loss of thiol, which is thought to increase susceptibility to apoptosis in response to oxidative stress (17, 18). Studies in chicken growth plates also demonstrated a maturation-dependent change in mitochondrial morphology accompanied by a decrease in staining with rhodamine 123, a fluorescent dye that binds to the mitochondrial membrane and inhibits transport (19). Our studies demonstrate a differentiation-dependent decrease in ΔΨ in primary murine chondrocyte cultures based on an increased percentage of JC-1 monomers associated with acquisition of the differentiated phenotype, characterized by nodule formation and expression of osteopontin mRNA. In addition, our investigations demonstrate that exposure of these differentiated hypertrophic chondrocytes, but not proliferative chondrocytes, to phosphate causes a further increase in the number of cells with low ΔΨ, confirming that this change in ΔΨ is an important determinant of the susceptibility of hypertrophic chondrocytes to phosphate-mediated apoptosis. Modulation of phosphate-induced apoptosis by Bcl family members also plays a critical role in the susceptibility of hypertrophic chondrocytes to this apoptotic stimulus. Mice with chondrocyte-specific ablation of the anti-apoptotic protein Bcl-x demonstrate growth retardation associated with reduction in the hypertrophic chondrocyte layer, which is postulated to be due to enhanced apoptosis of these cells (9). Although Bcl-x expression levels are not altered during chondrocyte differentiation, the expression of the pro-apoptotic protein Bnip3 increases with chondrocyte hypertrophy. Thus, the increase in expression of Bnip3 is thought to impair the anti-apoptotic function of Bcl-x in hypertrophic chondrocytes, contributing to their susceptibility to phosphate-mediated apoptosis (9).
Activation of caspase-9 as early as 1 h after exposure of hypertrophic chondrocytes to phosphate led us to investigate potential signal transduction pathways that could contribute to phosphate-mediated apoptosis of hypertrophic chondrocytes. Although phosphorylation of Akt was not altered by phosphate, Erk phosphorylation was induced in a time- and dose-dependent fashion in hypertrophic, but not proliferative, chondrocytes. Although it has been shown that induction of matrix Gla protein mRNA expression in chondrocytes in response to 10 mm phosphate requires activation of Erk, the role of Erk phosphorylation in hypertrophic chondrocyte apoptosis had not been addressed previously. Our investigations, both in vitro in primary murine chondrocytes and in vivo in wild-type mice treated with a MEK1/2 inhibitor that blocks Erk phosphorylation, demonstrate that activation of Erk is required for phosphate-mediated hypertrophic chondrocyte apoptosis. Furthermore, immunohistochemical analyses demonstrate a reduction in phospho-Erk in the growth plates of mice with X-linked hypophosphatemia. Interestingly, studies in murine metatarsal cultures also demonstrate that Erk phosphorylation is associated with hypertrophic chondrocyte maturation (13). Our studies extend these observations, demonstrating that phosphate restriction leads to an increase in PTHrP mRNA expression and a decrease in Erk phosphorylation in this model of endochondral bone formation. Thus, the role of phospho-Erk in growth plate maturation may be 2-fold: first, to promote chondrocyte differentiation, and second, to promote phosphate-mediated apoptosis of hypertrophic chondrocytes. The observation that PTHrP prevents Erk phosphorylation suggests that the growth plate phenotype of PTHrP-null mice may be due in part to enhanced Erk activation, leading to premature hypertrophy and enhanced apoptosis. Notable in this respect, the hypertrophic chondrocyte region of the PTHrP-null growth plate exhibits evidence of programmed cell death (16), and PTHrP-null chondrogenic cells have been shown to have an increased susceptibility to apoptosis (20). Although Erk activation has not been examined in hypertrophic chondrocytes lacking PTHrP, PTHrP has been shown to increase the expression of the anti-apoptotic protein Bcl-2, delaying hypertrophy as well as apoptotic cell death in normal mice (21) and in mice with constitutive activation of FGFR3 (22).
These investigations confirm the critical role for phosphate ions in the regulation of growth plate maturation. Although the precise mechanism underlying the rapid activation of Erk1/2 phosphorylation by phosphate remains to be elucidated, our studies point to an important role for phosphate in modulation of mitochondrial ΔΨ and of the PTHrP signaling pathway as well as in promoting hypertrophic chondrocyte apoptosis.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK46974, P50 AR054086, and T32 DK007028.
- Erk
- extracellular signal-regulated kinase
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- RT
- reverse transcription
- TUNEL
- terminal deoxynucleotidyltransferase dUTP nick end labeling
- PTHrP
- parathyroid hormone-related protein.
REFERENCES
- 1.Chang W., Tu C., Pratt S., Chen T. H., Shoback D. (2002) Endocrinology 143, 1467–1474 [DOI] [PubMed] [Google Scholar]
- 2.Sabbagh Y., Carpenter T. O., Demay M. B. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 9637–9642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergwitz C., Roslin N. M., Tieder M., Loredo-Osti J. C., Bastepe M., Abu-Zahra H., Frappier D., Burkett K., Carpenter T. O., Anderson D., Garabedian M., Sermet I., Fujiwara T. M., Morgan K., Tenenhouse H. S., Juppner H. (2006) Am. J. Hum. Genet. 78, 179–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donohue M. M., Demay M. B. (2002) Endocrinology 143, 3691–3694 [DOI] [PubMed] [Google Scholar]
- 5.Pettifor M. P., Daniels E. D. (1997) Vitamin D Deficiency and Nutritional Rickets in Children, Academic Press, San Diego, CA [Google Scholar]
- 6.Shimada T., Muto T., Urakawa I., Yoneya T., Yamazaki Y., Okawa K., Takeuchi Y., Fujita T., Fukumoto S., Yamashita T. (2002) Endocrinology 143, 3179–3182 [DOI] [PubMed] [Google Scholar]
- 7.Magne D., Bluteau G., Faucheux C., Palmer G., Vignes-Colombeix C., Pilet P., Rouillon T., Caverzasio J., Weiss P., Daculsi G., Guicheux J. (2003) J. Bone Miner. Res. 18, 1430–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mansfield K., Teixeira C. C., Adams C. S., Shapiro I. M. (2001) Bone 28, 1–8 [DOI] [PubMed] [Google Scholar]
- 9.Oshima Y., Akiyama T., Hikita A., Iwasawa M., Nagase Y., Nakamura M., Wakeyama H., Kawamura N., Ikeda T., Chung U. I., Hennighausen L., Kawaguchi H., Nakamura K., Tanaka S. (2008) J. Biol. Chem. 283, 26499–26508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefebvre V., Garofalo S., Zhou G., Metsäranta M., Vuorio E., De Crombrugghe B. (1994) Matrix Biol. 14, 329–335 [DOI] [PubMed] [Google Scholar]
- 11.Livak K. J., Schmittgen T. D. (2001) Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 12.Li Y., Je H. D., Malek S., Morgan K. G. (2004) Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R328–R335 [DOI] [PubMed] [Google Scholar]
- 13.Provot S., Nachtrab G., Paruch J., Chen A. P., Silva A., Kronenberg H. M. (2008) Mol. Cell. Biol. 28, 344–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zalutskaya A. A., Cox M. K., Demay M. B. (2009) J. Cell. Biochem. 108, 668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Julien M., Magne D., Masson M., Rolli-Derkinderen M., Chassande O., Cario-Toumaniantz C., Cherel Y., Weiss P., Guicheux J. (2007) Endocrinology 148, 530–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amizuka N., Henderson J. E., Hoshi K., Warshawsky H., Ozawa H., Goltzman D., Karaplis A. C. (1996) Endocrinology 137, 5055–5067 [DOI] [PubMed] [Google Scholar]
- 17.Teixeira C. C., Mansfield K., Hertkorn C., Ischiropoulos H., Shapiro I. M. (2001) Am. J. Physiol. Cell Physiol. 281, C833–C839 [DOI] [PubMed] [Google Scholar]
- 18.Teixeira C. C., Rajpurohit R., Mansfield K., Nemelivsky Y. V., Shapiro I. M. (2003) J. Bone Miner Res. 18, 662–668 [DOI] [PubMed] [Google Scholar]
- 19.Pucci B., Adams C. S., Fertala J., Snyder B. C., Mansfield K. D., Tafani M., Freeman T., Shapiro I. M. (2007) J. Cell. Physiol. 210, 609–615 [DOI] [PubMed] [Google Scholar]
- 20.Okoumassoun L. E., Russo C., Denizeau F., Averill-Bates D., Henderson J. E. (2007) J. Cell. Physiol. 212, 591–599 [DOI] [PubMed] [Google Scholar]
- 21.Amling M., Neff L., Tanaka S., Inoue D., Kuida K., Weir E., Philbrick W. M., Broadus A. E., Baron R. (1997) J. Cell Biol. 136, 205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueda K., Yamanaka Y., Harada D., Yamagami E., Tanaka H., Seino Y. (2007) Bone 41, 13–18 [DOI] [PubMed] [Google Scholar]