

Abstract
We have previously reported that the dynein light chain (DLC) km23-1 is required for Smad2-dependent TGFß signaling. Here we describe another member of the km23/DYNLRB/LC7/robl family of DLCs, termed km23-2, which is also involved in TGFβ signaling. We show not only that TGFβ stimulates the interaction of km23-2 (DYNLRB2) with TGFβ receptor II (TβRII), but also that TGFβ regulates the interaction between km23-2 and endogenous TβRII in vivo. In addition, TGFβ treatment causes km23-2 phosphorylation, whereas a kinase-deficient form of TβRII prevents km23-2 phosphorylation. In contrast to the km23-1 isoform, blockade of km23-2 expression using small interfering RNAs (siRNAs) decreased key TGFβ/Smad3-specific responses, including the induction of both plasminogen activator inhibitor -1 (PAI-1) gene expression and p21 protein expression. Blockade of km23-1 expression had no effect on these two major TGFβ/Smad3 responses under similar conditions. Further, km23-2 was required for TGFβ stimulation of Smad3-dependent Smad-binding element (SBE)2-Luc transcriptional activity, but not for TGFβ stimulation of Smad2-dependent activin responsive element (ARE)-Lux transcriptional activity. In order to assess the mechanisms underlying the preferential stimulation of Smad3- vs. Smad2-specific TGFβ responses, immunoprecipitation (IP)/blot analyses were performed, which demonstrate that TGFβ stimulated preferential complex formation of km23-2 with Smad3, relative to Smad2. Collectively, our findings indicate that km23-2 is required for Smad3-dependent TGFβ signaling. More importantly, we demonstrate that km23-2 has functions in TGFβ signaling that are distinct from those for km23-1. This is the first report to describe a differential requirement for unique isoforms of a specific DLC family in Smad-specific TGFβ signaling.
Keywords: TGFβ, Smad, km23-2, DYNLRB, dynein, signaling
INTRODUCTION
Transforming growth factor β (TGFβ) is the prototype for the TGFβ super-family of highly conserved growth regulatory polypeptides that are critically involved in various biological processes, including cell growth, differentiation, apoptosis, motility, and extracellular matrix production (Derynck and Akhurst, 2007; Siegel and Massague, 2003; Yue and Mulder, 2001). As a result, TGFβ superfamily members influence a variety of physiologic and pathologic processes, including embryogenesis, wound healing, fibrosis, growth control, and oncogenesis (Derynck and Akhurst, 2007; Goumans et al., 2009; Roberts and Wakefield, 2003; Shi and Massague, 2003; Yue and Mulder, 2001).
TGFβ initiates its signaling by the binding of extracellular TGFβ to TβRII, followed by transphosphorylation of TGFβ receptor I (TβRI). The activated receptor complex stimulates intracellular mediators of TGFβ signaling, including receptor-activated Smads (RSmads). These activated RSmads form a complex with the common partner Smad4 and translocate to the nucleus, where they regulate transcription of a wide range of target genes (Feng and Derynck, 2005; Goumans et al., 2009; Roberts and Wakefield, 2003; Schmierer and Hill, 2007; Shi and Massague, 2003; Yue and Mulder, 2001). These target genes include cell cycle regulatory genes such as p21(Datto et al., 1995; Moustakas and Kardassis, 1998) and genes involved in extracellular matrix formation such as PAI-1 (Li et al., 1998; Westerhausen et al., 1991).
Although Smad2 and Smad3 are structurally similar, accumulating evidence suggests that Smad2 and Smad3 play distinct roles in mediating the cellular responses induced by TGFβ (Kretschmer et al., 2003; Piek et al., 2001). For example, HaCaT human keratinocytes display a Smad3-dependent up-regulation of p21 by TGFβ at both the mRNA and protein levels (Datto et al., 1995; Pardali et al., 2000). In addition, previous results have shown that TGFβ strongly induces PAI-1 levels in HaCaT cells (Laiho et al., 1990a; Siegel and Massague, 2003), a response that is mediated through the Smad3-targeted PAI-1 promoter (Shen et al., 1998). Further, it has been shown that TGFβ-mediated induction of matrix metalloproteinase-2 is selectively dependent upon Smad2, whereas induction of c-fos and of Smad7 relies on Smad3 (Piek et al., 2001). While, it is clear that Smad2 and Smad3 have distinct roles in mediating specific TGFβ responses, little is known about the mechanisms underlying this differential regulation of TGFβ responses by Smad2 and Smad3. A potential mechanism underlying this differential Smad2 versus Smad3 utilization will be described in the current report.
km23-1 [also known as km23, mLC7-1, DNLC2A, robl, DYNLRB1(Jiang et al., 2001; Nikulina et al., 2004; Pfister et al., 2005; Tang et al., 2002)] was shown to be a TβRII-interacting protein that is also a DLC (Tang et al., 2002). Consistent with a role for km23-1 in TGFβ signaling, siRNA blockade of km23-1 expression resulted in a reduction in both total intracellular Smad2 levels and in nuclear levels of phosphorylated Smad2 after TGFβ treatment (Jin et al., 2007). Further, blockade of km23-1 decreased TGFβ/Smad2-dependent ARE-lux transcriptional activity, with no effect on TGFβ/Smad3-dependent SBE2-Luc transcriptional activity, indicating that km23-1 is required in Smad2-dependent TGFβ signaling (Jin et al., 2008). Here we describe km23-2 (DYNLRB2), another mammalian ortholog of Chlamydomonas LC7 and Drosophila robl (Ding and Mulder, 2004; Nikulina et al., 2004; Tang et al., 2002). As for km23-1, km23-2 interacts with the TβRs after TGFβ stimulation and kinase-active TβRII is required for km23-2 phosphorylation. However, in contrast to km23-1, siRNA blockade of km23-2 decreased the ability of TGFβ to induce both PAI-1 gene expression and p21 protein expression, two major TGFβ/Smad3-specific responses. In keeping with these results, siRNA-specific blockade of km23-2 also reduced transcriptional regulation of a TGFβ/Smad3-specific, but not a Smad2-specific, reporter. Finally, TGFβ stimulated a preferential interaction of km23-2 with Smad3 relative to Smad2, suggesting that differential utilization of km23 DLC isoforms by Smad2 and Smad3 might be partially responsible for Smad2-dependent versus Smad3-dependent TGFβ signaling. Thus, our results demonstrate for the first time that km23-2 has functions distinct from those for km23-1 and is required for Smad3-dependent TGFβ signaling.
MATERIALS AND METHODS
Reagents
The anti-Flag M2 (F3165) and anti-c-myc (M5546) antibodies (Abs) and mouse IgG were from Sigma-Aldrich (St. Louis, MO). The c-myc monoclonal Ab (9E10) developed by Dr. J. Michael Bishop was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA). The anti-dynein intermediate chain (DIC) monoclonal Ab was from Chemicon (Temecula, CA). The anti-V5 Ab (R960 25) and the Lipofectamine™ 2000 transfection reagent were obtained from Invitrogen (Carlsbad, CA). The TβRII Ab (SC-220), rabbit IgG, and protein A/G plus agarose were from Santa Cruz Biotech (Santa Cruz, CA). 32P-orthophosphate (NEX-053) was from Perkin Elmer (Boston, MA). TGFβ1 was purchased from R & D Systems (Minneapolis, MN). The Fugene 6 transfection reagent was from Roche Applied Science (Indianapolis, IN). The Dual-Luciferase Reporter Assay System (Cat. # E1960) was purchased from Promega (Madison, MI). The anti-p21 antibody (CP74) was obtained from Neomarkers (Fremond, CA).
Cell Culture
HaCaT cells and Mv1Lu cells (CCL-64) were obtained from ATCC (Rockville, MD) and were grown in DMEM supplemented with 10% FBS. R1B cells and DR26 cells were kindly provided by Dr. Joan Massague (Sloan-Kettering) and maintained as for Mv1Lu cells. 293T cells were obtained from T-W. Wong (Bristol-Myers Squibb) and were maintained as for Mv1Lu cells. Cultures were routinely screened for mycoplasma using Hoechst staining.
Construction of pCMV5-human km23-2 (hkm23-2)-Flag plasmid
To prepare human km23-2-Flag, hkm23-2 was polymerase chain reaction (PCR)-amplified from a plasmid (IMAGE cDNA clone: 781013, Invitrogen) containing the coding region of human km23-2, with primers containing additional suitable flanking restriction enzyme sites for BglII (5’ primer) and SalI (3’ primer), and inserted into pCMV5-Flag (Sigma) after digestion with BglII and SalI restriction enzymes. The correct DNA sequences were confirmed by sequencing in both directions.
Transient transfections, IP/blot, Western
were performed essentially as described previously (Jin et al., 2007; Tang et al., 2002).
siRNAs
For the p21 experiments, km23-2 siRNA was constructed as follows. Briefly, the sense strand of the hairpin km23-2 siRNA, corresponding to nucleotides 102-126 of the km23-2 coding region (5’-GGACAACTCAACAACTGTTCAATAT-3’), was cloned into the pSUPER plasmid vector (Oligoengine, Seattle, WA). The km23-1 siRNA and NC siRNA were prepared as described previously (Jin et al., 2005). For other experiments, km23-2 siRNAs (sense, 5’-GGACAACUCAACAACUGUUCAAUAU-3’ and antisense, 5’-AUAUUGAACAGUUGUUGAGUUGUCC- 3’) were designed by BLOCK-iT™ RNAi Designer (Invitrogen) and synthesized by Invitrogen. For the luciferase reporter assays, both km23-1 and km23-2 siRNAs were cloned into the same RNAi-ready pSIRED-DNR-DsRed express vector (Clontech), respectively.
Luciferase reporter assays
HaCaT cells were plated at 1×104 cells/cm2 in 24-well plates. 24 h after plating, the cells were transfected with the indicated siRNAs, together with ARE-Lux and FAST-1 (Yeo et al., 1999), or SBE2-Luc, (Zawel et al., 1998) or PAI-1-Luc (Abe et al., 1994). Renilla was used to normalize transfection efficiencies. 24 h after transfections, the cells were washed once with serum-free (SF) medium and incubated in SF medium for 1 h. Then the cells were cultured in the absence and presence of TGFβ (5 ng/ml) for another 18 h. The luciferase activity was measured using the Dual-Luciferase Reporter Assay System following the manufacturer’s instructions. All assays were performed in triplicate. Data are expressed as mean±SEM.
Reverse transcription (RT)- PCR
HaCaT cells were transfected with NC siRNA, km23-1 siRNA, or km23-2 siRNA and treated as for luciferase assays. RNA isolation and RT were performed as described previously (Ding et al., 2005). Knockdown of km23-1 was confirmed by RT-PCR as described previously (Ding et al., 2005). The forward primer for km23-2 was 5’-TAGCGTTTTTGACATCCCG-3’ and the reverse primer was 5’-TTCCAACCCAGTGTCTAAA -3’. The conditions used for km23-2 PCR were 28 cycles as follows: 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds. The forward primer for GAPDH was 5’-ATCACTGCCACCCAGAAGAC-3’; reverse primer, 5’-ATGAGGTCCACCACCCTGTT -3’). The conditions used for GAPDH PCR were 21 cycles as follows: 95°C for 15 seconds, 57°C for 30 seconds, and 72°C for 50 seconds.
Real-Time quantitative RT- PCR
HaCaT cells were transfected with NC siRNA, km23-1 siRNA, or km23-2 siRNA and treated as for luciferase assays. Total RNA was isolated from HaCaT cells using TRIzol reagent according to the supplied protocol (Invitrogen, Carlsbad, CA), and digested with DNase RQI (Promega) to remove any contaminating genomic DNA. For RT, first strand cDNA was synthesized from 1.0 μg of total RNA using random hexamer primers and the SuperScript III Reverse Transcription kit (Invitrogen; Carlsbad, CA) by standard methods. The concentration and quality of the resulting cDNA was quantified and analyzed using a NanoDrop ND-1000 Fluorospectrometer. 40 ng of cDNA per sample was then utilized as a template for real-time PCR in a SYBR Green Master Mix (Qiagen Corp, USA). The 18S rRNA primers (Eurogentec, San Diego, CA), and the human PAI-1 specific primers [forward primer: 5’-GAG ACA GGC AGC TCG GAT TC-3’; reverse primer: 5’-GGC CTC CCA AAG TGC ATT AC-3’] (Kurisaki et al., 2003) were designed with the computer program Primer Express (Applied Biosystems) and produced by Invitrogen (Carlsbad, CA). To exclude the possibility of genomic DNA contamination, control PCR reactions with no cDNA template were also included for each gene-specific primer set. PCR amplifications were performed as described previously (Kurisaki et al., 2003). Levels of PAI-1 expression in each sample were determined using the relative standard curve method, with the 18S rRNA gene as an endogenous control (Kurisaki et al., 2003).
RESULTS
As mentioned earlier, km23-2 is a member of the DYNLRB/km23/LC7/robl family dynein motor protein LCs in mammalian cells (Tang et al., 2002, Nikulina et al., 2004, Ding et al., 2005). Because we have demonstrated that km23-1 is a TβR-interacting protein and is required for Smad2-dependent TGFβ signaling, it was of interest to determine the function of km23-2 in TGFβ signaling. In order to assess whether km23-2 could interact with the TβRs, we performed IP/blot analyses in the presence of TGFβ after transient transfection of RI-V5, RII-HA, and km23-2-Flag, or empty vector (EV). As shown in Fig. 1A, an interaction was observed between km23-2 and TβRII (lane 3, top panel). No specific band was apparent after expression of only EV or km23-2 alone (lanes 1 and 2, top panel). The IgG control was also negative (lane 4, top panel). Equal expression and loading of RI-V5, RII-HA, and km23-2-Flag were confirmed by Western blotting (lower panels). Thus, km23-2 did interact with TβRII in the presence of TGFβ.
Fig. 1. km23-2 interacts with and is phosphorylated by the TβRs, and the kinase activity of TβRII is required for this phosphorylation.


A: 293T cells were transiently transfected with km23-2-Flag, RI-V5, RII-HA, and/or EV, followed by IP/blot analyses using an anti-Flag Ab as the IP Ab and an RII polyclonal Ab as the blotting Ab (top panel). Cells were incubated in SF medium for 1h prior to addition of TGFb (10 ng/ml) for 5 min. Lower panels, controls for expression and loading of RI (V5 blot), RII (RII blot), and km23-2 (Flag blot). The results shown are representative of two similar experiments. B: HaCaT cells were transiently transfected either EV (lane 1) or km23-2-Flag (lanes 2-7). 24h after transfection, HaCaT cells were then incubated with SF medium for I h prior to addition of TGFβ (5 ng/ml) for the indicated times. Top panel, cells were then lysed , and lysates were subjected to IP/blot analyses using a Flag Ab as the IP Ab and an RII Ab as the blotting Ab. Bottom panels, Western blot analysis to demonstrate equal loading and expression of endogenous RII and km23-2-Flag. C: 293T cells were transiently transfected with RI-V5, RII-HA or KNRII, and either EV or km23-2-Flag. 28 h after transfection, in vivo phosphorylation of km23-2 were performed as described previously (Tang et al., 2002). Lower panel, the gel from top panel was transferred to a PVDF membrane and blotted with anti-Flag Ab. D: Mv1Lu, R1B (Mv1Lu cells lacking functional TβRI), and DR26 (Mv1Lu cells containing a truncated, nonfunctional TβRII) (Laiho et al., 1991; Laiho et al., 1990b) were transiently transfected with km23-2-Flag. 28h after transfection, in vivo phosphorylation assays were performed as described previously (Tang et al., 2002). Lower panel, the gel from top panel was transferred to a PVDF membrane and blotted with an anti-Flag Ab. Results are representative of two experiments.
To ensure that the interaction was not the result of over-expression of the TβRs and to determine whether the interaction between the receptors and km23-2 was regulated by TGFβ, we performed IP/blot analyses in HaCaT cells expressing endogenous TGFβ receptors in the absence or presence of TGFβ. As shown in Fig. 1B, no interaction between km23-2 and TβRII was detected in the absence of TGFβ (lane 1). In contrast, TGFβ induced a rapid interaction of km23-2 with endogenous TβRII at 5 min after TGFβ addition (lane 3). Further, the association of between km23-2 and TβRII was significantly decreased at 10-30 min after TGFβ addition to HaCaT cells (lane 4-6). As expected, no specific RII band was apparent after expression of EV alone (lane 1). The IgG control lane (lane 7) indicated that the band noted was specific for RII. Thus, basal levels of km23-2 –TβRII interaction were undetectable, and TGFβ regulated the interaction between km23-2 and endogenous TβRII.
TβRs display serine/threonine kinase activity and can phosphorylate a number of intracellular proteins to initiate and propagate various TGFβ signaling events and responses (Shi and Massague, 2003; Yue and Mulder, 2001). Since we have shown that km23-2 interacts with TβRII in the presence of TGFβ, it is conceivable that km23-2 could be phosphorylated upon TβR activation. To determine whether this was the case, we performed in vivo phosphorylation assays (6, 8) after transient co-expression of either TβRII or a dominant-negative kinase deficient RII (KNRII) with RI and km23-2. As shown in Fig. 1C, in the absence of TGFβ, expression of both receptors with km23-2 resulted in no detectable phosphorylation of km23-2 (lane 1, top panel). However, TGFβ treatment for 5 min resulted in a significant increase in km23-2 phosphorylation (lane 2, top panel). Furthermore, this phosphorylation of km23-2 was completely blocked upon expression of the KNRII (lane 3, top panel). The IgG control (lane 4, top panel) indicated that the band noted was specific for km23-2. Equal expression of km23-2 was confirmed by blotting with an anti-Flag Ab (bottom panel). Thus, the results in Fig. 1C demonstrate not only that km23-2 is phosphorylated upon TβR activation, but also that the kinase activity of TβRII is required for km23-2 phosphorylation.
To further confirm whether the kinase activity of TβRII and/or TβRI is required for km23-2 phosphorylation, we performed in vivo phosphorylation assays (Jin et al., 2005; Tang et al., 2002) in Mv1Lu cells expressing endogenous TβRs and in various receptor mutant lines derived from Mv1Lu cells (Boyd and Massague, 1989; Laiho et al., 1991; Laiho et al., 1990b). For example, we utilized R1B cells, which lack functional TβRI, and DR26 cells, which contain a truncated, nonfunctional TβRII (Laiho et al., 1991; Laiho et al., 1990b). As shown in Fig. 1D, in the absence of TGFβ, km23-2 was not phosphorylated in either the parent Mv1Lu cells or in the two mutant cell lines used (lanes 1, 3, 5), demonstrating that km23-2 is not constitutively phosphorylated when expressed in these cells. Upon TGFβ treatment for 5 min, km23-2 was highly phosphorylated in the parental Mv1Lu cells (lane 2). In contrast, in DR26 cells, with aberrant TβRII receptors that cannot transduce TGFβ-induced signals, km23-2 was not phosphorylated (lane 4). In R1B cells lacking functional TβRI, km23-2 was phosphorylated, but to a reduced extent (lane 6). Equal expression of km23-2 was confirmed by blotting with anti-Flag (bottom panel). These results demonstrate that the kinase activity of TβRII is required for km23-2 phosphorylation. Further, these results suggest that the presence of TβRI may enhance the phosphorylation of km23-2 via TβRII, although the kinase activity of TβRI may not be absolutely required for km23-2 phosphorylation.
The results in Fig. 1 suggest that km23-2 may function as a TGFβ signaling intermediate. In order to more definitively establish whether km23-2 was required in the signaling of any of the known TGFβ responses, we chose PAI-1 and p21 as two major downstream target genes of TGFβ (Datto et al., 1995; Siegel and Massague, 2003) to examine after knockdown of km23-2 by siRNA approaches. Initially, we confirmed that km23-2 siRNAs could block exogenous km23-2 expression by transiently transfecting 293T cells with the indicated siRNAs. As shown in Fig. 2A, no band was detectable in the EV-transfected cells, as expected (lane 1, left panel). km23-2-specific siRNAs totally knocked down km23-2-Flag expression (lane 4, left panel), compared with the NC siRNA (lane 2, left panel). Further, km23-1-specific siRNAs had no effect on km23-2-Flag expression (lane 3, left panel). Thus, the km23-2 siRNAs completely and specifically blocked km23-2-flag expression. As a control, we have also confirmed that km23-1 siRNAs could specifically block km23-1-Flag expression, but had no effect on km23-2-Flag expression (Fig. 2A, right panel).
Fig. 2. km23-1 and km23-2 siRNAs selectively and specifically knock down both exogenous and endogenous km23-1 and km23-2.
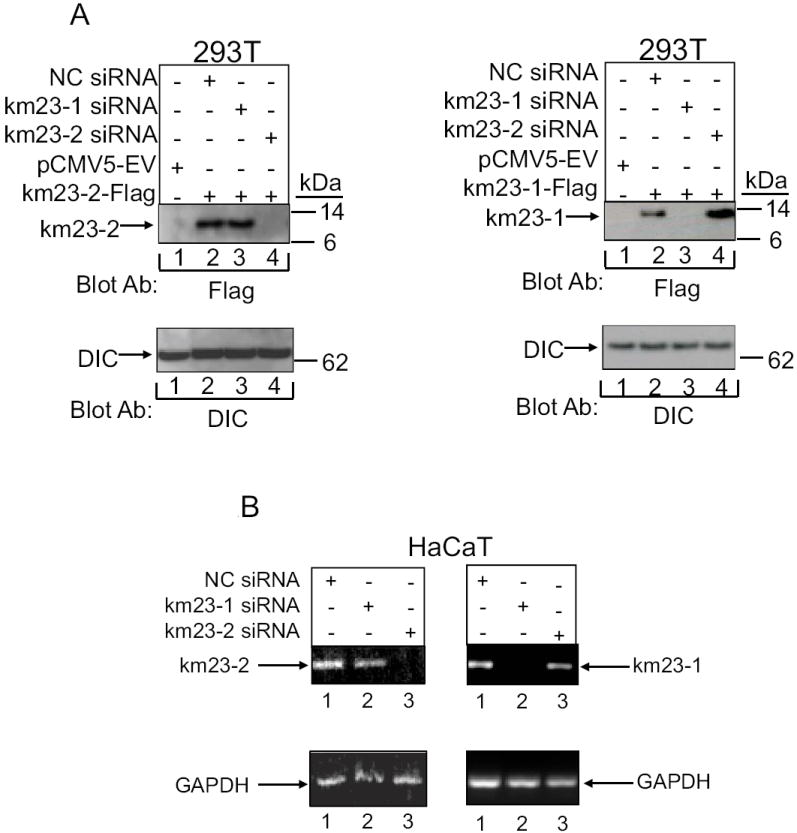
A: Left panel: 293T cells were transiently transfected with either EV (lane 1), or km23-2-Flag together with NC siRNA (lane 2), km23-1 siRNA (lane 3) or km23-2 siRNA (lane 4). Top panel, blockade of km23-1-Flag expression was analyzed via Western blotting with an anti-Flag M2 Ab. Bottom panel, equal loading was confirmed by blotting with an anti-DIC Ab. Right panel: 293T cells were transiently transfected with either EV (lane 1), or km23-1-Flag together with NC siRNA (lane 2), km23-1 siRNA (lane 3) or km23-2 siRNA (lane 4). Top panel, blockade of km23-1-Flag expression was analyzed via Western blotting with an anti-Flag M2 Ab. Bottom panel, equal loading was confirmed by blotting with an anti-DIC Ab. B: HaCaT cells were transfected with NC, km23-1-, or km23-2-specific siRNAs. Top panel, RT-PCR analysis of human km23-1 (left panel), and human km23-2 (right panel) mRNA expression from HaCaT cells were performed. Bottom panel, GAPDH was used as a loading control. Results are representative of two experiments.
In order to further establish that km23-2 siRNAs could specifically block endogenous km23-2, we transiently transfected HaCaT cells with the indicated siRNAs, and RT–PCR analyses were performed to monitor knockdown of km23-2 mRNA levels. Transfection efficiencies assessed using an RNAi-ready pSIRED-DNR-DsRed expression vector were about 60%-70% (data not shown). As shown in Fig. 2B, the km23-2 siRNAs specifically and completely blocked the expression of km23-2 mRNA, but not that of km23-1 mRNA (left panel). Our real time RT-PCR analyses also confirmed that km23-2 siRNA could significantly knockdown km23-2 mRNA (data not shown). Similarly, the km23-1 siRNAs specifically and completely knocked down the expression of km23-1 mRNA, but not that of km23-2 mRNA (right panel).
Because we have shown that km23-1 and km23-2 siRNAs could selectively and specifically block expression of both transfected and endogenous km23-1 vs. km23-2, respectively, in HaCaT cells, we used this approach to examine the effect of km23-2 knockdown on TGFβ-mediated transcription of the human PAI-1 promoter, which has been shown to be a Smad3-targeted promoter (Shen et al., 1998). Therefore, we transiently transfected HaCaT cells with NC siRNA, km23-1 siRNA, or km23-2 siRNA, and then performed PAI-1-luc luciferase reporter assays in the absence and presence of TGFβ. As shown in Fig. 3A, in the NC siRNA-transfected cells, TGFβ stimulated a 7-fold induction of PAI-1-Luc luciferase activity. A similar level of induction of the PAI-1-Luc luciferase activity was also seen in the km23-1 siRNA-transfected cells. In contrast, in the km23-2 siRNA-transfected cells, TGFβ-induction of the PAI-1-Luc luciferase activity was significantly decreased (to levels of only 3.5-fold) compared to those in the NC siRNA-transfected cells. Our results demonstrate that blockade of km23-2 specifically decreased TGFβ induction of PAI-1 gene transcription, whereas km23-1 was not required for this TGFβ/Smad3-specific response.
Fig. 3. TGFβ-mediated induction of PAI-Luc luciferase activity and endogenous PAI-1 gene expression in HaCaT cells specifically requires km23-2, but not km23-1.

A: HaCaT cells were transfected with NC siRNA, km23-1 siRNA, or km23-2 siRNA. 24h after transfection, the medium was replaced with SF medium for 1 h, followed by incubation of cells in the absence (open bar) and presence (black bar) of TGFβ (5ng/ml) for an additional 18 h. Luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega). The results are representative of at least two experiments, each performed in triplicate. B: HaCaT cells were transfected and treated as described for Fig. 3A. Real-time RT-PCR analysis of human PAI-1 mRNA expression from HaCaT cells was performed. Bars represent mean ± SD of PAI-1 mRNA levels normalized to control 18S rRNA levels. Results are representative of two experiments.
To further examine the km23-2 specificity of this TGFβ response, we examined PAI-1 mRNA expression levels in HaCaT cells using quantitative real-time RT-PCR analyses. For these studies, HaCaT cells were transiently transfected with NC siRNA, km23-1 siRNA, or km23-2 siRNAs in the absence and presence of TGFβ. As shown in Fig. 3B, in both NC siRNA-transfected cells and km23-1 siRNA-transfected cells, PAI-1 gene expression was induced approximately 6.3-fold after TGFβ stimulation, and the km23-1-specific siRNAs did not block these effects. In contrast, in the km23-2 siRNA-transfected cells, TGFβ induction of PAI-1 gene expression was significantly reduced to approximately 2.4-fold. Our results demonstrate that blockade of km23-2, but not of km23-1, specifically inhibited TGFβ induction of PAI-1 gene expression, thereby revealing differences in the ability of the km23 isoforms to mediate this biological response to TGFβ.
As mentioned earlier, another prominent biological effect of TGFβ in epithelial cells is induction of the cell cycle inhibitor p21 (Akhurst and Derynck, 2001; Roberts and Wakefield, 2003; Yue and Mulder, 2001). Thus, it was of interest of determine whether km23-2 was required for this biological response to TGFβ and whether the results would reveal a differential usage of the km23 isoforms as was seen for PAI-1. Therefore, we transiently transfected HaCaT cells with the indicated siRNAs and performed Western blot analyses to examine p21 induction by TGFβ. As shown in Fig. 4, in mock-transfected cells, there was no detectable p21 protein expression in the absence of TGFβ (lane 1, top panel). However, levels of p21 protein were greatly increased after TGFβ treatment (lane 2, top panel). Similar results were obtained in the both NC siRNA-transfected cells (lanes 3 and 4, top panel) and in the km23-1 siRNA-transfected cells (lanes 5 and 6, top panel). However, in the km23-2 siRNA-transfected cells, p21 protein levels were significantly decreased after TGFβ treatment (lane 8, top panel). Equal protein loading was confirmed by blotting with a DIC Ab. Thus, the results in Fig. 4 demonstrate that blockade of km23-2 specifically decreased TGFβ induction of p21 protein expression, whereas km23-1 was not required for this TGFβ/Smad3-specific response.
Fig. 4. TGFβ induction of p21 expression in HaCaT cells specifically requires km23-2, but not km23-1.
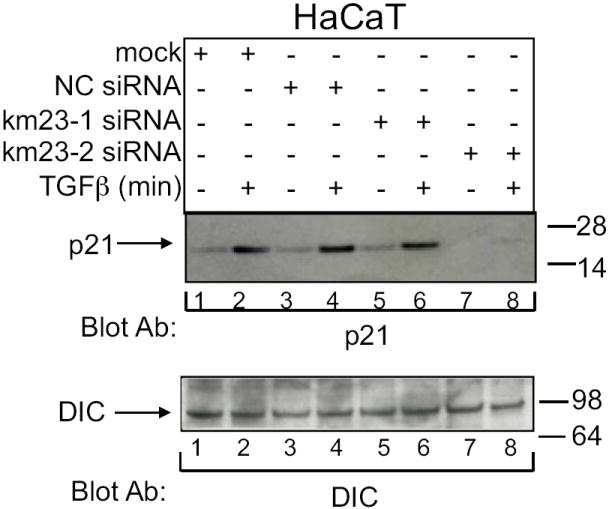
HaCaT cells were transiently transfected with the siRNA plasmids indicated: mock (lanes1-2), NC siRNA (3-4), km23-1 siRNA (lane 5-6), km23-2 siRNA (lane 7-8) and were treated with TGFβ as described in Fig. 3A. Top panel, p21 expression was detected by Western blot analysis using a mouse monoclonal anti-p21 Ab. Bottom panel, equal loading and expression of DIC was confirmed by blotting with a mouse monoclonal DIC Ab. Results are representative of two experiments.
Our results in Figs. 3-4 have shown that blockade of endogenous km23-2 specifically inhibited two Smad3-specific TGFβ targets, namely PAI-1 and p21. In addition, our previous results have shown that blockade of km23-1 inhibited TGFβ/Smad2-dependent ARE-lux, but had no effect on TGFβ/Smad3-dependent SBE2-Luc (Jin et al., 2007). Thus, it was of interest to determine whether blockade of km23-2 could preferentially influence a Smad3-specific TGFβ-dependent transcriptional reporter, as opposed to the Smad2-specific reporter regulated by km23-1. In this regard, the SBE2-Luc reporter has been used previously to demonstrate Smad3/4-dependent responses induced by TGFβ (Zawel et al., 1998). Accordingly, we performed SBE2-Luc luciferase reporter assays in the absence and presence of TGFβ after transiently transfecting HaCaT cells with the SBE2-Luc reporter and either EV, NC siRNA, km23-2 siRNA, or km23-1 siRNA. As shown in Fig. 5A, in the both EV and NC siRNA-transfected cells, TGFβ stimulated a 3.7-3.8-fold induction of the SBE2-Luc reporter. This induction level was significantly decreased (to levels that were 50% of NC siRNA values) upon km23-2 siRNA transfection to knock down km23-2. We also confirmed that blockade of km23-1 expression had no effect on TGFβ induction of SBE2-Luc as previously described (Jin et al., 2007). In these experiments, the down-regulation of km23-2 often decreased both basal and TGFβ-stimulated transcription, suggesting that km23-2 may also play a role in regulation of other signaling pathways, such as TGFβ production pathway, that contribute to autocrine TGFβ signaling in HaCaT cells. Thus, our results indicate that blockade of endogenous km23-2 expression impaired Smad3-specific transcriptional activity, whereas km23-1 siRNA did not.
Fig. 5. Knockdown of endogenous km23-2 expression results in inhibition of TGFβ-inducible Smad3-dependent (A), but not Smad2-dpendent (B), transactivation in HaCaT cells.

HaCaT cells were either EV transfected or transiently transfected with either NC siRNA, km23-1 siRNA, or km23-2 siRNA along with 0.1 μg SBE2-luc (A) or 0.1μg ARE-lux and 0.1 μg FAST-1 (B). Experiments were performed as in Fig. 3A. The results shown are representative of three similar experiments.
The ARE-lux reporter is known to be activated by TGFβ or activin in a Smad2-dependent manner (Yeo et al., 1999). As mentioned above, we have previously shown that blockade of km23-1 inhibits Smad2-dependent transcriptional activity of the ARE-Lux reporter (Jin et al., 2007). In order to examine whether knock-down of km23-2 could also affect this Smad2-specific reporter (Yeo et al., 1999), we performed ARE-Lux luciferase reporter assays in the absence and presence of TGFβ, after transiently transfecting HaCaT cells with the ARE-Lux reporter and FAST1, along with EV, NC siRNA, km23-2 siRNA or km23-1 siRNA. As shown in Fig. 5B, blockade of km23-1 significantly decreased TGFβ-induction of the ARE-Lux reporter in HaCaT cells, which is consistent with our previous results (Jin et al., 2007). In contrast, km23-2 and NC siRNAs had no effect on TGFβ-stimulated ARE-Lux activity. These results demonstrate that blockade of km23-2 had no effect on Smad2-dependent transcriptional activation, further revealing differences between km23-1 and km23-2 in regulating TGFβ-mediated events. These findings also support a specific role for km23-2 in mediating Smad3-dependent TGFβ signaling events.
Based upon our observations here that the km23 isoforms could differentially regulate Smad2- versus Smad3-specific TGFβ responses in HaCaT cells, it is conceivable that there may be selective complexes formed between the km23 isoforms and the different RSmads. Thus, we examined km23-2 complexes with Smad2/3 by IP/blot analyses in the absence and presence of TGFβ, after co-expression of Smad2-Myc (lanes 1-4), Smad3-Myc (lanes 5-9), and RI-V5, RII-HA, or EV, with or without km23-2-Flag as indicated. As shown in Fig. 6A, there was a strong interaction observed between km23-2 and Smad3 in the presence of TGFβ stimulation (lane 6), while in the absence TGFβ stimulation, there was a very low level of interaction (lane 7). In contrast, the interaction between Smad2 and km23-2 was very week even in the presence TGFβ stimulation (lane 2). No bands were observed in the lysates from cells expressing the receptors alone (lanes 4 and 8) or when using normal mouse IgG as the IP antibody (lane 9). In addition, no specific band was apparent after expression of EV and km23-2 (lanes 1 and 5). Thus, our results demonstrate that TGFβ preferentially stimulates the interaction of km23-2 with Smad3, over that with Smad2. In addition, since Smad3-Myc expression levels were lower than those for Smad2-Myc in the bottom panel, the Smad3-km23-2 specific interactions are actually even greater than those depicted.
Fig. 6. km23 isoforms selectively associate with the different R-Smads in IP/blot assays.

A: km23-2 interacts preferentially with Smad3, relative to Smad2. 293T cells were transiently co-transfected with Smad2-Myc (lanes 1-4), Smad3-Myc (lanes 5-9), and RI-V5, RII-HA, or EV, together with or without km23-2-Flag as indicated. 28h after transfection, cells were treated as for Fig. 1A. Top panel, 293T cells were lysed, followed by IP/blot analyses using an anti-Myc Ab as the IP Ab and an anti-Flag Ab as the blotting Ab. Middle panel, the same membrane was blotted with an anti-Myc Ab to indicate expression of the IP’d Smads. Bottom panel, equal expression of transfected km23-2-Flag was verified by immunoblot analysis. The results shown are representative of two similar experiments. B: km23-1 interacts preferentially with Smad2, relative to Smad3. 293T cells were transiently co-transfected with Smad3-Myc (lanes 1-4), Smad2-Myc (lanes 5-9), and RI-V5, RII-HA, or EV, together with or without km23-1-Flag as indicated. Top panel, experiments were performed as in Fig. 6A. Bottom panels, equal expression of transfected Smad2-myc, Smad3-myc, and km23-1-Flag were verified by immunoblot analysis.
Our previous results have shown that km23-1 is required for a Smad2-dependent TGFβ signaling (Jin et al., 2007). To further test our hypothesis that the km23 isoforms may selectively form complexes with the different RSmads, we have also performed IP/blot analyses after co-expression of Smad3-Myc (lanes 1-4), Smad2-Myc (lanes 5-9), and RI-V5, RII-HA, or EV, with or without km23-1-Flag as indicated. As shown in Fig. 6B, there was a strong interaction observed between km23-1and Smad2 in the presence of TGFβ stimulation (lane 6). Although there was some basal interaction between km23-1 and Smad2, the association was increased after treatment with TGFβ for 5 min. In contrast, km23-1 had a very week interaction with Smad3 in both the absence and the presence of TGFβ (lanes 2-3), further demonstrating Smad isoform specificity for the interaction with km23. As expected, no bands were observed in the lysates from cells expressing the receptors alone (lanes 4 and 8) or when using normal mouse IgG as the IP antibody (lane 9). In addition, no specific band was apparent after expression of EV and km23-1 (lanes 1 and 5). Thus, these results demonstrate that TGFβ preferentially stimulates the interaction of km23-1 with Smad2, over that with Smad3.
DISCUSSION
We have previously shown that km23-1 plays an important role in Smad2-dependent TGFβ signaling (Jin et al., 2007). To further define the function of the km23/DYNLRB/LC7/robl family of DLCs in TGFβ signaling, we have examined a second member of this family in mammalian cells. In the current report, we found that in similarity to km23-1, TGFβ regulates the interaction of km23-2 with TβRII and the TβRII kinase activity was required for the phosphorylation of km23-2 after TGFβ stimulation. However, we also demonstrate that blockade of endogenous km23-2 attenuated TGFβ stimulation of the Smad3-specific target genes PAI-1 and p21, while blockade of km23-1 had no effect on either of these TGFβ responses. Thus, in contrast to km23-1, the km23-2 isoform has unique functions in mediating Smad3-dependent TGFβ responses. Collectively, our findings indicate for the first time that the km23-2 DLC is required for Smad3-dependent TGFβ signaling.
In the current report, we have also demonstrated that Smad2- versus Smad3-specific responses are differentially regulated by km23-1 versus km23-2 in that blockade of endogenous km23-2 expression reduced TGFβ stimulation of the Smad3-dependent transcriptional reporter SBE2-Luc, while km23-1 knockdown did not. Conversely, a reduction in TGFβ stimulation of Smad2-dependent transcription was not observed by knockdown of km23-2, as we have observed for km23-1 knockdown. Finally, we have demonstrated that TGFβ regulated the interaction of km23-2 preferentially with Smad3, relative to Smad2, while TGFβ regulated the interaction of km23-1 with Smad2, but not with Smad3. Thus, the km23 DLC isoforms form selective complexes with Smad2 and Smad3 as a potential mechanism for the differential regulation of Smad2 versus Smad3 TGFβ signaling.
PAI-1 induction by TGFβ is a classic target of TGFβ signaling via the Smad pathway and is Smad3-specific in a variety of cell types (Kortlever et al., 2008; Kretzschmar and Massague, 1998). For example, Smad3 and Smad4 have been shown to mediate TGFβ signaling through direct binding to a novel TGFβ-responsive element in the PAI-1 promoter (Song et al., 1998). Silencing of Smad3, but not silencing of Smad2, blocked TGFβ-stimulated PAI-1 protein levels in HaCaT cells, providing further support that PAI-1 is a Smad3-dependent target gene in HaCaT cells (Kretschmer et al., 2003). Here we show that blockade of km23-2, but not blockade of km23-1, diminished TGFβ induction of both PAI-1 promoter luciferase activity and PAI-1 gene expression. Thus, km23-2, unlike km23-1, is required for TGFβ induction of PAI-1 via a Smad3-specific TGFβ pathway. In addition, we suggest that the specific requirement for km23-2 in this regard may be explained by the preferential formation of km23-2-Smad3 complexes after TGFβ treatment.
TGFβ induction of p21 is a critical step in mediating the inhibitory effects of TGFβ on cell growth. Further, Smad pathway components, and especially Smad3, are indispensable for the induction of p21 by TGFβ in HaCaT cells (Feng et al., 2000; Kretschmer et al., 2003; Pardali et al., 2000). For example, overexpression of Smad3 and Smad4 in HaCaT cells enhanced TGFβ induction of p21 gene expression, whereas dominant-negative mutants of Smad3 and Smad4 inhibited p21 induction by TGFβ in a dose-dependent manner (Pardali et al., 2000). Further, Smad3 has been shown to be the predominant Smad for mediating transcriptional induction of p21 by TGFβ (Pardali et al., 2000). Moreover, a previous report has shown that silencing of Smad3 diminished the increase in p21 protein expression in HaCaT cells (Kretschmer et al., 2003). In the current report, we demonstrate that silencing of km23-2, but not silencing of km23-1, blocked the ability of TGFβ to mediate induction of p21 expression. Therefore, in contrast to km23-1, km23-2 appears to be required for TGFβ induction of p21 expression via a Smad3-specific TGFβ pathway.
It is known that Smad2 and Smad3 associate with a number of cytoplasmic and membrane–bound proteins to differentially activate Smad2- or Smad3-specific signaling downstream (Brown et al., 2007; Feng and Derynck, 2005; Roberts et al., 2003; ten Dijke and Hill, 2004; Zhang and Derynck, 1999). For example, after TGFβ stimulation, the adaptor protein embryonic liver fodrin interacts with Smads3 and 4, but not with Smad2, in facilitating TGFβ transcriptional responses (Tang et al., 2003). More recently, a number of putative Smad2 or Smad3-associated proteins have been identified (Brown et al., 2008). It has been suggested that protein–protein interactions among these components may aid in the selective activation of Smad2 versus Smad3, thereby modulating Smad2- and/or Smad3-specific functions (Brown et al., 2007; Roberts et al., 2003; ten Dijke and Hill, 2004). Similarly, we show herein that km23-2 interacts with TβRII and preferentially with Smad3 after TGFβ stimulation. In contrast, our previous results have shown that km23-1 selectively interacts with TGFβ/Smad2 complexes (Jin et al., 2007). We suggest that the differential regulation of Smad2- versus Smad3-depedent TGFβ responses by the km23-1 and km23-2 DLCs may be due, in part, to the formation of selective Smad3-km23-2 and Smad2-km23-1 complexes. Collectively, our findings herein point to a novel paradigm for TGFβ signaling, namely one invoking DLCs in the modulation and specification of RSmad-specific signaling pathways.
The fact that Smad2 and Smad3 are not be phosphorylated in the same endocytic locale (Felici et al., 2003), suggests that there is some mechanism to divert the Smad isoforms to distinct early endosome compartments (Felici et al, 2003). The formation of the selective Smad2-km23-1 and Smad3-km23-2 complexes described above may help to selectively direct Smad2 and Smad3 to the appropriate subdomains or subcompartments in early endosomes. Along these lines, it is has been reported that TβR-associated proteins play roles in directing vesicular trafficking. For example, SARA has been shown to be enriched in early endosomes, bind both TβR and R-Smads, and promote Smad phosphorylation and TGFβ signaling (Di Guglielmo et al., 2003). Our previous results have shown that km23-1 selectively interacts with TGFβ/Smad2 complexes in early endosomes (Jin et al., 2007). Similarly, herein we show that km23-2 interacts with TβRII and preferentially with Smad3 at 5 min after TGFβ addition, when TβR and Smads are internalized in early endosomes (Di Guglielmo et al., 2003; Jin et al., 2007). The presence of km23-2 in early endosomes at 5 min after TGFβ treatment was also confirmed by sucrose gradient analyses (data not shown). Overall then, our data suggest that the km23 isoforms may play an important role in the early endosome compartment specification process for Smad 2 and 3.
It is also of interest that specific Smad TGFβ signaling components form selective complexes with light chain components of the dynein motor complex. Aside from our previous report regarding km23-1 complex formation with Smad2 (Jin et al, 2007), dynein motor regulation in TGFβ signaling has not been widely studied. However, there is one report describing the regulation of Smad2 signaling by the plus-end directed motor kinesin-1 (Batut et al., 2007). Similar to our findings herein, the kinesin studies demonstrate that a light chain of kinesin-1 is required for both interaction with Smad2 and trafficking Smad2 to the cell periphery in response to activin/nodal signaling (Batut et al., 2007). Other reports have described interactions of other DLCs with receptors for TGFβ or with those for other TGFβ superfamily members (Machado et al., 2003; Meng et al., 2006). However, the regulation of Smad-specific TGFβ responses by these interactions is still unclear. In addition, while reports of DLC binding to specific cargoes (ie, growth factors, receptors, binding partners), presumably to regulate specific intracellular responses, are abundant in the literature, all of these reports pertain to DLCs other than km23/DYNLRB (Lo et al., 2007; Machado et al., 2003; Meng et al., 2006; Tai et al., 1999; Vallee et al., 2004). Moreover, there are no reports implicating unique isoforms of a specific DLC family in the differential regulation of TGFβ signaling pathways. Accordingly, our findings are novel and contribute uniquely and significantly not only to the TGFβ field, but also to the dynein field. Future studies will address the precise nature of the intracellular regulation of TGFß/Smad3 signaling complexes by km23-2.
Acknowledgments
We thank Dr. Scott E. Kern (John Hopkins Oncology Center, Baltimore, MD) for the SBE2-Luc; Dr. Malcolm Whitman (Harvard Medical School, Boston, MA) for the ARE-Lux and FAST-1 constructs; Dr. Joan Massague (Memorial Sloan-Kettering Cancer Center, New York, NY) for the R1B and DR26 cells. We also thank Dr. Jeffery L. Wrana (Samuel Lunenfeld Research Institute, Toronto, Canada) for pCMV5-HA-TβRII, pCMV5B-Smad2-myc and pCMV5B-Smad3-myc constructs. This project is funded, in part, under a grant with the Pennsylvania Department of Health using Tobacco Settlement Funds. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
Contract grant sponsor: National Institutes of Health; Contract grant numbers: CA90765, CA92889, and CA100239
References
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216(2):276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11(11):S44–51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- Batut J, Howell M, Hill CS. Kinesin-mediated transport of Smad2 is required for signaling in response to TGF-beta ligands. Dev Cell. 2007;12(2):261–274. doi: 10.1016/j.devcel.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Boyd FT, Massague J. Transforming growth factor-beta inhibition of epithelial cell proliferation linked to the expression of a 53-kDa membrane receptor. J Biol Chem. 1989;264(4):2272–2278. [PubMed] [Google Scholar]
- Brown KA, Ham AJ, Clark CN, Meller N, Law BK, Chytil A, Cheng N, Pietenpol JA, Moses HL. Identification of novel Smad2 and Smad3 associated proteins in response to TGF-beta1. J Cell Biochem. 2008;105(2):596–611. doi: 10.1002/jcb.21860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J Cell Biochem. 2007;101(1):9–33. doi: 10.1002/jcb.21255. [DOI] [PubMed] [Google Scholar]
- Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci U S A. 1995;92(12):5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol. 2007;9(9):1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGFbeta family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5(5):410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- Ding W, Mulder KM. km23: a novel TGFbeta signaling target altered in ovarian cancer. Cancer Treat Res. 2004;119:315–327. doi: 10.1007/1-4020-7847-1_15. [DOI] [PubMed] [Google Scholar]
- Ding W, Tang Q, Espina V, Liotta LA, Mauger DT, Mulder KM. A transforming growth factor-beta receptor-interacting protein frequently mutated in human ovarian cancer. Cancer Res. 2005;65(15):6526–6533. doi: 10.1158/0008-5472.CAN-04-4385. [DOI] [PubMed] [Google Scholar]
- Felici A, Wurthner JU, Parks WT, Giam LR, Reiss M, Karpova TS, McNally JG, Roberts AB. TLP, a novel modulator of TGF-beta signaling, has opposite effects on Smad2- and Smad3-dependent signaling. Embo J. 2003;22(17):4465–4477. doi: 10.1093/emboj/cdg428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Feng XH, Lin X, Derynck R. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15(Ink4B) transcription in response to TGF-beta. Embo J. 2000;19(19):5178–5193. doi: 10.1093/emboj/19.19.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19(1):116–127. doi: 10.1038/cr.2008.326. [DOI] [PubMed] [Google Scholar]
- Hayes S, Chawla A, Corvera S. TGF beta receptor internalization into EEA1- enriched early endosomes: role in signaling to Smad2. J Cell Biol. 2002;158(7):1239–1249. doi: 10.1083/jcb.200204088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Yu L, Huang X, Chen X, Li D, Zhang Y, Tang L, Zhao S. Identification of two novel human dynein light chain genes, DNLC2A and DNLC2B, and their expression changes in hepatocellular carcinoma tissues from 68 Chinese patients. Gene. 2001;281(1-2):103–113. doi: 10.1016/s0378-1119(01)00787-9. [DOI] [PubMed] [Google Scholar]
- Jin Q, Ding W, Mulder KM. Requirement for the dynein light chain km23-1 in a Smad2-dependent transforming growth factor-beta signaling pathway. J Biol Chem. 2007;282(26):19122–19132. doi: 10.1074/jbc.M609915200. [DOI] [PubMed] [Google Scholar]
- Jin Q, Ding W, Staub CM, Gao G, Tang Q, Mulder KM. Requirement of km23 for TGFbeta-mediated growth inhibition and induction of fibronectin expression. Cell Signal. 2005;17(11):1363–1372. doi: 10.1016/j.cellsig.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Jin Q, Gao G, Mulder KM. Involvement of km23 dynein light chain in TGF beta signaling. In: Jakowlew SB, editor. Transforming growth Factor -beta in cancer therapy. Totowa NJ: Humana Press Inc.; 2008. pp. 169–181. [Google Scholar]
- Kortlever RM, Nijwening JH, Bernards R. Transforming growth factor-beta requires its target plasminogen activator inhibitor-1 for cytostatic activity. J Biol Chem. 2008;283(36):24308–24313. doi: 10.1074/jbc.M803341200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer A, Moepert K, Dames S, Sternberger M, Kaufmann J, Klippel A. Differential regulation of TGF-beta signaling through Smad2, Smad3 and Smad4. Oncogene. 2003;22(43):6748–6763. doi: 10.1038/sj.onc.1206791. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Massague J. SMADs: mediators and regulators of TGF-beta signaling. Curr Opin Genet Dev. 1998;8(1):103–111. doi: 10.1016/s0959-437x(98)80069-5. [DOI] [PubMed] [Google Scholar]
- Kurisaki K, Kurisaki A, Valcourt U, Terentiev AA, Pardali K, Ten Dijke P, Heldin CH, Ericsson J, Moustakas A. Nuclear factor YY1 inhibits transforming growth factor beta- and bone morphogenetic protein-induced cell differentiation. Mol Cell Biol. 2003;23(13):4494–4510. doi: 10.1128/MCB.23.13.4494-4510.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiho M, DeCaprio JA, Ludlow JW, Livingston DM, Massague J. Growth inhibition by TGF-beta linked to suppression of retinoblastoma protein phosphorylation. Cell. 1990a;62(1):175–185. doi: 10.1016/0092-8674(90)90251-9. [DOI] [PubMed] [Google Scholar]
- Laiho M, Weis FM, Boyd FT, Ignotz RA, Massague J. Responsiveness to transforming growth factor-beta (TGF-beta) restored by genetic complementation between cells defective in TGF-beta receptors I and II. J Biol Chem. 1991;266(14):9108–9112. [PubMed] [Google Scholar]
- Laiho M, Weis MB, Massague J. Concomitant loss of transforming growth factor (TGF)-beta receptor types I and II in TGF-beta-resistant cell mutants implicates both receptor types in signal transduction. J Biol Chem. 1990b;265(30):18518–18524. [PubMed] [Google Scholar]
- Li JM, Datto MB, Shen X, Hu PP, Yu Y, Wang XF. Sp1, but not Sp3, functions to mediate promoter activation by TGF-beta through canonical Sp1 binding sites. Nucleic Acids Res. 1998;26(10):2449–2456. doi: 10.1093/nar/26.10.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo KW, Kogoy JM, Pfister KK. The DYNLT3 light chain directly links cytoplasmic dynein to a spindle checkpoint protein, Bub3. J Biol Chem. 2007;282(15):11205–11212. doi: 10.1074/jbc.M611279200. [DOI] [PubMed] [Google Scholar]
- Machado RD, Rudarakanchana N, Atkinson C, Flanagan JA, Harrison R, Morrell NW, Trembath RC. Functional interaction between BMPR-II and Tctex-1, a light chain of Dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2003;12(24):3277–3286. doi: 10.1093/hmg/ddg365. [DOI] [PubMed] [Google Scholar]
- Meng Q, Lux A, Holloschi A, Li J, Hughes JM, Foerg T, McCarthy JE, Heagerty AM, Kioschis P, Hafner M, Garland JM. Identification of Tctex2beta, a novel dynein light chain family member that interacts with different transforming growth factor-beta receptors. J Biol Chem. 2006;281(48):37069–37080. doi: 10.1074/jbc.M608614200. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Kardassis D. Regulation of the human p21/WAF1/Cip1 promoter in hepatic cells by functional interactions between Sp1 and Smad family members. Proc Natl Acad Sci U S A. 1998;95(12):6733–6738. doi: 10.1073/pnas.95.12.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulina K, Patel-King RS, Takebe S, Pfister KK, King SM. The Roadblock light chains are ubiquitous components of cytoplasmic dynein that form homo- and heterodimers. Cell Motil Cytoskeleton. 2004;57(4):233–245. doi: 10.1002/cm.10172. [DOI] [PubMed] [Google Scholar]
- Pardali K, Kurisaki A, Moren A, ten Dijke P, Kardassis D, Moustakas A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. J Biol Chem. 2000;275(38):29244–29256. doi: 10.1074/jbc.M909467199. [DOI] [PubMed] [Google Scholar]
- Penheiter SG, Mitchell H, Garamszegi N, Edens M, Dore JJ, Jr, Leof EB. Internalization-dependent and -independent requirements for transforming growth factor beta receptor signaling via the Smad pathway. Mol Cell Biol. 2002;22(13):4750–4759. doi: 10.1128/MCB.22.13.4750-4759.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister KK, Fisher EM, Gibbons IR, Hays TS, Holzbaur EL, McIntosh JR, Porter ME, Schroer TA, Vaughan KT, Witman GB, King SM, Vallee RB. Cytoplasmic dynein nomenclature. J Cell Biol. 2005;171(3):411–413. doi: 10.1083/jcb.200508078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, Deng C, Kucherlapati R, Bottinger EP, Roberts AB. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276(23):19945–19953. doi: 10.1074/jbc.M102382200. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Russo A, Felici A, Flanders KC. Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann N Y Acad Sci. 2003;995:1–10. doi: 10.1111/j.1749-6632.2003.tb03205.x. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100(15):8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8(12):970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- Shen X, Hu PP, Liberati NT, Datto MB, Frederick JP, Wang XF. TGF-beta-induced phosphorylation of Smad3 regulates its interaction with coactivator p300/CREB-binding protein. Mol Biol Cell. 1998;9(12):3309–3319. doi: 10.1091/mbc.9.12.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3(11):807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Song CZ, Siok TE, Gelehrter TD. Smad4/DPC4 and Smad3 mediate transforming growth factor-beta (TGF-beta) signaling through direct binding to a novel TGF-beta-responsive element in the human plasminogen activator inhibitor-1 promoter. J Biol Chem. 1998;273(45):29287–29290. doi: 10.1074/jbc.273.45.29287. [DOI] [PubMed] [Google Scholar]
- Tai AW, Chuang JZ, Bode C, Wolfrum U, Sung CH. Rhodopsin’s carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell. 1999;97(7):877–887. doi: 10.1016/s0092-8674(00)80800-4. [DOI] [PubMed] [Google Scholar]
- Tang Q, Staub CM, Gao G, Jin Q, Wang Z, Ding W, Aurigemma RE, Mulder KM. A novel transforming growth factor-beta receptor-interacting protein that is also a light chain of the motor protein dynein. Mol Biol Cell. 2002;13(12):4484–4496. doi: 10.1091/mbc.E02-05-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299(5606):574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29(5):265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Williams JC, Varma D, Barnhart LE. Dynein: An ancient motor protein involved in multiple modes of transport. J Neurobiol. 2004;58(2):189–200. doi: 10.1002/neu.10314. [DOI] [PubMed] [Google Scholar]
- Westerhausen DR, Jr, Hopkins WE, Billadello JJ. Multiple transforming growth factor-beta-inducible elements regulate expression of the plasminogen activator inhibitor type-1 gene in Hep G2 cells. J Biol Chem. 1991;266(2):1092–1100. [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massague J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. Embo J. 1995;14(10):2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo CY, Chen X, Whitman M. The role of FAST-1 and Smads in transcriptional regulation by activin during early Xenopus embryogenesis. J Biol Chem. 1999;274(37):26584–26590. doi: 10.1074/jbc.274.37.26584. [DOI] [PubMed] [Google Scholar]
- Yue J, Mulder KM. Transforming growth factor-beta signal transduction in epithelial cells. Pharmacol Ther. 2001;91(1):1–34. doi: 10.1016/s0163-7258(01)00143-7. [DOI] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1(4):611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Derynck R. Regulation of Smad signalling by protein associations and signalling crosstalk. Trends Cell Biol. 1999;9(7):274–279. doi: 10.1016/s0962-8924(99)01579-2. [DOI] [PubMed] [Google Scholar]