

SUMMARY
High Gleason grade prostate carcinomas are aggressive, poorly differentiated tumors that exhibit diminished estrogen receptor β (ERβ) expression. We report that a key function of ERβ and its specific ligand 5α-androstane-3β,17β-diol (3β-adiol) is to maintain an epithelial phenotype and repress mesenchymal characteristics in prostate carcinoma. Stimuli (TGF-β and hypoxia) that induce an epithelial-mesenchymal transition (EMT) diminish ERβ expression, and loss of ERβ is sufficient to promote an EMT. The mechanism involves ERβ-mediated destabilization of HIF-1α and transcriptional repression of VEGF-A. The VEGF-A receptor neuropilin-1 drives the EMT by promoting Snail1 nuclear localization. Importantly, this mechanism is manifested in high Gleason grade cancers, which exhibit significantly more HIF-1α and VEGF expression, and Snail1 nuclear localization compared to low Gleason grade cancers.
INTRODUCTION
The Gleason grading system for prostate carcinoma (PCa) is a key parameter that is extremely valuable for assessing prognosis and choice of therapy (Gleason and Mellinger, 1974; Egevad, 2008a; Egevad, 2008b). This system is based on the grade of histological and cytological differentiation within a tumor and provides a score that ranges from 1 (well-differentiated) to 5 (poorly differentiated). The combined total Gleason score for a tumor, which is used to predict prognosis, reflects the sum of the predominant and secondary grades observed in that cancer. Grade 5 patterns are relatively uncommon and are more frequently found as tertiary foci admixed with grade 4 and to a lesser extent with grade 3 cancers. The presence of tertiary grade 5 cancers within a tumor confers a poor outcome (Trpkov et al., 2009), most likely because these cancers exhibit highly invasive characteristics in histological sections (Gleason and Mellinger, 1974). For this reason, the International Society of Urological Pathology has recommended that biopsy Gleason score be generated by adding tertiary grade 5 to the primary score to provide a more accurate assessment of prognosis (Epstein et al., 2005). A key biological issue that emerges from these observations is the molecular basis for the histological differentiation and range of invasiveness that underlies the Gleason grading system. Although high Gleason grade PCa is characterized by a de-differentiated morphology, the possibility that the progression of Gleason grade reflects, in part, the differential expression of EMT pathways has not been pursued rigorously.
The potential role of estrogen receptors (ERs) in regulating the epithelial-mesenchymal transition (EMT) and aggressive behavior in PCa merits investigation. Although ERα can regulate E-cadherin and the EMT in breast cancer (Dhasarathy et al., 2007; Wang et al., 2007), breast and prostate are different with respect to ER expression and function (Morani et al., 2008). In fact, ERα is predominantly localized in prostatic stroma where its effects on epithelia are considered to be indirect (Prins and Korach, 2008). In contrast, ERβ (Kuiper and Gustafsson, 1997; Lau et al., 2000; Leung et al., 2006; Prins et al., 1998) is expressed in the epithelial compartment of the gland and may regulate epithelial proliferation and differentiation (Imamov et al., 2004). The expression pattern of ERβ in human PCa is of interest because there is an inverse relationship between the expression of ERβ and the progression of PCa to highly aggressive Gleason grades (Leav et al., 2001; Zhu et al., 2004). Given these data, we hypothesized that ERβ functions as a ‘gatekeeper’ of the epithelial phenotype and a repressor of invasion and sought to elucidate the mechanisms involved in ERβ-mediated regulation of EMT in PCa.
RESULTS
ERβ1 sustains an epithelial phenotype and represses mesenchymal characteristics
Gleason grade 5 PCa is distinguished from grade 3 PCa by a merger of neoplastic glands and cytological de-differentiation (Gleason and Mellinger, 1974) with diminished expression of ERβ (Horvath et al., 2001; Leav et al., 2001; Zhu et al., 2004), as well as E-cadherin (Gravdal et al., 2007; Tomita et al., 2000) (Figure 1A). Thus, we hypothesized that ERβ actually regulates the EMT in PCa and that high Gleason grade cancers exhibit EMT characteristics associated with diminished ERβ1 expression. To address this hypothesis, we used PC3 cells initially because these androgen-independent cells express E-cadherin and ERβ, and exhibit epithelial characteristics (Figure 1B). We focused on ERβ1 because it is the only functional ERβ isoform (Leung et al, 2006). Treatment of PC3 cells with TGF-β (Figure 1B) or exposure to hypoxia (Figure 1C) resulted in the transition to a dispersed, fusiform morphology, significant loss of E-cadherin and increased expression of vimentin and N-cadherin. These results were substantiated by immunofluorescence microscopy (Figure S1). Similar data were obtained with LNCaP cells (Figure 1D), which also exhibit epithelial features but are androgen-dependent, indicating that the ability of microenvironmental stimuli to induce an EMT is independent of androgen receptor status (Figure 1D).
Figure 1. ERβ1 and EMT of PCa.
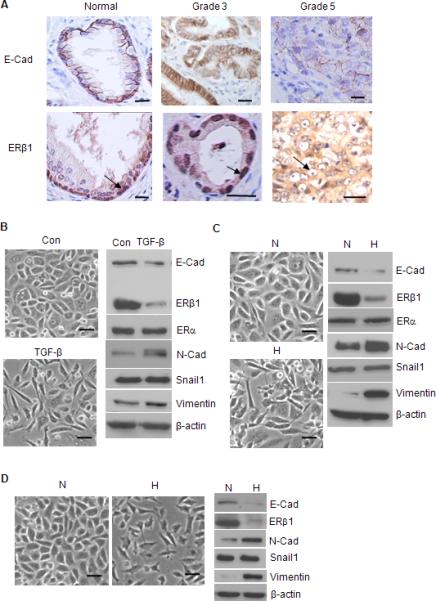
(A) Specimens of normal glandular epithelium, Gleason grade 3 and 5 PCa were stained for E-cadherin and ERβ1 and photographed. ERβ1 is localized in the nuclei of basal cells in the normal prostate and nuclei of grade 3 tumor cells (arrow). In contrast, nuclear ERβ1 staining is absent in grade 5 PCa (arrow). The data are representative of 3 separate specimens for each classification. Scale bars = 20 μm. (B) PC3 cells were treated with PBS (con) or TGF-β for 3 days, photographed and extracts were analyzed for the expression of EMT markers and ERβ1 by immunoblotting. PC3 (C) or LNCaP (D) cells were maintained in either normoxia (N) or hypoxia (H) (0.5% O2) for 24 hrs, photographed and extracts from these cells were immunoblotted as described above. Scale bars = 50 μm. See also Figure S1.
A striking observation was that both TGF-β and hypoxia markedly decreased ERβ1 expression without affecting ERα (Figure 1B-C) suggesting that loss of this ER promotes an EMT in PCa. To assess whether ERβ1 plays a causal role in the EMT, stable clones of PC3 cells were generated that express an ERβ1 shRNA and exhibit diminished ERβ1 expression (Figures 2A-B). These cells have a fusiform morphology, diminished E-cadherin and increased expression of vimentin and N-cadherin in comparison to parental cells or cells that express a scrambled shRNA (Figures 2A-B). In contrast, siRNA-mediated repression of ERα did not affect morphology (data not shown) or the expression of EMT markers (Figure 2C). Quantitative real time-PCR (qPCR) revealed that loss of ERβ1 increased expression of vimentin mRNA and decreased E-cadherin mRNA significantly (Figure 2D). To establish that ERβ1 regulates E-cadherin transcription, we assayed E-cadherin promoter activity in cells that expressed either ERβ1 shRNA (‘knockdown’ cells) and or a scrambled shRNA (control cells) using a reporter construct containing the E-cadherin promoter. As shown in Fig. 2E, ERβ1 knockdown cells had substantially diminished promoter activity compared to control cells.
Figure 2. ERβ1 Sustains An Epithelial Phenotype and Impedes EMT in PCa.

(A) PC3 cells that express either an ERβ1 shRNA (shERβ1) or scrambled shRNA (shCon) or parental cells were photographed. (B) Extracts of these cells were immunoblotted for ERβ1 and EMT markers. (C) Extracts of PC3 cells that express either an ERα siRNA (siERα) or a scrambled siRNA (Scr) were immunoblotted for E-cadherin, N-cadherin, ERα and ERβ1. (D) The relative expression of E-cadherin and vimentin was assayed in PC3 cells that stably express either a control shRNA or ERβ1 shRNA by qPCR using PGK1 as an internal control. The data represent the average of 2 experiments. (E) PC3 cells expressing a scrambled shRNA (Scr) or an ERβ1 shRNA (shERβ1) were transfected with an E-cadherin promoter reporter construct (Src+ and shERβ1+) or pGL2 Basic vector as a control (Scr and shERβ1) and assayed for luciferase activity. The data represent the mean of Firefly luciferase activity normalized to Renilla from 3 separate experiments (±SEM) with P-value (*) <0.05. (F) PC3 cells that express either a control shRNA (shCon) or ERβ1 shRNA (shERβ1) were assessed for their ability to either migrate or invade. The data represent the mean of 3 separate experiments (±SEM) with P-value (*) <0.05. (G) PC3 cells transfected with either control siRNA (siCon) or ERβ1 SMARTpool siRNA (siERβ1) and the parental cells were examined for morphology and EMT marker expression after 3 days. Scale bars = 50 μm.
Loss of ERβ1 expression also resulted in a significant increase in migration and invasion (Figure 2F), functions characteristic of an EMT (Yang and Weinberg, 2008). To exclude the possibility of clonal artifacts during the selection of stable cell lines, we used an siRNA pool for ERβ1 that yielded similar effects on morphology and expression of mesenchymal markers as did the shRNA (Figure 2G). These RNAi data were substantiated using PHTPP (Figure 3A), a highly selective ERβ antagonist (Compton et al., 2004). Treatment of PC3 cells with PHTPP resulted in the acquisition of a spindle-shaped morphology, diminished expression of E-cadherin and increased expression of N-cadherin and vimentin (Figure 3A). Collectively, these data indicate that ERβ1 expression is required to maintain an epithelial phenotype in PCa cells and that an endogenous ERβ1 ligand for is engaged in this process.
Figure 3. The ERβ1 Ligand 3β-Adiol Sustains An Epithelial Phenotype in PCa.

PC3 cells were incubated with either DMSO (Con), the ERβ1 antagonist PHTPP (A) or 3β-Adiol (B) for 3 days, and morphology and EMT marker expression were examined. PC3 cells were treated with TGF-β in the absence or presence of estradiol-17β (E2) (10 nM) or 3β-Adiol (1 μM) and examined for morphology (C) and expression of ERβ1 and EMT markers (D: left panel). Cells treated in the absence or presence of TGF-β were also examined for ERβ1 transcripts by RT-PCR (D: right panel). ERβ1 knockdown cells (shERβ1) untreated or treated with either 3β-Adiol (1 μM) or E2 (10 nM) were examined for morphology and expression of EMT markers (E). Scale bars = 50 μm.
5α-androstane-3β, 17β-diol (3β-Adiol), an ERβ ligand, sustains an epithelial phenotype and impedes a mesenchymal transition in PCa cells
An important issue is the identification of the ERβ1–specific ligand that sustains an epithelial phenotype and impedes an EMT in PCa cells. Although ERβ1 binds estradiol-17β (E2), there is evidence that 3β-Adiol, a metabolite of dihydrotestosterone, is the natural ligand of ERβ1 in the prostate (Guerini et al., 2005). To evaluate the function of this ligand, PC3 cells were treated with either DMSO or 3β-Adiol for 3 days. As shown in Fig. 3B, 3β-Adiol treatment resulted in a more compact, epithelial morphology, consistent with increased expression of E-cadherin, and diminished expression of N-cadherin and vimentin. In contrast, treatment with DMSO had no significant effect on either morphology or expression of EMT markers. Moreover, cells treated with 3β-Adiol but not E2 were unable to undergo TGF-β-induced EMT as evidenced by morphology and expression of EMT markers (Figures 3C-D). We also observed that 3β-Adiol prevented the dimunition in ERβ1 expression that occurs in response to TGF-β stimulation (Figure 3D) suggesting that 3β-Adiol prevents an EMT by stabilizing ERβ1 and enabling it to function to sustain an epithelial phenotype. This hypothesis is further supported by the observation that 3β-Adiol treatment did not affect expression of ERβ1 mRNA during TGF-β-induced EMT (Figure 3D) and that methylation of the ERβ1 promoter, which has been shown to regulate its expression (Zhu et al., 2004) did not change during this EMT (data not shown). The specificity of 3β-Adiol for ERβ1 in maintaining an epithelial phenoptype is evidenced by the inability of this ligand to impede an EMT in cells lacking ERβ1 (Figure 3E).
HIF-1α is destabilized by ERβ1 via proteosomal degradation
Based on our findings that either loss of ERβ1 expression or hypoxia induce EMT and that hypoxia diminishes ERβ1 expression, we assessed a possible relationship between ERβ1 and HIF-1. HIF-1α protein expression is low in PC3 cells but it increased markedly in response to either hypoxia, TGF-β stimulation, knockdown of ERβ1 with both shRNA and siRNA, or PHTPP (Figure 4A). However, expression of HIF-1α transcripts did not change under these conditions (Figures 4A) suggesting that ERβ1 destabilizes HIF-1α protein via proteasomal degradation. To test this hypothesis, we examined HIF-α protein expression in control PC3 cells and ERβ1 knockdown cells in either the absence or presence of the proteasome inhibitor MG132. MG132 increased HIF-β expression dramatically in control cells but neither E2 nor 3β-Adiol had an effect (Figure 4B). In comparison, the elevated level of HIF-1α in ERβ1 knockdown cells was not affected by MG132, E2 or 3β-Adiol (Figure 4B). Importantly, control cells but not ERβ1 knockdown cells treated with MG132 converted from an epithelial to a mesenchymal phenotype, as evidenced by morphology and expression of EMT markers (Figure 4C). These data strongly suggest that ERβ1 destabilizes HIF-1α protein via proteasomal degradation.
Figure 4. ERβ1 Destabilizes HIF-1α Protein and Represses HIF-1-mediated transcription of VEGF-A.
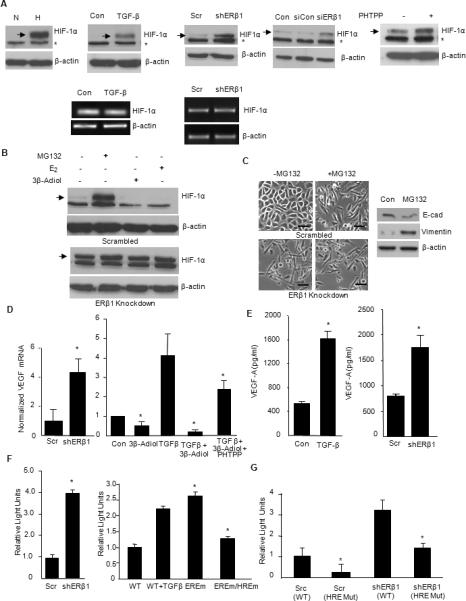
(A) PC3 cells maintained in either normoxia (N) or hypoxia (H) for 24 hrs, treated with PBS (Con) or TGF-β, transfected with control or ERβ1 shRNA or siRNA, or treated with PHTPP were analyzed for the expression of HIF-1α by immunoblotting. * denotes a non-specific band. HIF-1α mRNA was detected by RT-PCR in TGF-β–stimulated cells and shRNA transfected cells. (B) PC3 cells (scrambled control cells) or ERβ1 knockdown cells were treated in the absence or presence of MG132 (1 μM), 3β-Adiol (1 μM) or E2 (10 nM) for 6 hours and immunoblotted for HIF-1α. (C) PC3 cells (scrambled control cells) or ERβ1 knockdown cells were treated in the absence or presence of MG132 (1 μM) for 6 hours and photographed. Scale bars = 50 μm. Extracts of the control cells treated with MG132 were immunoblotted for E-cadherin, vimentin and β-actin. (D) PC3 cells expressing a scrambled shRNA (Scr) or an ERβ1 shRNA (shERβ1) were analyzed for VEGF-A mRNA expression by qPCR (left graph). PC3 cells were treated with PBS (Con) or TGF-β in the absence or presence of 3β-Adiol (1 μM) or 3β-Adiol (1 μM) plus PHTPP (5 μM). After 3 days, cells were analyzed for VEGF-A mRNA expression by qPCR (right graph). (E) VEGF-A secretion in culture medium from PC3 cells treated with PBS (Con) or TGF-β or transfected with control or ERβ1 shRNAs was quantified by ELISA. (F) Scrambled control cells (Scr) or ERβ1 knockdown cells (shERβ1) were transfected with a VEGF promoter reporter construct and luciferase activity normalized to Renilla was measured (left graph). PC3 cells were transfected with a wild type VEGF promoter reporter construct in the absence (Wt) or presence of TGF-β (WT+TGFβ). Concurrently, cells were transfected with the reporter construct containing either a mutated ERE (EREm) or both a mutated ERE and HRE (EREm/HREm) and normalized luciferase activity was measured (right graph). (G) Scrambled control cells (Scr) or ERβ1 knockdown cells (shERβ) were transfected either with a wild-type HRE reporter construct: Scr (Wt) or shERβ (Wt) or with a mutated version of the HRE reporter construct: Scr (mut) or shERβ (mut) under hypoxic conditions for 16-18 hours and normalized luciferase activity was measured. All data are the mean of 3 separate experiments with SEM and P value (*) < 0.05 indicated.
ERβ1 and 3β-Adiol repress HIF-1-mediated transcription of VEGF-A
We hypothesized that VEGF-A is a HIF-1 target gene important for the EMT of PCa that is regulated by 3β-Adiol/ERβ1. To pursue this possibility, we assessed VEGF-A expression by qPCR in either control PC3 cells or ERβ1 knockdown cells and observed that ERβ1 suppresses VEGF-A expression significantly (Figure 4D). TGF-β also induces a dramatic increase in VEGF-A expression (Figure 4D). Interestingly, 3β-Adiol attenuated VEGF expression in PC3 cells and it prevented the ability of TGF-β to increase VEGF-A expression (Figure 4D). These effects of 3β-Adiol were partially blocked by PHTPP further supporting the notion that the interaction of 3β-Adiol with ERβ1 represses VEGF-A expression. To confirm that ERβ1 regulates VEGF-A secretion, we quantified VEGF-A expression in culture medium by ELISA. Indeed, both TGF-β and loss of ERβ1 increased VEGF-A secretion markedly (Fig. 4E).
To elucidate the mechanism of how ERβ1 suppresses VEGF-A expression, we measured VEGF-A promoter activity in PC3 cells using a reporter construct containing the full-length VEGF promoter, which contains an estrogen response element (ERE) and a hypoxia response element (HRE) (Stevens et al., 2003). The latter element is a key regulator of VEGF transcription (Liao and Johnson, 2007). The luciferase activity of this reporter construct was significantly higher in ERβ1 knockdown cells compared to control cells suggesting that ERβ1 is required to suppress promoter activity (Figure 4F). Cells treated with TGF-β also had elevated promoter activity compared to untreated cells (Figure 4F). Interestingly, mutating the ERE in this promoter construct increased luciferase activity supporting our hypothesis that ERβ1 acts as a repressor of VEGF transcription via the ERE (Figure 4F). Moreover, mutating both the ERE and HRE abrogated the increase in transcription observed with the ERE mutant alone (Figure 4F), arguing that the HRE contributes to the de-repression of transcription that occurs when the ERE is mutated. We then examined the role of ERβ1 itself in regulating HRE-mediated transcription of VEGF-A in hypoxia by expressing reporter constructs containing only wild-type or mutated HRE and no ERE in both control and ERβ1 knockdown cells. As shown in Figure 4G, loss of ERβ1 under hypoxic conditions stimulated transcription significantly as compared to the control cells. However, promoter activity in both cell lines was attenuated when the HRE was mutated. Together with the other data shown in Figure 4, we conclude that ERβ1 represses VEGF-A transcription directly using the ERE and indirectly by destabilizing HIF-1α and repressing HIF-1-mediated transcription.
VEGF-A and Neuropilin-1 promote an EMT
The possibility that VEGF-A promotes an EMT is demonstrated by the finding that treatment of PC3 cells with recombinant VEGF165 resulted in a fusiform morphology (Figure 5A), decreased E-cadherin and increased expression of N-cadherin and vimentin (Figure 5A). The expression of ERβ1 did not change indicating that the regulation of VEGF-A is downstream of ERβ1 signaling. Autocrine VEGF signaling in tumor cells is a non-angiogenic mechanism that contributes to their autonomy and aggressive behavior (Bachelder et al., 2001; Bates et al., 2003; Cao et al., 2008; Castro-Rivera et al., 2004). A key VEGF-A receptor implicated in autocrine signaling is neuropilin-1 (NRP1) (Bachelder et al., 2001; Soker et al., 1998). In contrast to VEGF-A, expression of NRP1 did not change in response to either EMT stimuli or loss of ERβ1 expression (data not shown). To elucidate the function of NRP1 during an EMT, we generated NRP1 knockdown PC3 cells using shRNA (Figure 5B). Strikingly, cells with diminished NRP1 expression were resistant to EMT induction by TGF-β treatment compared to control cells as evidenced by their morphology and expression of EMT markers (Figures 5B).
Figure 5. VEGF-A Promotes EMT and the VEGF Receptor NRP1 is Necessary for EMT and Regulates GSK-3β Activity.

(A) PC3 cells were grown in RPMI medium in the absence or presence of recombinant VEGF165 (50 ng/ml) for 24 hours. Cells were photographed and extracts were immunoblotted to assess expression of EMT markers. (B) Photomicrographs of PC3 cells that express either an empty vector (shCon), GFP shRNA (shGFP) or 2 different NRP1 shRNAs (shNRP1A and shNRP1B) were treated with or without TGF-β for 3 days. Extracts from these cells were immunoblotted for NRP1, as well as EMT markers. (C) Extracts from PC3 cells stimulated with either TGF-β, normoxia (N) or hypoxia (H), or ERβ1 knockdown cells (shERβ1) were immunoblotted with Abs specific for pAkt (Ser473), pGSK-3β (Ser9), Akt, GSK-3β and β-actin. (D) Extracts of LNCaP cells maintained in either normoxia (N) or hypoxia (H) for 24 hrs were immunoblotted with the same Abs. (E) PC3 cells were treated in the absence or presence of 3β-Adiol or PHTPP and subsequently analyzed for phospho-GSK3β, total GSK3β and Snail1 expression. Scale bars = 50 μm.
The activities of Akt and GSK-3β, which regulate the EMT, are controlled by ERβ1 and 3β-adiol
Given that NRP1 can regulate Akt/GSK-3β activity (Bachelder et al., 2001) and that GSK-3β impedes an EMT (Zhou et al., 2004; Bachelder et al., 2005; Yook et al., 2005; Yook et al., 2006), we examined the relationship between ERβ1 and GSK-3β activation. Phosphorylation of GSK-3β on Ser9 by Akt inactivates its kinase activity (Doble and Woodgett, 2003). Treatment of PC3 cells with TGF-β or exposure to hypoxia resulted in a significant increase in the relative phosphorylation of both Akt and GSK-3β as assessed by immunoblotting (Figure 5C). Similar results were obtained with LNCaP cells and ERβ1 knockdown cells (Figures 5C-D). These data indicate that ERβ1 sustains GSK-3β activation and that loss of its expression during an EMT activates the pAkt/pGSK-3β pathway. To show that this signaling is ligand-dependent, cells were treated with either 3β-Adiol or PHTPP, and subsequently analyzed for GSK-3β activation. Cells treated with 3β-Adiol had a decrease in GSK-3β phosphorylation compared to control, whereas cells treated with PHTPP had a significant increase in GSK-3β phosphorylation (Figure 5E). In contrast, E2 did not affect GSK-3β phosphorylation (data not shown). These data strongly suggest that 3β-Adiol is the endogenous ligand for ERβ1 that sustains GSK-3β activation.
Snail1 nuclear localization is regulated by 3β-adiol/ERβ1 and VEGF-A/NRP1
Given that Snail1 expression in tumors often correlates with aggressive disease and poor outcome (Blanco et al., 2002; Moody et al., 2005; Wu et al., 2009), we were surprised that Snail1 expression did not change in response to either TGF-β, hypoxia, VEGF-A, loss of ERβ1 or NRP1 (Figures 1, 2, 5). To assess the potential role of Snail1 in the EMT, we used siRNA to decrease its expression in ERβ1 knockdown cells. Indeed, reduction of Snail1 expression caused a reversion to a more epithelial morphology and decreased the expression of vimentin and N-cadherin with a concomitant increase in E-cadherin (Figure 6A). These observations and the finding that Snail1 stability and nuclear localization can be regulated by phosphorylation and EMT pathways (Dominguez et al., 2003; Yook et al., 2005; Zhou et al., 2004) prompted us to assess the intracellular localization of Snail1. Surprisingly, the EMT induced by either hypoxia, VEGF-A or TGF-β was coincident with a significant translocation of Snail1 from the cytoplasm to the nucleus as assessed by immunofluorescence microscopy using a Snail1 Ab (Figures 6B-C). This conclusion was strengthened by the finding that loss of ERβ1 resulted in a significant increase in the nuclear localization of a GFP-Snail1 construct (Zhou et al., 2004) (Figure 6D). In contrast, treatment of PC3 cells with 3β-adiol reduced the basal localization of Snail1 in the nucleus significantly and it prevented the increase in Snail1 nuclear localization that occurs in response to TGF-β stimulation (Figure 6E). Importantly, treatment with LiCl2, a GSK-3β inhibitor, increased the nuclear localization of Snail1 significantly providing evidence that the signaling pathway that is repressed by ERβ1 regulates Snail1 localization (Figure 6C).
Figure 6. ERβ1 and EMT Regulate Snail1 Nuclear Localization.
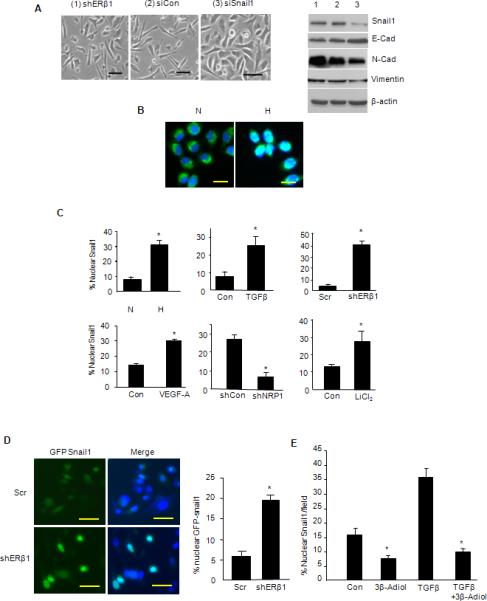
(A) PC3 cells that express an ERβ1 shRNA were transfected with either a control siRNA (siCon) or Snail1 siRNA (siSnail1), and analyzed for morphology and expression of EMT markers. (B) Snail1 was visualized by immunofluorescence microscopy in PC3 cells maintained in either normoxia (N) or hypoxia (H) for 48 hrs. The photomicrographs shown represent the merged images obtained from Snail1 staining (green; FITC) and nuclear staining (blue; DAPI). Note that in normoxia, Snail1 staining is predominantly cytoplasmic and excluded from nuclei. In hypoxia, however, Snail1 localization in nuclei is evidenced by ‘whitish-blue’ staining. (C) The percentage of nuclei that had Snail1 staining was quantified in PC3 cells maintained in normoxia (N) and hypoxia (H), and in cells stimulated with TGF-β or VEGF-A, as well as in PC3 cells in which ERβ1 or NRP1 expression was depleted by shRNA. Snail1 nuclear localization was also quantified in PC3 cells treated with LiCl2, a GSK-3β inhibitor. The data represent the mean of 3 separate experiments with SEM and P value (*) <0.05 indicated. (D) PC3 cells that express either a scrambled shRNA (Scr) or ERβ1 shRNA (shERβ1) were transfected with a GFP-Snail1 construct. GFP and DAP1 were visualized and the images merged. Note the nuclear localization of GFP-Snail1 as evidenced by the whitish blue staining that is associated with loss of ERβ1 expression. The bar graph represents the quantification of nuclear GFP-Snail1 from 3 independent experiments (±SEM) and P value (*) <0.05 indicated. (E) PC3 cells were treated with TGB-β in the absence or presence of 3β-Adiol (1 μM) and nuclear Snail1 was quantified. The data represent the mean of 3 separate experiments with SEM and P-value (*) <0.05 indicated.
HIF-1α/VEGF/Snail1 Pathway is Manifested in High Gleason Grade PCa
A critical question that arises from our in vitro data is whether the expression of HIF-1α, VEGF-A and nuclear Snail1 correlates with Gleason grade in human PCa, and whether the expression of these proteins correlates inversely with ERβ1 in the same specimens as predicted by our hypothesis. To address this question, we used a semi-quantitative analysis of IHC staining to assess expression of these proteins in specimens from 30 PCa patients of which 20 were Gleason grade 3 and 10 were grade 5. Expression of ERβ1 was significantly higher in the nuclei of grade 3 compared to grade 5 PCa (Figure 7A), confirming previous studies (Horvath et al., 2001; Leav et al., 2001; Zhu et al., 2004). In marked contrast, however, we observed intense, widespread nuclear HIF-1α expression in grade 5 cells that was significantly less in grade 3 cells (Figure 7B). Intense VEGF-A immunostaining was evident in Gleason grade 5 tumor cells when compared to grade 3 tumor cells, and semi-quantitative analysis of multiple specimens revealed that this difference is significant (Figure 7C). This observation was strengthened by qPCR analysis of VEGF-A expression from microdissected specimens of human PCa, which demonstrated that grade 5 tumor cells had significantly higher VEGF-A mRNA expression than did grade 3 tumor cells (Figure 7D).
Figure 7. HIF-1α/VEGF/Snail1 Pathway is Manifested in High Gleason Grade PCa.

Thirty specimens of human PCa including 20 Gleason grade 3 tumors and 10 Gleason grade 5 tumors were immunostained for ERβ1 (A), HIF-1α (B), VEGF-A (C) and Snail1 (E). Semi-quantitative analysis of IHC staining was performed for all samples that assessed both the percentage of cells stained and the intensity of the staining, and this analysis is reported as the Quotient (Q) of these two parameters (±SD). The significance of the difference in Q between Gleason grade 3 and 5 as determined by Students t test is shown for each bar graph. Photomicrographs representative of the mean Q for each IHC staining are shown. (D) Microdissected samples from grade 3 and grade 5 PCa were analyzed for the expression of VEGF-A mRNA by qPCR and the data represent the average of 7 separate specimens for each grade. Red scale bars = 25 μm; black scale bars = 50 μm.
Our data on Snail1 localization in vitro and its regulation by ERβ1 prompted us to compare Snail1 nuclear localization in Gleason grade 3 and 5 PCa. In grade 3 PCa, only a scattered number of positively stained nuclei were apparent. However, intense widespread nuclear Snail1 staining was evident in the majority of grade 5 tumor cells (Figure 7E) accompanied by a decline in ERβ1 expression. These results are consistent with the hypothesis that ERβ1 restricts Snail1 to the cytoplasm but it becomes translocated to the nucleus when receptor levels decline.
We conclude from our data that a major function of the 3β-adiol/ERβ1 complex in PCa is to impede a mesenchymal transition and consequent invasive behavior by a mechanism that involves its ability to destabilize HIF-1α and repress transcription of VEGF-A, which drives an EMT by enhancing nuclear localization of Snail1 (Figure 8). Most importantly, key features of this pathway are manifested in high Gleason grade PCa.
Figure 8. Proposed model for how ERβ1 sustains an epithelial phenotype and represses a mesenchymal phenotype.

The interaction of ERβ1 with its ligand 3β-Adiol represses an EMT by destabilizing HIF-1α and inhibiting VEGF-A transcription. Stimuli that induce an EMT diminish ERβ1 expression resulting in increased VEGF-A expression and the consequent activation of a VEGF-A/NRP1 signaling pathway that inhibits GSK-3β and promotes Snail1 nuclear localization.
DISCUSSION
Our data highlight a pivotal role for ERβ1 and its natural ligand 3β-adiol in sustaining an epithelial phenotype and repressing the acquisition of mesenchymal characteristics and invasive behavior in PCa. The key mechanism that we elucidate to account for this function of ERβ1 is that it destabilizes HIF-1α and represses transcription of VEGF-A, a growth factor that can promote an EMT (Wanami et al., 2008; Yang et al., 2006). The significance of our in vitro data is strengthened by the fact that key components of this pathway we describe are detected in high Gleason-grade PCa, which is characterized by highly invasive and aggressive behavior. Moreover, other studies support the existence and clinical relevance of EMT-like processes in PCa, e.g., (Acevedo et al., 2007). Of note, a recent study that compared gene expression during prostate development to PCa suggested that those tumors with a transcript profile consistent with branching morphogenesis, which involves EMT, were likely to be invasive and have an early relapse after surgical resection (Pritchard et al., 2009).
We identify 3β-adiol, a 5α-DHT metabolite, as a specific ligand for ERβ1 that mediates the ability of this ER to sustain an epithelial phenotype and repress EMT and invasion. 3β-adiol binds to ERβ but not ERα or the androgen receptor (Kuiper et al., 1997). Interestingly, E2, a ubiquitous ligand for both ERα and ERβ, was ineffective in regulating E-cadherin and the EMT. Our data also suggest that an important function of 3β-adiol is to maintain ERβ expression, an observation also made in normal rat prostate (Oliveira et al., 2007). These data strengthen the hypothesis that 3β-adiol is the primary ligand for ERβ in the prostate and that its major function is to maintain a differentiated, epithelial phenotype. This conclusion is supported by the fact that the concentration of 3β-adiol in the prostate gland is 100–fold higher than that of E2 (Voigt and Bartsch, 1986).
The key mechanistic finding in our study is that ERβ1 represses VEGF-A transcription by a complex mechanism that involves its ability to regulate two key response elements within the VEGF promoter: the ERE and the HRE. This finding is distinct from studies demonstrating that E2 stimulates VEGF transcription in the breast and uterus (Buteau-Lozano et al., 2002; Hyder, 2006; Stoner et al., 2004). Our data suggest that ERβ1 represses VEGF transcription directly via the ERE, a function that may require recruitment of co-repressors such as NCoR (Girault et al., 2003). Importantly, we also conclude that ERβ1 represses VEGF-A transcription indirectly by destabilizing HIF-1α and impeding HIF-1-mediated transcription of VEGF. This conclusion is supported by our proteasome inhibitor data and mutational analysis of the HRE and ERE in the VEGF-A promoter, as well as the observation that ERβ and HIF-1α can associate physically (Lim et al., 2009).
We implicate VEGF-A as an ERβ1-regulated HIF-1 target gene that links 3β-adiol/ERβ1 to Snail1 localization and the EMT. This function of VEGF-A in PCa cells is of interest because the hypothesis that VEGF and its receptors impact the behavior of tumor cells directly is gaining prominence (Bachelder et al., 2001; Bates et al., 2003; Cao et al., 2008; Castro-Rivera et al., 2004; Su et al., 2006). The significance of our results is that we provide a mechanism for how VEGF expression is regulated pathophysiologically by ERβ1 and establish the relevance of this mechanism to PCa by demonstrating that VEGF-A expression in PCa correlates with Gleason grade. Moreover, our data indicate that the expression of HIF-1α itself and HIF-1α target genes is associated with a mesenchymal, aggressive phenotype. We note also an emerging relationship among ERβ1, VEGF and hypoxia. Hypoxia selects for the survival of more aggressive tumor cells (Brown, 1999) and it induces an EMT as shown here and previously (Higgins et al., 2007; Lester et al., 2007). Hypoxia also stimulates VEGF expression (Harris, 2002) and diminishes ERβ1 expression as we demonstrate. Thus, hypoxia emerges as one mechanism that facilitates the acquisition of mesenchymal characteristics in PCa cells by suppressing ERβ1 and stimulating HIF-1α-mediated VEGF expression. Interestingly, a recent study concluded that cells from PCa patients with a poor prognosis exhibited a hypoxic phenotype (Nanni et al., 2009).
The ability of ERβ1 to control NRP1 function by regulating VEGF-A expression establishes a connection between this ER and VEGF receptor signaling. Since the seminal observation that NRP1 can function as a VEGF receptor (Soker et al., 1998), studies have demonstrated its functional importance in angiogenesis and cancer (Guttmann-Raviv et al., 2006). However, the ability of NRP1 to regulate Snail1 localization is unexpected and may contribute to the reported association of NRP1 with PCa progression (Latil et al., 2000; Miao et al., 2000) and tumor de-differentiation (Cao et al., 2008). Our observation that ERβ1 can impact NRP1 function by controlling VEGF expression adds to our understanding of how this VEGF receptor can be regulated in cancer. In addition, our identification of a VEGF-A/NRP1 pathway that is regulated by ERβ1, promotes an EMT and distinguishes high Gleason grade PCa may be appropriate and feasible for therapeutic targeting. Adjuvant therapy aimed at targeting VEGF (bevacizumab) is being used for the clinical management of several tumors (Ferrara, 2005) and recent data suggest that NRP antibodies have the potential to have clinical efficacy (Caunt et al., 2008; Gray et al., 2008; Pan et al., 2007). Although the overarching assumption had been that such drugs function by blocking tumor angiogenesis, it is likely that they also target tumor cells directly, and that patients with high Gleason grade PCa may benefit from such anti-VEGF/NRP therapy.
The essence of our study is that 3β-adiol/ERβ1 sustain E-cadherin transcription and prevent an EMT in PCa cells by sequestering Snail1 in the cytoplasm. Although other transcription factors that regulate E-cadherin may be important for PCa progression such as SIP1 (ZEB2) (Kong et al., 2009), our data indicate that ERβ1 regulates Snail1. The mechanism involved is linked to the regulation of GSK-3β activity by 3β-adiol/ERβ1, an enzyme that is critical for regulating Snail1 localization and stability and, as a consequence, the EMT (Bachelder et al., 2005; Yook et al., 2006; Zhou et al., 2004). The mechanism we propose for the regulation of Snail1 by ERβ1 is distinct from the regulation of Snail1 by ERα in breast cancer, which involves the ERα-dependent regulation of MTA3, a repressor of Snail1 transcription (Fearon, 2003; Fujita et al., 2003). ERα has also been reported to impede the EMT and invasiveness of breast cancer cells by inhibiting the synthesis of RelB (Wang et al., 2007).
Our finding that Snail1 localization is predominantly nuclear in Gleason grade 5 PCa but largely cytoplasmic in grade 3 PCa is distinct from other studies that have shown that Snail1 expression but not localization in other cancers differs among tumor subtypes or stages, e.g., (Blanco et al., 2002; Moody et al., 2005). A potentially important and useful implication of our data is that nuclear Snail1 could serve as a biomarker to predict the propensity of a given tumor to progress to advanced disease. This possibility is particularly relevant and timely given the uncertainty and ineffectiveness of PSA screening in predicting outcome (Andriole et al., 2009; Schroder et al., 2009).
In summary, our data contribute to an understanding of the molecular basis for the Gleason grading system and suggest a mechanism that promotes for the aggressive and invasive nature of high Gleason grade tumors.
EXPERIMENTAL PROCEDURES
Clinical specimens
Tissue samples of defined Gleason grades were obtained from the UMASS Cancer Center Tissue Bank with approval of the Institutional Review Board (IRB) of UMASS Medical School. The IRB granted a waiver for obtaining patient consent in accordance with NIH guidelines because these were pre-existing, de-identified specimens. Tissue microarrays containing Gleason grade 5 PCa were kindly provided by Dr M. Loda of the Dana Farber Cancer Institute, Boston MA. Specimens were stained with Abs specific for ERβ1 (Gene Tex), E-cadherin (Abcam) and Snail1 (Abcam), VEGF-A (R&D Systems) and HIF-1α (Novus). Frozen specimens were microdissected by laser capture microscopy (Arcturus PixCell 2) as described (Loric et al., 2001) to obtain pure populations of tumor cells of defined Gleason grades. RNA was isolated from these specimens using the RNeasy kit (Qiagen). Additional details on the clinical specimens used and method of analysis are provided in Supplementary Experimental Procedures.
Cells and Reagents
The human prostate cancer cell lines (LNCaP and PC3) were obtained from American Type Culture Collection (ATCC: Manassas, VA). TGF-β experiments were performed by incubating cells with recombinant human TGF-β (5 ng/ml; Peprotech) for 3-4 days. For hypoxia experiments, cells were grown in a Ruskinn Hypoxic Chamber (0.5% O2; 5% CO2) for 18-20 hours. The psiSTRIKE™ U6 Hairpin Cloning System (Promega) was used for DNA-directed shRNA expression using sequences optimized for ERβ1 (Mak et al., 2006). Cells were also transfected transiently with On-Target Plus SMARTpool siRNAs (Dharmacon, CO) for ERβ1, ERβ and Snail1. These target sequences have been published by Dharmacon. Non-targeting pools were used as negative controls. Lentiviruses (pLKO.1) containing the NRP1 shRNA Oligonucleotide ID TRCN0000063527 (Open Biosystems, Huntsville, AL) and pLKO.1 empty vector or pLKO-GFP controls were generated and used to infect PC3 cells following standard protocols. The reporter gene, p11w, which contains the wild type HRE (hypoxia responsive element) and the mutated version, p11m fused to luciferase, were obtained from ATCC. The E-cadherin promoter reporter gene (pGL2Basic-EcadK1) and GFP Snail WT plasmids were obtained from Addgene. The Renillaluciferase plasmid was purchased from Promega.
Biochemical analyses
Total RNA was isolated using the TRI reagent (Sigma) and reverse-transcribed to cDNA using Superscript II reverse transcriptase (Invitrogen). Details on primers used and PCR methods are provided in Supplementary Methods. For immunoblots, cells were extracted with RIPA buffer containing EDTA and EGTA (Boston BioProducts) with a protease inhibitor cocktail, and blots were performed as described previously (Bae et al., 2008) using primary Abs against ERα, ERβ1, E-cadherin, N-cadherin, vimentin, Snail1, pAkt (Ser473), total Akt, pGSK3β (Ser9), total GSK3β, NRP1, HIF-1α and β-actin, which were obtained from Santa Cruz BioTechnology (CA), Sigma (MO), Abcam (MA) or Gene Tex, Inc. (CA). Estradiol-17β (E2), 3β-androstane-diol (3β-adiol) and PHTPP were obtained from Tocris. The proteasome inhibitor MG132 was obtained from Calbiochem. VEGF-A levels in culture medium were quantified by ELISA (R&D Systems).
Snail1 localization
For immunofluorescence microscopy, cells were maintained under the conditions described in Fig. 3B, fixed with para-formaldehyde and incubated with a Snail1 Ab (Abcam, MA) and a fluorescein isothiocynate-conjugated secondary Ab (Jackson Immunoresearch, West Grove, PA). Coverslips were mounted on slides with SlowFade Antifade reagent containing DAPI (Molecular Probes). For localization of exogenous Snail1, cells were transfected with a GFP-Snail1 construct (Addgene) using Lipofectamine™2000 and analyzed as described above.
Analysis of VEGF-A promoter
The VEGF promoter was PCR amplified from human genomic DNA. The PCR amplified fragment was confirmed by restriction mapping and cloned at the Xho1-Hind III site into the pGL3 basic vector (Promega). Site-directed mutagenesis was used to mutate the ERE sequence from (AATCAGACTGACT) to (AACTGGACCAACT) and the HRE sequence from (TACGTG) to (TAAAAG). Details on this analysis are provided in Supplementary Experimental Procedures.
Luciferase assays
PC3 cells were transfected with the desired plasmids and the Renilla luciferase construct to normalize for transfection efficiency. Relative Light Units were calculated as the ratio of Firefly luciferase to Renilla luciferase activity (normalized luciferase activity). The protocol used for transfection and measurement of luciferase activity has been described previously (Mak et al., 2006).
Migration and invasion assays
Assays were performed using 6.5 mm Transwell chambers (8 μm pore size) that had been coated with either collagen I or Matrigel (BD Biosciences, Bedford, MA) for migration and invasion, respectively, as described previously (Shaw et al., 1997). After 5 hours, the cells that had translocated to the lower surface of the filters were fixed in methanol. The fixed membranes were mounted on glass slides using Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). Assays were quantified by counting the number of stained nuclei in 5 independent fields in each Transwell.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants CA CA89209, CA1107548 (A.M.M.) and RO3CA121351 (P.M.), NIDDK P30DK32520 and an ACS Postdoctoral Fellowship (X.Y.). We thank Dr. Shaolei Lu and Cheng Chang for expert assistance, and Drs. Zhong Jiang (UMASS Medical School) and Massimo Loda (Dana Farber Cancer Institute, Boston, MA) for providing clinical specimens.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SIGNIFICANCE: The Gleason grading system for prostate cancer is based on the degree of histological differentiation and it is valuable for assessing prognosis and choice of therapy. Although high Gleason grades portend poor prognosis, the molecular basis for the differentiation that underlies Gleason grading has not been defined. We now report that loss of ERβ, which distinguishes high from low Gleason grade tumors, is associated with the expression of an EMT program of de-differentiation that involves HIF-1 and VEGF/neuropilin signaling. These findings should facilitate our understanding of the mechanisms responsible for the aggressive behavior exhibited by these high-grade cancers and the development of effective methods for their therapeutic intervention.
References
- Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, Ayala GE, Peterson LE, Ittmann M, Spencer DM. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12:559–571. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, Chia D, Church TR, Fouad MN, Gelmann EP, Kvale PA, Reding DJ, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009;360:1310–1319. doi: 10.1056/NEJMoa0810696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelder RE, Crago A, Chung J, Wendt MA, Shaw LM, Robinson G, Mercurio AM. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res. 2001;61:5736–5740. [PubMed] [Google Scholar]
- Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29–33. doi: 10.1083/jcb.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae D, Lu S, Taglienti CA, Mercurio AM. Metabolic stress induces the lysosomal degradation of neuropilin-1 but not neuropilin-2. J Biol Chem. 2008 doi: 10.1074/jbc.M804203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates RC, Goldsmith JD, Bachelder RE, Brown C, Shibuya M, Oettgen P, Mercurio AM. Flt-1 (VEGFR-1)-dependent survival characterizes the epithelial-mesenchymal transition of colonic organoids. Current Biology. 2003;13:1721–1727. doi: 10.1016/j.cub.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, Nieto MA. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21:3241–3246. doi: 10.1038/sj.onc.1205416. [DOI] [PubMed] [Google Scholar]
- Brown JM. The hypoxic cell: a target for selective cancer therapy--eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res. 1999;59:5863–5870. [PubMed] [Google Scholar]
- Buteau-Lozano H, Ancelin M, Lardeux B, Milanini J, Perrot-Applanat M. Transcriptional regulation of vascular endothelial growth factor by estradiol and tamoxifen in breast cancer cells: a complex interplay between estrogen receptors alpha and beta. Cancer Res. 2002;62:4977–4984. [PubMed] [Google Scholar]
- Cao Y, Wang L, Nandy D, Zhang Y, Basu A, Radisky D, Mukhopadhyay D. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res. 2008;68:8667–8672. doi: 10.1158/0008-5472.CAN-08-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Rivera E, Ran S, Thorpe P, Minna JD. Semaphorin 3B (SEMA3B) induces apoptosis in lung and breast cancer, whereas VEGF165 antagonizes this effect. Proc Natl Acad Sci U S A. 2004;101:11432–11437. doi: 10.1073/pnas.0403969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caunt M, Mak J, Liang WC, Stawicki S, Pan Q, Tong RK, Kowalski J, Ho C, Reslan HB, Ross J, et al. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell. 2008;13:331–342. doi: 10.1016/j.ccr.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Compton DR, Sheng S, Carlson KE, Rebacz NA, Lee IY, Katzenellenbogen BS, Katzenellenbogen JA. Pyrazolo[1,5-a]pyrimidines: estrogen receptor ligands possessing estrogen receptor beta antagonist activity. J Med Chem. 2004;47:5872–5893. doi: 10.1021/jm049631k. [DOI] [PubMed] [Google Scholar]
- Dhasarathy A, Kajita M, Wade PA. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol Endocrinol. 2007;21:2907–2918. doi: 10.1210/me.2007-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Montserrat-Sentis B, Virgos-Soler A, Guaita S, Grueso J, Porta M, Puig I, Baulida J, Franci C, Garcia de Herreros A. Phosphorylation regulates the subcellular location and activity of the snail transcriptional repressor. Mol Cell Biol. 2003;23:5078–5089. doi: 10.1128/MCB.23.14.5078-5089.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egevad L. Recent trends in Gleason grading of prostate cancer: I. Pattern interpretation. Anal Quant Cytol Histol. 2008a;30:190–198. [PubMed] [Google Scholar]
- Egevad L. Recent trends in gleason grading of prostate cancer. II. Prognosis, reproducibility and reporting. Anal Quant Cytol Histol. 2008b;30:254–260. [PubMed] [Google Scholar]
- Epstein JI, Allsbrook WC, Jr., Amin MB, Egevad LL. The 2005 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Am J Surg Pathol. 2005;29:1228–1242. doi: 10.1097/01.pas.0000173646.99337.b1. [DOI] [PubMed] [Google Scholar]
- Fearon ER. Connecting estrogen receptor function, transcriptional repression, and E-cadherin expression in breast cancer. Cancer Cell. 2003;3:307–310. doi: 10.1016/s1535-6108(03)00087-4. [DOI] [PubMed] [Google Scholar]
- Ferrara N. VEGF as a therapeutic target in cancer. Oncology. 2005;69(Suppl 3):11–16. doi: 10.1159/000088479. [DOI] [PubMed] [Google Scholar]
- Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207–219. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- Girault I, Lerebours F, Amarir S, Tozlu S, Tubiana-Hulin M, Lidereau R, Bieche I. Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin Cancer Res. 2003;9:1259–1266. [PubMed] [Google Scholar]
- Gleason DF, Mellinger GT. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J Urol. 1974;111:58–64. doi: 10.1016/s0022-5347(17)59889-4. [DOI] [PubMed] [Google Scholar]
- Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007;13:7003–7011. doi: 10.1158/1078-0432.CCR-07-1263. [DOI] [PubMed] [Google Scholar]
- Gray MJ, Van Buren G, Dallas NA, Xia L, Wang X, Yang AD, Somcio RJ, Lin YG, Lim S, Fan F, et al. Therapeutic targeting of neuropilin-2 on colorectal carcinoma cells implanted in the murine liver. J Natl Cancer Inst. 2008;100:109–120. doi: 10.1093/jnci/djm279. [DOI] [PubMed] [Google Scholar]
- Guerini V, Sau D, Scaccianoce E, Rusmini P, Ciana P, Maggi A, Martini PG, Katzenellenbogen BS, Martini L, Motta M, Poletti A. The androgen derivative 5alpha-androstane-3beta,17beta-diol inhibits prostate cancer cell migration through activation of the estrogen receptor beta subtype. Cancer Res. 2005;65:5445–5453. doi: 10.1158/0008-5472.CAN-04-1941. [DOI] [PubMed] [Google Scholar]
- Guttmann-Raviv N, Kessler O, Shraga-Heled N, Lange T, Herzog Y, Neufeld G. The neuropilins and their role in tumorigenesis and tumor progression. Cancer Lett. 2006;231:1–11. doi: 10.1016/j.canlet.2004.12.047. [DOI] [PubMed] [Google Scholar]
- Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath LG, Henshall SM, Lee CS, Head DR, Quinn DI, Makela S, Delprado W, Golovsky D, Brenner PC, O'Neill G, et al. Frequent loss of estrogen receptor-beta expression in prostate cancer. Cancer Res. 2001;61:5331–5335. [PubMed] [Google Scholar]
- Hyder SM. Sex-steroid regulation of vascular endothelial growth factor in breast cancer. Endocr Relat Cancer. 2006;13:667–687. doi: 10.1677/erc.1.00931. [DOI] [PubMed] [Google Scholar]
- Imamov O, Morani A, Shim GJ, Omoto Y, Thulin-Andersson C, Warner M, Gustafsson JA. Estrogen receptor beta regulates epithelial cellular differentiation in the mouse ventral prostate. Proc Natl Acad Sci U S A. 2004;101:9375–9380. doi: 10.1073/pnas.0403041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong D, Li Y, Wang Z, Banerjee S, Ahmad A, Kim HR, Sarkar FH. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells. 2009;27:1712–1721. doi: 10.1002/stem.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Gustafsson JA. The novel estrogen receptor-beta subtype: potential role in the cell- and promoter-specific actions of estrogens and anti-estrogens. FEBS Lett. 1997;410:87–90. doi: 10.1016/s0014-5793(97)00413-4. [DOI] [PubMed] [Google Scholar]
- Latil A, Bieche I, Pesche S, Valeri A, Fournier G, Cussenot O, Lidereau R. VEGF overexpression in clinically localized prostate tumors and neuropilin-1 overexpression in metastatic forms. Int J Cancer. 2000;89:167–171. doi: 10.1002/(sici)1097-0215(20000320)89:2<167::aid-ijc11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Lau KM, LaSpina M, Long J, Ho SM. Expression of estrogen receptor (ER)-alpha and ER-beta in normal and malignant prostatic epithelial cells: regulation by methylation and involvement in growth regulation. Cancer Res. 2000;60:3175–3182. [PubMed] [Google Scholar]
- Leav I, Lau KM, Adams JY, McNeal JE, Taplin ME, Wang J, Singh H, Ho SM. Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. Am J Pathol. 2001;159:79–92. doi: 10.1016/s0002-9440(10)61676-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178:425–436. doi: 10.1083/jcb.200701092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung YK, Mak P, Hassan S, Ho SM. Estrogen receptor (ER)-beta isoforms: a key to understanding ER-beta signaling. Proc Natl Acad Sci U S A. 2006;103:13162–13167. doi: 10.1073/pnas.0605676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- Lim W, Cho J, Kwon HY, Park Y, Rhyu MR, Lee Y. Hypoxia-inducible factor 1 alpha activates and is inhibited by unoccupied estrogen receptor beta. FEBS Lett. 2009;583:1314–1318. doi: 10.1016/j.febslet.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Loric S, Paradis V, Gala JL, Berteau P, Bedossa P, Benoit G, Eschwege P. Abnormal E-cadherin expression and prostate cell blood dissemination as markers of biological recurrence in cancer. Eur J Cancer. 2001;37:1475–1481. doi: 10.1016/s0959-8049(01)00143-5. [DOI] [PubMed] [Google Scholar]
- Mak P, Leung YK, Tang WY, Harwood C, Ho SM. Apigenin suppresses cancer cell growth through ERbeta. Neoplasia. 2006;8:896–904. doi: 10.1593/neo.06538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao HQ, Lee P, Lin H, Soker S, Klagsbrun M. Neuropilin-1 expression by tumor cells promotes tumor angiogenesis and progression. Faseb J. 2000;14:2532–2539. doi: 10.1096/fj.00-0250com. [DOI] [PubMed] [Google Scholar]
- Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8:197–209. doi: 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Morani A, Warner M, Gustafsson JA. Biological functions and clinical implications of oestrogen receptors alfa and beta in epithelial tissues. J Intern Med. 2008;264:128–142. doi: 10.1111/j.1365-2796.2008.01976.x. [DOI] [PubMed] [Google Scholar]
- Nanni S, Benvenuti V, Grasselli A, Priolo C, Aiello A, Mattiussi S, Colussi C, Lirangi V, Illi B, D'Eletto M, et al. Endothelial NOS, estrogen receptor beta, and HIFs cooperate in the activation of a prognostic transcriptional pattern in aggressive human prostate cancer. J Clin Invest. 2009;119:1093–1108. doi: 10.1172/JCI35079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AG, Coelho PH, Guedes FD, Mahecha GA, Hess RA, Oliveira CA. 5alpha-Androstane-3beta,17beta-diol (3beta-diol), an estrogenic metabolite of 5alpha-dihydrotestosterone, is a potent modulator of estrogen receptor ERbeta expression in the ventral prostrate of adult rats. Steroids. 2007;72:914–922. doi: 10.1016/j.steroids.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. 2007;11:53–67. doi: 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Prins GS, Korach KS. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids. 2008;73:233–244. doi: 10.1016/j.steroids.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins GS, Marmer M, Woodham C, Chang W, Kuiper G, Gustafsson JA, Birch L. Estrogen receptor-beta messenger ribonucleic acid ontogeny in the prostate of normal and neonatally estrogenized rats. Endocrinology. 1998;139:874–883. doi: 10.1210/endo.139.3.5827. [DOI] [PubMed] [Google Scholar]
- Pritchard C, Mecham B, Dumpit R, Coleman I, Bhattacharjee M, Chen Q, Sikes RA, Nelson PS. Conserved Gene Expression Programs Integrate Mammalian Prostate Development and Tumorigenesis. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-07-6817. [DOI] [PubMed] [Google Scholar]
- Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, Nelen V, Kwiatkowski M, Lujan M, Lilja H, Zappa M, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320–1328. doi: 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform- specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- Stevens A, Soden J, Brenchley PE, Ralph S, Ray DW. Haplotype analysis of the polymorphic human vascular endothelial growth factor gene promoter. Cancer Res. 2003;63:812–816. [PubMed] [Google Scholar]
- Stoner M, Wormke M, Saville B, Samudio I, Qin C, Abdelrahim M, Safe S. Estrogen regulation of vascular endothelial growth factor gene expression in ZR-75 breast cancer cells through interaction of estrogen receptor alpha and SP proteins. Oncogene. 2004;23:1052–1063. doi: 10.1038/sj.onc.1207201. [DOI] [PubMed] [Google Scholar]
- Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY, Chou CH, Jeng YM, Wang MY, Chang KJ, et al. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell. 2006;9:209–223. doi: 10.1016/j.ccr.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET, Jansen CF, Bussemakers MJ, Schalken JA. Cadherin switching in human prostate cancer progression. Cancer Res. 2000;60:3650–3654. [PubMed] [Google Scholar]
- Trpkov K, Zhang J, Chan M, Eigl BJ, Yilmaz A. Prostate cancer with tertiary Gleason pattern 5 in prostate needle biopsy: clinicopathologic findings and disease progression. Am J Surg Pathol. 2009;33:233–240. doi: 10.1097/PAS.0b013e31817fb3bd. [DOI] [PubMed] [Google Scholar]
- Voigt KD, Bartsch W. Intratissular androgens in benign prostatic hyperplasia and prostatic cancer. J Steroid Biochem. 1986;25:749–757. doi: 10.1016/0022-4731(86)90304-3. [DOI] [PubMed] [Google Scholar]
- Wanami LS, Chen HY, Peiro S, Garcia de Herreros A, Bachelder RE. Vascular endothelial growth factor-A stimulates Snail expression in breast tumor cells: implications for tumor progression. Exp Cell Res. 2008;314:2448–2453. doi: 10.1016/j.yexcr.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F, Chalbos D, Sonenshein GE. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat Cell Biol. 2007;9:470–478. doi: 10.1038/ncb1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AD, Camp ER, Fan F, Shen L, Gray MJ, Liu W, Somcio R, Bauer TW, Wu Y, Hicklin DJ, Ellis LM. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006;66:46–51. doi: 10.1158/0008-5472.CAN-05-3086. [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005;280:11740–11748. doi: 10.1074/jbc.M413878200. [DOI] [PubMed] [Google Scholar]
- Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- Zhu X, Leav I, Leung YK, Wu M, Liu Q, Gao Y, McNeal JE, Ho SM. Dynamic regulation of estrogen receptor-beta expression by DNA methylation during prostate cancer development and metastasis. Am J Pathol. 2004;164:2003–2012. doi: 10.1016/s0002-9440(10)63760-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.