

Abstract
The neurotransmitter dopamine (DA) modulates brain circuits involved in attention, reward, and motor activity. Synaptic DA homeostasis is primarily controlled via two presynaptic regulatory mechanisms, DA D2 receptor (D2R)-mediated inhibition of DA synthesis and release, and DA transporter (DAT)-mediated DA clearance. D2Rs can physically associate with DAT and regulate DAT function, linking DA release and reuptake to a common mechanism. We have established that the attention-deficit hyperactivity disorder-associated human DAT coding variant Ala559Val (hDAT A559V) results in anomalous DA efflux (ADE) similar to that caused by amphetamine-like psychostimulants. Here, we show that tonic activation of D2R provides support for hDAT A559V-mediated ADE. We determine in hDAT A559V a pertussis toxin-sensitive, CaMKII-dependent phosphorylation mechanism that supports D2R-driven DA efflux. These studies identify a signaling network downstream of D2R activation, normally constraining DA action at synapses, that may be altered by DAT mutation to impact risk for DA-related disorders.
Introduction
Dopamine (DA) acts in brain to modulate behaviors including motor activity, attention, and reward (Giros and Caron, 1993; Bannon, 2004; Palmiter, 2008). Disrupted DA signaling is implicated in brain disorders such as Parkinson's disease and attention-deficit hyperactivity disorder (ADHD) (Bannon et al., 2000; Volkow et al., 2007). The implications of dysregulated DA signaling in these diseases have fueled research into the mechanisms by which DA signaling is controlled and integrated (Nestler and Carlezon, 2006). Two main determinants of dopaminergic tone are the D2 DA receptor (D2R) and the DA transporter (DAT), both of which are localized on presynaptic terminals (Nirenberg et al., 1997; Missale et al., 1998). D2Rs are a family of Gi/Go G-protein-coupled receptors that inhibit vesicular DA release (Missale et al., 1998), whereas DAT proteins are Na+-coupled transport proteins that maintain low extracellular [DA] and limit synaptic DA spillover (Giros et al., 1996). Together, D2R and DAT proteins provide control over synaptic DA signaling, as evidenced by behavioral abnormalities that accompany D2R and DAT gene ablation (Giros et al., 1996; Jones et al., 1998). Recently, D2Rs have been found to associate with DAT, and support DAT trafficking (Bolan et al., 2007; Lee et al., 2007). Stimulation of D2R also results in the elevation of intracellular Ca2+ concentration and activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Nishi et al., 1997; Takeuchi et al., 2002). Importantly, CaMKII enhances the reverse transport of DA by DAT (Fog et al., 2006). These findings underscore the coordinated nature of DA homeostasis that, once perturbed, may lead to multiple neuropsychiatric disorders.
To date, limited evidence exists linking endogenous alterations in DA homeostasis to ADHD. Recently, we identified the hDAT coding variant A559V in two male siblings with ADHD, which results in anomalous DA efflux (ADE) without affecting DA transport (Mazei-Robison et al., 2008). Importantly, DA efflux in hDAT A559V can be blocked by the two DA-directed medications used for the treatment of ADHD, methylphenidate and amphetamine (AMPH) formulations (Mazei-Robison et al., 2008), both of which target DAT. The hDAT A559V variant was also found in a third unrelated subject with bipolar disorder (Grünhage et al., 2000).
The anomalous properties of the hDAT A559V variant have focused our attention on the mechanisms by which DAT-mediated DA efflux is elicited. Both CaMKII and protein kinase C (PKC) promote reverse transport of DA through DAT phosphorylation (Kantor and Gnegy, 1998; Fog et al., 2006). Additionally, hDAT N-terminal phosphorylation site mutants can suppress or induce DA efflux (Khoshbouei et al., 2004), consistent with phosphorylation as a mechanism stabilizing DAT in an “efflux-willing” state. Indeed, direct phosphorylation of DAT on N-terminal serines has been observed (Vaughan et al., 1997; Foster et al., 2002), and deletion of the N-terminal 22 aa of DAT eliminates DAT phosphorylation in response to kinase activation (Cervinski et al., 2005; Fog et al., 2006).
Here we uncover a novel mechanism linking D2R/CaMKII activation to N-terminal DAT phosphorylation as well as to ADE, and possibly ADHD.
Materials and Methods
Cell culture and transfection.
The hDAT-pcDNA3 expression vectors containing hDAT, hDAT A559V, hDAT A559V S/A, or hDAT S/D sequence were generated, confirmed, and transiently transfected into human embryonic kidney cells housing a stably integrated plasmid containing the SV40 large T antigen (HEK 293T) and maintained in a 5% CO2 incubator at 37°C (Mazei-Robison et al., 2005). HEK 293T cells were used unless otherwise noted and were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Importantly, neither hDAT A559V, hDAT A559V S/A, nor hDAT S/D significantly alter DA transport, as evidenced by uptake assays using [3H]DA (data not shown). For single-cell recordings, cells were cotransfected with enhanced green fluorescence protein (EGFP) to aid in transfected cell identification. Fugene-6 (Roche Molecular Biochemicals) in serum-free media was used to transfect cells ∼24 h after plating using a 1:3 DNA:lipid ratio. Assays were conducted ∼24 h after transfection.
Mouse striatum preparation.
C57BL/6J 15- to 20-week-old male mice from The Jackson Laboratory were anesthetized with isoflurane and rapidly decapitated. Following brain removal, the brain was chilled in oxygenated 4°C sucrose solution (sucrose 210 mm; NaCl 20 mm; KCl 2.5 mm; MgCl2 1 mm; NaH2PO4·H2O 1.2 mm), and the striatum was quickly removed while on ice. Striatum was lysed in 1% Triton buffer (25 mm HEPES, 150 mm NaCl, 2 mm sodium orthovanadate, and 2 mm NaF, plus a cocktail of protease inhibitors), and lysates were then centrifuged at 17,000 × g for 30 min at 4°C. The samples were processed for protein concentration using Bio-Rad's protein assay and spectrometry at 595 nm. Approximately 20 μg of lysate was resuspended in 2× SDS-PAGE sample loading buffer, heated at 95°C for 5 min, and then subjected to SDS-PAGE and immunodetection for D2R. The blots were then stripped and reprobed for actin content.
Amperometry.
Twenty thousand HEK 293T cells plated in 35 mm MaTek plates were transfected as described above and recorded ∼24 h later. To preload cells with DA, cells were washed twice with KRH assay buffer (130 mm NaCl, 1.3 mm KCl, 2.2 mm CaCl2, 1.2 mm MgSO4, 1.2 mm KH2PO4, and 10 mm HEPES, pH 7.4) containing 10 mm d-glucose, 100 μm pargyline, 10 μm tropolone, and 100 μm ascorbic acid, and then incubated with 1 μm DA in assay buffer for 45 min at 37°C. For nonclamped, amperometric experiments, a carbon fiber electrode (ProCFE; fiber diameter is 5 μm; obtained from Dagan Corporation) apposed to the plasma membrane and held at +700 mV with respect to the bath ground (a potential greater than the oxidizing potential of DA) was used to measure DA flux through oxidation reactions. The amperometric electrode measures electrical currents (in picoamperes) as a result of DA molecule oxidation, which, after integration, can be converted when required to number of DA molecules. Cells were not voltage clamped to permit measurement of basal efflux under resting membrane potential conditions. Amperometric currents were recorded using Axopatch 200B with a low-pass Bessel filter set at 100 Hz and digitally filtered offline at 1 Hz before analysis. Anomalous DAT-mediated DA efflux (ADE) was defined as amperometric baseline current minus current present after the addition of 10 μm cocaine (COC; Sigma) to the bath solution. The dotted horizontal lines in the figures represent the amperometric currents recorded after addition of cocaine when we achieved the maximum DAT blockade. Data were acquired by averaging a 15 s interval of current directly before cocaine application (baseline current), and 20 min following COC application (COC current). Cells were washed twice before recording with the external solution containing the following: 130 mm NaCl, 10 mm HEPES, 34 mm dextrose, 1.5 mm CaCl2, 0.5 mm MgSO4, and 1.3 mm KH2PO4 adjusted to pH 7.35. For CaMKII inhibition, cells were pretreated before amperometric recording for 20 min in external solution containing a 5 μm concentration of either KN-93 or the inactive analog KN-92. For CaMKII inhibitory peptide studies, cells were pretreated for 20 min in external solution containing a 5 μm concentration of the membrane-permeant form of the noncompetitive peptide inhibitor of CaMKII (CaMKIINtide) (Chang et al., 1998), antennapedia-CaMKIINtide (antCaMKIINtide; RQIKIWFQNRRMKWK-KRPPKLGQIGRSKRV-VIEDDRIDDVLK) (Sanhueza et al., 2007). As control, cells were pretreated for 20 min with a 5 μm concentration of the membrane permeant peptide, reversed Ant-Tirap138-151 (Ant-Tirap-R; RQIKIWFQNRRMKW-KKSVIAGGPAADRLQL) (EMD Biosciences). For D2R inhibition, cells were treated for 20 min with a 1 μm concentration of the D2R antagonists: raclopride, sulpiride, or eticlopride (Sigma) before amperometric recording. For Gi/Go inhibition, cells were pretreated for 4 h with 200 ng/ml pertussis toxin (PTX) (Bolan et al., 2007) (Calbiochem). Amperometric currents recorded from unloaded mouse midbrain DA neurons were obtained using neuronal external solution containing the following: 146 mm NaCl, 30 mm dextrose, 5 mm KCl, 5 mm HEPES, 2.5 mm CaCl2, and 1.2 mm MgCl2 adjusted to pH 7.35.
Amperometric analysis.
Average amperometric current was obtained as described above for hDAT and hDAT A559V cells. The mean amperometric currents ± SEM were normalized to the currents of the appropriate controls and converted into a percentage.
Electrophysiology.
Patch electrodes with a resistance of 4 MΩ were pulled from quartz pipettes on a P-2000 puller (Sutter Instruments) and filled with the pipette solution containing the following: 130 mm KCl, 0.1 mm CaCl2, 2 mm MgCl2, 1.1 mm EGTA, 10 mm HEPES, and 30 mm dextrose adjusted to pH 7.35 and 270 mOsm plus 2 mm DA. Cells were washed twice before recording with the external solution. For the nonclamped perfusion assays, we used a peptide comprising the first 27 residues of hDAT with the five most distal N-terminal serines (Ser-2, Ser-4, Ser-7, Ser-12, and Ser-13) substituted by aspartates (hDAT S/D peptide) or substituted by alanines (hDAT S/A peptide). Three micromolar hDAT S/D or S/A N-terminal peptide was added to pipette solution containing 2 mm DA, and cells were patched and held at −20 mV. The patch electrode was then lifted off the cell following 10 min of perfusion, while the cell remained intact. Nonclamped amperometric data were then recorded using a second, amperometric electrode held at +700 mV. For single-channel experiments, cells were patched in the outside-out configuration with the inner membrane exposed to the pipette solution plus 2 mm DA and held at −20 mV. DAT channels were recorded using an Axopatch 200B with a low-pass Bessel filter set at 1000 Hz and were later refiltered for analysis at 500 Hz. Channels were recorded before cocaine application (control, CTR) and following 3 min cocaine application (cocaine, COC) to the bath. The same 30 s of data in CTR and COC conditions were analyzed in QuB (software package for single-channel analysis, www.qub.buffalo.edu), and open probability (NPo) was calculated using amplitude histogram analysis as previously described (Kahlig et al., 2005).
Antibodies.
For immunoblots, CaMKII antibody (Affinity BioReagents #MA1-048) was used at 1:2000, phospho-CaMKII antibody (Cell Signaling Technology #3361) used at 1:1000, D2R antibody (Santa Cruz #sc-5303) used at 1:100, DAT antibody (Millipore Bioscience Research Reagents #MAB369) used at 1:1000, and β-actin antibody (Sigma #A5441) used at 1:5000.
Immunoblot assays.
HEK 293T cells were transiently transfected with hDAT or hDAT A559V. Forty-eight hours after transfection, cells were lysed in a Tris-HCl-based CaMKII lysis buffer (1 mm EDTA, 0.5 mm PMSF, 1% SDS, 1 mm microcystin-LR, 1 mm sodium orthovanadate, and 1 mm sodium pyrophosphate in the presence of protease inhibitors). The lysate was spun at 13,000 × g for 30 min, and the supernatant was separated by SDS-PAGE after being assayed for protein (Bradford). Activated CaMKII was measured using a phosphospecific antibody raised against a synthetic phosphopeptide corresponding to amino acid residues surrounding the phosphorylated Thr-286 (the autophosphorylation site associated with CaMKII activation) (Cell Signaling Technology). For raclopride assays, cells were pretreated with 1 μm raclopride for 20 min. Quantitation of the band density detected by Western blot was normalized to control conditions as well as to the total (unphosphorylated) CaMKII and expressed as a percentage of hDAT.
DAT phosphorylation assays.
Affinity-purified rabbit polyclonal phosphospecific antibodies targeting DAT serines 7, 12, and 13 of the DAT N terminus (pSer7, pSer12, and pSer13, respectively) were used (N. Sen and J. A. Javitch, unpublished work). Flp-In CHO cells (Invitrogen) stably expressing hDAT or hDAT A559V were washed in 5 ml of ice-cold buffer [25 mm Tris-HCl, pH 7.6, with 150 mm NaCl, 1 mm EDTA, 1 mm Na3VO4, 50 mm sodium pyrophosphate, 50 mm NaF, 10 mm glycerophosphate, and 10 mm NEM supplemented with protease inhibitors (Pefabloc 2 mg/ml, aprotinin 2 mg/ml, leupeptin 1 mg/ml, and pepstatin A 1 mg/ml)]. Cells were lysed in the same buffer plus 1% Triton X-100. The lysate was immunoprecipitated using a goat antibody directed against the C-terminal DAT epitope (Santa Cruz Biotechnology), resolved by SDS-PAGE, transferred to a PVDF membrane, and blotted with the rabbit anti-pSer7, -pSer12, and -pSer13 antibodies and in parallel with a rat anti-DAT N-terminal antibody (Millipore Bioscience Research Reagents). Bands were visualized using goat anti-rabbit and goat anti-rat horseradish peroxidase-conjugated secondary antibody and detected by ECL with data capture achieved on a FluorChem 8900 imaging system (Alpha Innotech).
[32P] phosphorylation assays.
HEK 293T cells were transiently transfected with YFP-HA-EL2-hDAT/A559V-hDAT. Cells were incubated at 37°C in phosphate-free DMEM for 1 h to deplete intracellular phosphate. Cells were then supplemented with [32P] orthophosphate-containing medium for 4 h at 37°C. All the treatments were performed in the medium at the end of this incubation period. Cells were washed three times with ice-cold PBS and lysed with 400 μl of RIPA buffer containing protease and phosphatase inhibitors for 1 h at 4°C. Cells were centrifuged for 30 min at 13,000 × g and supernatants incubated overnight with prewashed HA affinity matrix at 4°C. HA-affinity matrix was washed three times with RIPA buffer containing protease and phosphatase inhibitors before eluting the bound protein in Laemmli sample buffer and analysis using SDS-PAGE.
Neuronal cultures.
Mouse midbrain neuronal cultures were obtained from a transgenic mouse strain generated as previously described (Zhang et al., 2004) where midbrain DA neurons carry the red florescent protein (RFP) marker driven by the tyrosine hydroxylase (TH) promoter. The TH promoter:RFP transgene (TH::RFP) was constructed by ligating a 4.5 kb HindIII/EcoRI fragment of the rat tyrosine hydroxylase promoter. To create a source of DA neurons that do not express DAT (DAT-null) as a background for transfection with DAT mutants, we developed a line of mice in which midbrain DA neurons express the TH::RFP marker in a DAT knock-out (KO) background (Giros et al., 1996). We bred female heterozygous TH::RFP mice with male homozygous DAT knock-out mice to generate double heterozygous mice. Offspring heterozygous for both alleles was confirmed using PCR genotyping with DAT KO and TH probes. These animals were then intercrossed to generate mice that express TH::RFP and are homozygous for the DAT deletion. Male and females mice of this genotype were crossed to create offspring that are DAT null where ∼75% of these also carry TH::RFP. Poly-d-lysine-coated glass bottom culture dishes (MatTek #P35GC-1.5-14-C) were coated with 10 μg/ml laminin. A monolayer of rat C6 glial cells was plated 2–3 d before culturing (Rayport et al., 1992) neurons and maintained in Neurobasal-A media (Invitrogen #10888-022) supplemented with 10% fetal bovine serum, 30 U/ml penicillin, 30 mg/ml streptomycin, and 0.6 mm l-glutamine. Midbrains from 1- to 4-d-old mouse pups were dissected, cut into small pieces (∼1 mm3), and digested with papain solution consisting of 15 U/ml papain, 1.25 mm cysteine, 1.9 mm Ca2+, 100 U/ml DNase I, 0.5 mm kynurenic acid. The papain digestion was performed for 5–20 min while stirring in the presence of 5% CO2 at 37°C. Cells were rinsed three times with neuronal media consisting of Neurobasal-A media supplemented with 10% fetal bovine serum (heat-inactivated), 30 U/ml penicillin, 30 mg/ml streptomycin, 0.6 mm l-glutamine, B-27 supplement, and 0.5 mm kynurenic acid. Neurons were dissociated by titration, suspended in neuronal media additionally supplemented with 10 ng/ml GDNF, and plated onto glial cell cultures. The following day 25 μm 5-fluorodeoxyuridine (FDU) with 70 μm uridine was added to the culture medium to suppress glial cell growth. DAT-null neurons were transiently transfected with either hDAT, hDAT A559V, or hDAT S/D constructs and were cotransfected with enhanced green fluorescence protein (EGFP) for selection. Lipofectamine 2000 (Invitrogen) in serum-free media was used to transfect neurons ∼5 d after culturing using a 1:3 DNA:Lipofectamine 2000 ratio. Assays were conducted ∼24 h after transfection. Only neurons expressing both red fluorescence (DA neuron) and green fluorescence (successful transfection) were used for analysis.
Quantitative PCR.
RNA was isolated (Qiagen RNeasy) and reverse transcription reaction was performed with a kit from Applied Biosystems (catalog #4368814). qPCR was performed using the following primers pairs against human D2R (long and short): CTACTCCTCCATCGTCTCCTTC and GTTTGGTGTTGACTCGCTTG. For primers against the reference house keeping enzyme (expressed to a moderate degree) phosphoglycerate kinase (PKG), we used GGGTCGAGCTAAGCAGATTG and GCTTTCACCACCTCATCCAT. For PGK, we obtained Ct values of 23.15 (SE 0.1611; n = 8). qPCR was performed with an Applied Biosystems, 7300 real-time PCR system.
Immunostaining.
HEK 293T cells were transiently cotransfected with vectors for hDAT and eGFP. Forty-eight hours after transfection, cells were fixed, processed for immunofluorescence, and immunostained with a rat polyclonal antibody against DAT (Millipore Bioscience Research Reagents MAN369) and a mouse monoclonal antibody recognizing D2R (Santa Cruz Biotechnology, sc-5303). Cells were washed and labeled with the appropriate secondary antibodies (Invitrogen).
Results
D2R signaling sustains hDAT A559V-mediated ADE through a Gi/Go-dependent pathway
Aberrant D2R signaling has been linked to numerous brain disorders, including schizophrenia, bipolar disorder, drug abuse, and ADHD (Seeman and Niznik, 1990; Volkow et al., 1993; Pearlson et al., 1995; Wang et al., 1997; Volkow et al., 2007). In addition, D2R associates with DAT and regulates its function (Bolan et al., 2007; Lee et al., 2007). Recently, we identified the hDAT coding variant A559V in two male siblings with ADHD, which results in anomalous DA efflux, or ADE (Mazei-Robison et al., 2008). ADE is defined as the amperometric current (DA efflux) inhibited by cocaine (see Materials and Methods). Here we demonstrate that blockade of D2R signaling inhibits ADE in HEK 293T cells expressing hDAT A559V (hDAT A559V cells) (Fig. 1A). Cells were loaded with DA (see Materials and Methods) and treated with D2R antagonist raclopride (1 μm) for 20 min. Raclopride treatment reduces ADE with respect to vehicle-treated control (Fig. 1A). Importantly, in wild-type hDAT-expressing cells (hDAT cells, see Materials and Methods), ADE is not observed (Mazei-Robison et al., 2008), and raclopride has no effect on the amperometric trace (data not shown). Similar results were obtained using the D2R antagonists eticlopride (1 μm) and sulpiride (1 μm) (data not shown). Figure 1B (top) verifies the presence of endogenous D2R mRNA in the heterologous expression system used in our experiments (HEK 293T cells, see Materials and Methods) by quantitative PCR. Figure 1B (middle) confirms the presence of endogenous D2R in the HEK 293T cells used in our experiments by immunoblot. HEK 293T cells transfected with either hDAT (first lane) or hDAT A559V (second lane) exhibit endogenous D2R expression. Striatal extracts (third lane) were used as positive controls, and actin bands are shown as a loading control. Figure 1B (bottom) validates the presence and coexpression of both endogenous D2R and transfected hDAT in HEK 293T cells.
Figure 1.
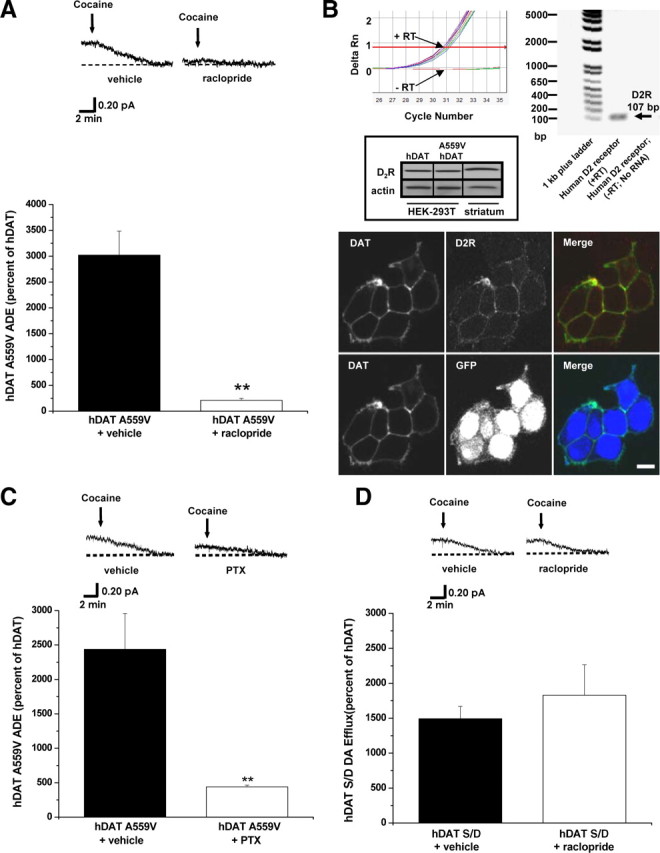
D2R signaling sustains hDAT A559V-mediated ADE through a Gi/Go-dependent pathway. A, Top, Representative oxidative currents, filtered at 1 Hz, in hDAT A559V cells following pretreatment with vehicle (left) or 1 μm raclopride (right). Arrows indicate application of cocaine. Bottom, Data reported as mean amperometric current ± SEM in hDAT A559V cells expressed as a percentage of current recorded in hDAT cells, both for vehicle control conditions (n = 7) and with raclopride (n = 6) (t = 5.6, df = 11, p = 0.0002, Student's t test). B, Top, qPCR demonstrating the presence of endogenous D2R in HEK 293T cells. Traces from real-time PCR (top, left) and representative PCR gel (top, right) demonstrating that when RNA isolated from HEK 293T cells is reverse transcribed (+RT), PCR product crosses threshold near cycle 30 (arrow) (cT = 31.2 ± 0.1, n = 8), whereas if no reverse transcription is performed (−RT), no product reaches threshold (arrow). Middle, Representative immunoblot for D2R (top) and actin (bottom) in HEK 293T cells transfected with the indicated cDNA, confirming D2R expression. Mouse striatum, heavily enriched with endogenous D2R, was used as a positive control (n = 3). Bottom, Representative confocal images of single sections demonstrating that endogenous D2R and transfected hDAT are expressed in HEK 293T cells (upper row) and that cotransfection of hDAT and GFP leads to their coexpression (lower row). C, Top, Representative oxidative currents recorded from hDAT A559V cells pretreated for 4 h with vehicle control (left) or with 200 ng/ml PTX (right). Bottom, Data reported as mean amperometric current ± SEM in hDAT A559V cells expressed as a percentage of current in hDAT cells, both in vehicle control conditions (n = 3) and with PTX (n = 5) (t = 5.2, df = 6, p = 0.0019, Student's t test). D, Top, Representative oxidative currents in hDAT S/D cells [5 most distal hDAT N-terminal serines (Ser-2, Ser-4, Ser-7, Ser-12, and Ser-13) substituted by aspartates to mimic phosphorylated state] following pretreatment with vehicle (left) or 1 μm raclopride (right). Bottom, Data reported as mean amperometric current ± SEM in hDAT S/D cells, expressed as a percentage of current in hDAT cells both in control conditions (hDAT, n = 6; hDAT S/D, n = 6) and with raclopride (hDAT, n = 6; hDAT S/D n = 4) (t = 0.8, df = 8, p = 0.44, Student's t test). **p < 0.05.
D2Rs signal through a PTX-sensitive Gi/Go-dependent pathway (Missale et al., 1998). Therefore, we demonstrate that ADE in hDAT A559V cells is inhibited by PTX. PTX treatment (200 ng/ml) for 4 h robustly reduces ADE in hDAT A559V cells with respect to vehicle-treated cells (Fig. 1C) with no effect in hDAT cells. Phosphorylation of the hDAT N terminus, specifically a subset of the five N-terminal serine residues in positions 2, 4, 7, 12, and 13, has been shown to regulate DAT-mediated DA efflux (Khoshbouei et al., 2004; Fog et al., 2006). Additionally, N-terminal pseudophosphorylation achieved by substituting aspartate for the five N-terminal serines (hDAT S/D) is sufficient to drive DA efflux (Fog et al., 2006) resembling ADE in hDAT A559V cells. To examine the possibility that D2R signaling sustains ADE via N-terminal phosphorylation, we tested whether inhibition of D2R signaling reduces efflux in the hDAT S/D cells. In contrast to hDAT A559V, Figure 1D shows that raclopride (1 μm) has no effect on hDAT S/D-mediated DA efflux.
D2R-stimulated ADE is mediated by CaMKII
CaMKII has been shown to stimulate reverse transport of DA through a mechanism associated with hDAT N-terminal phosphorylation (Fog et al., 2006). Therefore, we examined whether D2R signaling regulates ADE through CaMKII activation and CaMKII-dependent DAT phosphorylation. First, we used a phosphospecific antibody raised against the amino acid residues surrounding the phosphorylated Thr-286 (the autophosphorylation site associated with CaMKII activation). We show by immunoblot that basal CaMKII autophosphorylation is enhanced in cells expressing hDAT A559V (Fig. 2A) with no changes of total CaMKII or DAT levels. Next, we determine whether D2R signaling mediates the increase in CaMKII activity in hDAT A559V cells. Figure 2C shows that CaMKII autophosphorylation is decreased in hDAT A559V cells treated for 20 min with a 1 μm concentration of the D2R antagonist raclopride. Similar results are obtained using 1 μm sulpiride (data not shown). Importantly, in hDAT cells, raclopride treatment has no effect on either CaMKII activity or total levels of CaMKII (data not shown), suggesting that the absence of ADE in hDAT cells under basal conditions is due to a lack of D2R signaling through CaMKII.
Figure 2.

D2R-mediated ADE is CaMKII-dependent in hDAT A559V cells. A, Representative immunoblot for autophosphorylated CaMKII, total CaMKII, and total DAT in hDAT and hDAT A559V cells. B, hDAT (n = 5) and hDAT A559V (n = 5) cell extracts were immunoblotted for CaMKII autophosphorylated at Thr-286 to measure CaMKII activity under basal conditions. The immunoprecipitated band densities were quantified, normalized to total CaMKII, and expressed as a percentage of autophosphorylated CaMKII in hDAT cells (t = 2.1, df = 8, p = 0.036, Student's t test comparing hDAT A559V to hDAT). C, Representative immunoblot for autophosphorylated CaMKII and total CaMKII following 20 min treatment with a 1 μm concentration of the D2R antagonist raclopride or vehicle control. D, hDAT A559V (n = 5) cell extracts were immunoblotted for autophosphorylated CaMKII with and without raclopride treatment. The immunoprecipitated band densities were quantified and normalized to the corresponding density of total CaMKII. Data reported as a percentage of vehicle control (t = 2.3, df = 8, p = 0.047, Student's t test). E, Top, Representative oxidative currents in hDAT A559V cells following 20 min pretreatment with vehicle (left), or a 5 μm concentration of the cell-permeable CaMKII peptide inhibitor, antCaMKIINtide (right). Arrows indicate addition of cocaine. Bottom, Data reported as mean amperometric current ± SEM in hDAT A559V cells expressed as a percentage of current recorded in hDAT cells both for control conditions (n = 6) and with antCaMKIINtide (n = 8) (t = 6.8, df = 12, p < 0.0001, Student's t test). Control peptide, ant-Tirap-R, has no effect on basal DA efflux in either hDAT A559V (n = 4) or hDAT cells (n = 4) (data not shown). F, Top, Representative oxidative currents in hDAT S/D cells following pretreatment with vehicle (left) or 5 μm antCaMKIINtide (right). Bottom, Data reported as mean amperometric current ± SEM in hDAT S/D cells expressed as a percentage of current in hDAT cells both in control conditions (n = 6) and with antCaMKIINtide (n = 5) (t = 0.7, df = 9, p = 0.48, Student's t test). *p < 0.05; **p < 0.01.
To more definitively link CaMKII activation to ADE, we selectively inhibited CaMKII with a membrane-permeant form of the noncompetitive peptide inhibitor of CaMKII, antCaMKIINtide. Figure 2E demonstrates that pretreatment (20 min) of hDAT A559V cells with 5 μm antCaMKIINtide significantly decreases ADE. Importantly, pretreatment for 20 min with a 5 μm concentration of the cell-permeant control peptide, reversed Ant-Tirap138-151 (Ant-Tirap-R) has no effect on either ADE in hDAT A559V cells or DA efflux in hDAT cells (data not shown). Furthermore, inhibition of CaMKII with 5 μm KN-93 (20 min) significantly reduces ADE in hDAT A559V cells. KN-93 has no effect on either amperometric currents in hDAT cells or cell surface expression of hDAT A559V or hDAT as measured by cell surface biotinylation (data not shown). In addition, the KN-93 inactive analog KN-92 (5 μm) has no effect on basal efflux in either hDAT A559V or hDAT cells (data not shown).
To determine whether N-terminal phosphorylation of DAT supports ADE as a consequence of elevated CaMKII activity, we used hDAT S/D cells. Because hDAT S/D is pseudophosphorylated at the N terminus, it should bypass the need for kinase activation in inducing DA efflux and be insensitive to CaMKII inhibition. Figure 2F demonstrates that S/D cells are insensitive to pharmacological inhibition of CaMKII, consistent with CaMKII and N-terminal DAT phosphorylation being linked through a common pathway to trigger ADE.
N-terminal phosphorylation of hDAT A559V supports ADE
Previous studies indicate that CaMKII-dependent N-terminal phosphorylation supports reverse transport of DA under physiological conditions and upon AMPH treatment (Fog et al., 2006). To examine whether hDAT A559V must be N-terminally phosphorylated to produce its anomalous activity, we mutated the distal five N-terminal serines to alanines in the hDAT A559V construct (hDAT A559V S/A) to prevent phosphorylation. Whereas hDAT A559V exhibits ADE as noted above, ADE in hDAT A559V S/A cells is significantly blunted (Fig. 3A,B); hence the ability of hDAT A559V to sustain ADE is dramatically impaired by preventing N-terminal phosphorylation. Thus, N-terminal phosphorylation is required for ADE in hDAT A559V cells.
Figure 3.

N-terminal phosphorylation of hDAT A559V supports ADE. A, Representative oxidative currents recorded from hDAT, hDAT A559V, and hDAT A559V S/A cells [5 most distal N-terminal serines (Ser-2, Ser-4, Ser-7, Ser-12, and Ser-13) substituted by alanine in A559V background to prevent phosphorylation]. Arrows indicate addition of cocaine. B, Data reported as mean amperometric current ± SEM from hDAT (n = 5), hDAT A559V (n = 6), and hDAT A559V S/A (n = 7) cells (F(2,15) = 18.2, p = 0.0005, **p < 0.01, one-way ANOVA followed by Bonferroni's multiple-comparison test). C, Representative immunoblot in hDAT and hDAT A559V cells for phosphorylated serines 7, 12, and 13 (pSer7, pSer12, and pSer13, respectively) and for total DAT. Inset, Immunoblot for D2R (top) and CaMKII (bottom) confirming their presence in Flp-In CHO cells; lanes are shown in duplicate (n = 6). D, Quantification of pSer DAT intensities normalized to total DAT. Data are reported as percentage of phosphorylation of pSer7, pSer12, and pSer13 in hDAT A559V with respect to hDAT (n = 5–7) (F(3,22) = 4.1, p = 0.018, *p < 0.05 **p < 0.01, one-way ANOVA followed by Dunnett's multiple-comparison test).
To verify that hDAT A559V shows increased N-terminal phosphorylation, we used phosphospecific antibodies targeted to DAT N-terminal serines 7, 12, and 13 (pSer7, pSer12, and pSer13 respectively). We selected these three phosphorylation sites based on evidence that they regulate reverse transport of DA (Khoshbouei et al., 2004). Immunoblots reveal increased basal phosphorylation at two of the three serines in hDAT A559V stably transfected Flp-In CHO cells compared to hDAT (Fig. 3C), indicating that phosphorylation at serines 7 and 13 specifically may regulate the anomalous activity of hDAT A559V. The Flp-In CHO cells express both D2R and CaMKII (inset). For quantitation, immunoreactivity of pSer7, pSer12, and pSer13 for hDAT and hDAT A559V is normalized to total DAT. In Figure 3D, hDAT A559V immunoreactivity (pSer7, pSer12, and pSer13) is expressed as a percentage of hDAT. The Flp-In CHO cells stably transfected with hDAT A559V show ADE that is significantly different from cells stably transfected with hDAT. However, the magnitude of the ADE observed in the stable cell line (0.05 ± 0.02 pA; n = 4) is not sufficient to allow pharmacological and molecular manipulation. Consistent with the phosphorylation results obtained in stably transfected Flp-In CHO cells, we found that basal 32P incorporation is increased in HEK 293T cells transiently transfected with hDAT A559V compared to hDAT (281 ± 60%, n = 3; t = 3.0, df = 4, p = 0.039, Student's t test).
Phosphorylated DAT N terminus forms critical interactions that underlie the ability of A559V to cause ADE
Recent evidence suggests that reverse transport of DA mediated by DAT is regulated by physical association of DAT with CaMKII (Fog et al., 2006; Binda et al., 2008). We demonstrate that disruption of hDAT A559V N-terminal protein/protein interactions could alter ADE. A peptide comprising the first 27 residues of hDAT with the five most distal N-terminal serines substituted by aspartates (S/D peptide) was synthesized and used to disrupt the interaction of the hDAT A559V phosphorylated N terminus with possible interacting proteins. Because the S/D peptide is not cell permeable, we combined the whole-cell voltage-clamp technique with amperometry to allow for perfusion of S/D peptide into the cytoplasm via the whole-cell pipette. hDAT or hDAT A559V cells were perfused for 10 min. with either the S/D peptide (3 μm) or vehicle. Following perfusion, ADE was measured in intact, nonclamped cells (see Materials and Methods). Figure 4, A and B, shows that perfusion with the S/D peptide significantly decreases ADE in hDAT A559V cells with respect to control. In contrast, intracellular perfusion with the S/A peptide (in which the N-terminal serines are substituted by alanines to mimic the absence of phosphorylation) has no effect on ADE (Fig. 4C,D).
Figure 4.

Phosphorylated DAT N terminus forms critical interactions that underlie the ability of A559V to cause ADE. A, Representative oxidative currents in hDAT A559V cells following whole-cell perfusion with vehicle (left) or a peptide corresponding to the first 27 residues of hDAT with the N-terminal serines substituted for aspartates (hDAT S/D peptide, right). Arrows indicate addition of cocaine. Cells were whole-cell patch clamped and perfused for 10 min to allow for loading of 2 mm DA and either 3 μm hDAT S/D peptide or vehicle control, and measured for ADE as described in Materials and Methods. B, Data reported as mean amperometric current ± SEM in hDAT A559V cells expressed as a percentage of current recorded in hDAT cells both in control conditions (n = 3) and with hDAT S/D peptide (n = 5) (t = 5.9, df = 6, p = 0.001, Student's t test). C, Representative oxidative currents in hDAT A559V cells following whole-cell perfusion with vehicle (left) or a peptide corresponding to the first 27 residues of hDAT with the N-terminal serines substituted for alanines (hDAT S/A peptide, right). Cells were whole-cell patch clamped and perfused as described above. D, Data reported as mean amperometric current ± SEM in hDAT A559V cells expressed as a percentage of current recorded in hDAT cells both in control conditions (n = 3) and with hDAT S/A peptide (n = 3) (t = 0, df = 4, p = 1.0, Student's t test). E, Representative oxidative currents in hDAT S/D cells following whole-cell perfusion with vehicle (left) or hDAT S/D peptide (right). Cells were whole-cell patch clamped and perfused as described above. F, Data reported as mean amperometric current ± SEM in hDAT S/D cells expressed as a percentage of current recorded in hDAT cells both in control conditions (n = 3) and with hDAT S/D peptide (n = 3) (t = 3.5, df = 4, p = 0.02, Student's t test). *p < 0.05; **p < 0.01.
ADE could be sustained by the A559V mutation itself or could be a consequence of an A559V-mediated increase in N-terminal phosphorylation and the resulting protein/protein interactions. If the latter is true, then interactions of the phosphorylated hDAT N terminus may be a general requirement for reverse transport of DA. To test this, we performed intracellular perfusion experiments with the S/D peptide in hDAT S/D cells, which exhibit constitutive DA efflux. Our data demonstrate that intracellular perfusion of the S/D peptide, as above, in hDAT S/D cells reduces DA efflux with respect to control (Fig. 4E,F). These data strongly support our hypothesis that the phosphorylated DAT N terminus must form critical interactions to cause reverse transport of DA and that these interactions underlie the ability of A559V to cause ADE.
hDAT A559V N-terminal phosphorylation promotes a D2R-dependent channel-like mode of DAT
Recently, we determined that DA efflux is sustained in part by a novel mode of conduction that consists of rapid (on the order of milliseconds) bursts of DA through DAT (channel-like mode of DAT) (Kahlig et al., 2005). We hypothesize that hDAT A559V-mediated ADE is promoted, in part, by increased channel-like activity of hDAT A559V triggered by N-terminal phosphorylation. To record DAT channel activity in isolated membrane patches, we used the patch-clamp technique in the outside-out configuration (Galli et al., 1996; Kahlig et al., 2005) in hDAT, hDAT A559V, hDAT A559V S/A, and hDAT S/D cells. In this configuration, the intracellular side of the plasma membrane faces the pipette solution containing 30 mm NaCl and 2 mm DA (see Materials and Methods), while the extracellular side faces the bath solution. The channel-like activity mediated by DAT was confirmed by its sensitivity to the DAT blocker, cocaine. Figure 5A shows DAT channel-like activity recorded at −20 mV from outside-out patches of hDAT A559V, hDAT, hDAT A559V S/A, and hDAT S/D cells under control conditions (CTR) and 3 min after bath application of 10 μm cocaine (COC). In Figure 5B, we show the NPo of the DAT channel-like mode calculated from 30 s of data (Galli et al., 1996; Kahlig et al., 2005). Consistent with previous reports (Kahlig et al., 2005), under CTR conditions in hDAT cells DAT channel-like openings are low probability and are blocked by application of COC (Fig. 5B, open bars). Importantly, hDAT A559V exhibits significantly increased channel-like activity in CTR conditions compared to hDAT, and this increase is blocked by cocaine (Fig. 5B, filled bars). The increased hDAT A559V channel-like activity correlates with previously documented nonclamped DA efflux data (Mazei-Robison et al., 2008) and suggests that the increased channel activity in hDAT A559V is responsible, at least in part, for A559V-mediated ADE. Moreover, preventing phosphorylation of hDAT A559V by the hDAT A559V S/A mutation significantly reduces channel-like activity to a level comparable to that of hDAT (Fig. 5B, sparse diagonal stripes). Therefore hDAT A559V N-terminal phosphorylation is required not only for ADE, but also for the increased channel-like activity.
Figure 5.
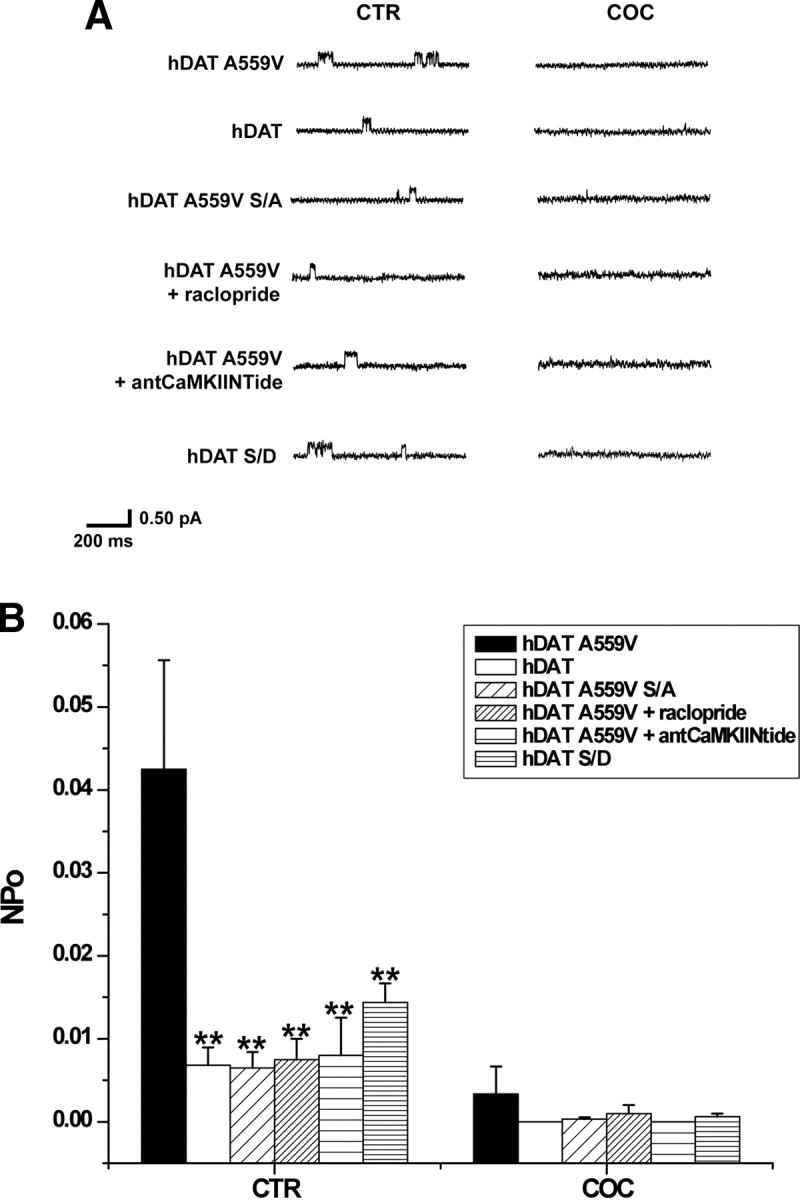
D2R-stimulated hDAT A559V N-terminal phosphorylation promotes a channel-like mode of DAT. A, Representative traces of DAT-mediated channel-like activity in hDAT A559V, hDAT, hDAT A559V S/A, and hDAT S/D cells. hDAT A559V cells were also treated with raclopride (1 μm) and antCaMKIINtide (5 μm) as described above. Traces shown both for control condition (CTR) and after bath application of 10 μm cocaine (COC) to confirm DAT specificity. Isolated membrane patches were excised using the outside-out patch-clamp technique and held at −20 mV. B, Data were quantified using amplitude histogram analysis of 30 s of data to calculate the NPo of the DAT channel-like mode. For both CTR and COC conditions, data are reported as mean NPo ± SEM calculated for hDAT A559V cells (filled bars; n = 4), hDAT cells (open bars; n = 5), hDAT A559V S/A cells (sparse diagonal striped bars; n = 6), hDAT A559V cells pretreated for 20 min with 1 μm raclopride (dense diagonal striped bars; n = 4), hDAT A559V cells pretreated for 20 min with 5 μm antCaMKIINtide (sparse horizontal striped bars; n = 4), and hDAT S/D cells (dense horizontal striped bars; n = 5) (F(5,20) = 5.9, p = 0.0016, **p < 0.01, one-way ANOVA followed by Dunnett's test compared to hDAT A559V).
In Figures 1 and 2, we show that hDAT A559V-mediated ADE is regulated by CaMKII through D2R signaling. Thus, in an effort to determine whether the increase in DAT channel activity in hDAT A559V cells is regulated by D2R, we measured channel activity in hDAT A559V cells following 20 min pretreatment with a 1 μm concentration of the D2R antagonist raclopride. Under control conditions, pretreatment with raclopride significantly reduces the NPo of hDAT A559V with respect to vehicle control (Fig. 5B, dense diagonal striped bar). Inhibition of CaMKII using 5 μm antCaMKIINtide significantly reduces the NPo of hDAT A559V with respect to vehicle-treated hDAT A559V (Fig. 5B, sparse horizontal striped bar). Importantly, inhibition of either D2R or CaMKII attenuates hDAT A559V channel-like activity to a level comparable to that of hDAT.
While DAT channel activity has been previously reported (Carvelli et al., 2004; Kahlig et al., 2005), the molecular mechanism involved in the regulation of DAT channel activity has yet to be identified. In Figure 5, we show that N-terminal phosphorylation is necessary for the increased channel activity exhibited by hDAT A559V. Therefore to address the possibility that N-terminal phosphorylation is also responsible for regulating the channel mode of DAT independently of the A559V mutation, we measured channel activity in hDAT S/D cells. One-way ANOVA followed by Dunnett's multiple-comparison test did not reveal a significant increase in NPo in hDAT S/D cells compared to hDAT cells. However, our data do suggest a trend for increased hDAT S/D NPo. Nonetheless, the NPo for hDAT S/D is significantly less than that of hDAT A559V, indicating that A559V supports a channel-like state of DAT not only through N-terminal phosphorylation, but also through another mechanism.
Expression of hDAT A559V in DAT-null DA neurons confers ADE properties
To assess the impact of the A559V mutation and the D2R pathway on ADE in neurons, we developed a line of mice where an RFP transgene is expressed in DA neurons via the TH promoter in a DAT knock-out background (see Materials and Methods) (Lute et al., 2008). Mouse midbrain DA neurons cultured from these mice (RFP:DAT-null neurons) were transiently cotransfected with EGFP in combination with either hDAT, hDAT A559V, or hDAT S/D and visually selected for amperometric recording based on both RFP and EGFP fluorescence. Figure 6A (inset) shows the merged confocal image of RFP and GFP fluorescence from a RFP:DAT-null neuron expressing EGFP. Figure 6A shows amperometric recordings of ADE from hDAT A559V-transfected RFP:DAT-null neurons demonstrating the presence of cocaine-sensitive ADE. Such neurons treated with raclopride (20 min, 1 μm) show significantly diminished ADE. Importantly, in wild-type hDAT-expressing neurons, ADE is not observed, and raclopride has no effect on the amperometric trace (data not shown). Importantly, because dopaminergic neurons contain DA vesicular machinery, in these experiments we do not preload the cells with DA.
Figure 6.
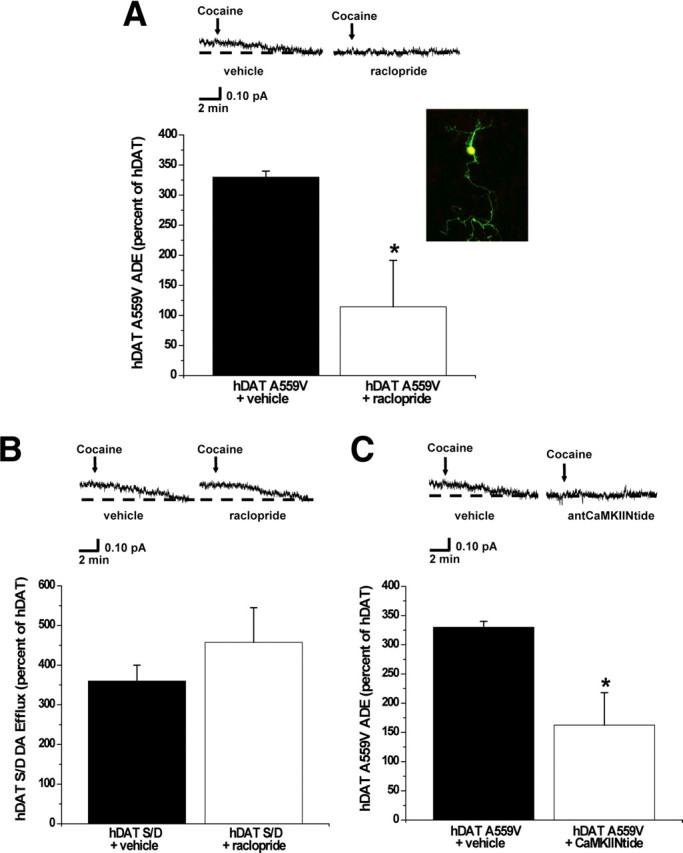
Expression of hDAT A559V in DAT-null DA neurons confers ADE properties. A, Inset, Mouse midbrain neuron expressing both red fluorescence (selection for DA neuron) and green fluorescence (successful transfection with DAT). Confocal image represents the merging (yellow) of both green and red fluorescence, indicating areas where both red and green fluorescence are dually expressed. Top, Representative oxidative currents in unloaded hDAT A559V-transfected neurons following 20 min pretreatment with vehicle (left), or 1 μm raclopride (right). Arrows indicate addition of cocaine. Bottom, Data reported as mean amperometric current ± SEM in hDAT A559V neurons expressed as a percentage of current recorded in hDAT neurons both for control conditions (n = 4) and with raclopride (n = 4) (t = 2.8, df = 6, p = 0.0327, Student's t test). B, Top, Representative oxidative currents in unloaded hDAT S/D-transfected neurons following 20 min pretreatment with vehicle (left) or 1 μm raclopride (right). Bottom, Data reported as mean amperometric current ± SEM in hDAT S/D neurons expressed as a percentage of current recorded in hDAT neurons both for control conditions (hDAT, n = 4; hDAT S/D, n = 3) and with raclopride (hDAT, n = 4; hDAT S/D, n = 3) (t = 1, df = 4, p = 0.37, Student's t test). C, Top, Representative oxidative currents in unloaded hDAT A559V-transfected neurons following 20 min pretreatment with vehicle (left) or 5 μm antCaMKIINtide (right). Bottom, Data reported as mean amperometric current ± SEM in hDAT A559V neurons expressed as a percentage of current recorded in hDAT neurons both for control conditions (n = 4) and with antCaMKIINtide (hDAT, n = 3; hDAT A559V, n = 4) (t = 2.9, df = 6, p = 0.0248, Student's t test). *p < 0.05.
As with transfected, non-neuronal cells, the hDAT S/D mutant expressed in DA neurons also displays enhanced DA efflux in transfected neurons (Fig. 6B). However, raclopride is ineffective in inhibiting DA efflux in these cells (Fig. 6B). Moreover, treatments of neurons with antCaMKIINtide (20 min, 5 μm) inhibits ADE in these neurons (Fig. 6C). Thus, D2R signaling via CaMKII supports the ADE of hDAT A559V in DA neurons, most likely through phosphorylation of the hDAT N terminus.
Discussion
Dysregulation of DA tone in the midbrain has been shown to underlie several neuropsychiatric disorders, including bipolar disorder, schizophrenia, drug abuse, obesity, and ADHD (Seeman and Niznik, 1990; Wise, 1998; Volkow et al., 2007). With respect to ADHD, DA signaling is believed to be critical both for control of motor activity and for mechanisms involving attention, which are controlled by both subcortical and cortical circuits. Recently, we identified ADE as a prominent phenotype associated with the rare hDAT coding variant A559V (Mazei-Robison et al., 2008). Our data support a model in which ADE is a direct result of the signaling events elicited by the A559V mutation. Our findings reveal that, rather than arising as an autonomous feature of altered hDAT structure, D2R signaling sustains hDAT A559V-mediated ADE. This action is supported by CaMKII-dependent activation and signaling that leads to N-terminal phosphorylation. The increase in N-terminal phosphorylation enhances the probability of DAT channel-like activity, which has been shown to provide an effective pathway for DA efflux (Kahlig et al., 2005). Our model of ADE is further supported by enhanced DA efflux and a trend for increased channel-like activity in pseudophosphorylated hDAT (hDAT S/D). N-terminal phosphorylation has been shown to regulate reverse transport of DA mediated by hDAT (Khoshbouei et al., 2004). These findings reveal the critical role played by the DAT N terminus in regulating functional states of the transporter, which can be altered by genetic and pharmacological mechanisms.
In wild-type hDAT, reverse transport of DA requires an increase in intracellular Na+, above physiological (low Na+) conditions (Khoshbouei et al., 2003). Strikingly, this increase is not required for hDAT A559V, due to an increased affinity of the transporter for intracellular Na+ (Mazei-Robison et al., 2008). Because D2R receptor antagonists and PTX treatment attenuate ADE, we propose that an initial Na+-dependent DA efflux triggers a feedforward mechanism mediated by D2R activation and subsequent Gi/Go-dependent signaling. Consistent with this hypothesis, neither blockade of D2R signaling, CaMKII inhibition, nor the S/A mutation completely abolishes ADE (Figs. 1–3). Further studies are needed to establish whether A559V structural perturbations lead to the increased affinity of hDAT A559V for intracellular Na+. Additionally, we demonstrate that D2R provides critical support for hDAT A559V-mediated ADE; however D3R, which is also in the D2R family of Gi/Go G-protein-coupled receptors, cannot be ruled out and warrants further study. While we focus on D2R activation of CaMKII, we acknowledge that other kinases may also contribute to ADE, such as ERK1/2 and PKC, which have been shown to regulate DAT function (Giambalvo, 1992; Bolan et al., 2007).
D2Rs, through PTX-sensitive G-proteins, signal to a number of downstream effectors that can trigger the release of Ca2+ from intracellular stores (Takeuchi et al., 2002), possibly by activation of phospholipase Cβ and IP3 production (Morris and Scarlata, 1997). This increase in intracellular Ca2+ leads to activation of kinases, including CaMKII (Takeuchi et al., 2002), which has been shown both to associate with DAT and to stimulate DAT-mediated DA efflux (Fog et al., 2006). Additionally, CaMKII can phosphorylate serine residues in the distal N terminus of DAT in vitro, and mutation of these serines to alanines eliminates the ability of CaMKII to enhance DA efflux (Fog et al., 2006). Here, we provide evidence that the hDAT A559V mutation initiates a D2R-mediated intracellular signaling cascade that leads to an increase in basal CaMKII activity and a subsequent increase in DAT N-terminal phosphorylation. In support of this mechanism, mutation of the N-terminal serines to alanines in hDAT A559V significantly reduces ADE. Previously, we have proposed that an increase in N-terminal phosphorylation shifts DAT from a “reluctant” to a “willing” state for DA efflux (Khoshbouei et al., 2004). However, how N-terminal phosphorylation of hDAT A559V translates to ADE remains to be determined. Protein/protein interactions of the DAT N terminus have been demonstrated to play a role in AMPH-induced DA efflux (Binda et al., 2008). Thus, ADE may also be sustained by interaction of the phosphorylated N terminus with an associated protein and/or an intracellular domain of DAT. Indeed, intracellular perfusion of an N-terminal hDAT S/D peptide reduces ADE in hDAT A559V as well as DA efflux in hDAT S/D cells (Fig. 4). In contrast, the S/A peptide has no effect on ADE, suggesting that the phosphorylated hDAT A559V N terminus requires interactions to cause ADE. These results could lead to new molecular interventions and the design of drugs that restore normal DA signaling in individuals carrying the A559V mutation, or a similarly acting mutation.
The question remains, however, as to how DAT N-terminal phosphorylation leads to changes in the functional status of a single transporter to promote DA efflux. One possibility is that N-terminal phosphorylation stabilizes an inward-facing conformation of the DAT and therefore stimulates DA efflux by increasing the probability of intracellular DA binding to DAT. Alternatively, N-terminal phosphorylation could promote a channel-like mode of DAT, which has been shown to support outward fluxes of DA (Kahlig et al., 2005). Here we show, for the first time, that N-terminal phosphorylation of hDAT A559V stabilizes a channel-like mode of DAT (Fig. 5). Indeed, A559V, which increases phosphorylation of DAT, increases basal channel-like activity of DAT, possibly supporting A559V-mediated ADE (Fig. 5). In nonclamped hDAT A559V cells, ADE is triggered by D2R-mediated activation of CaMKII (Figs. 1, 2). Here, we demonstrate that hDAT A559V channel-like activity is regulated by the same pathways involved in ADE (Fig. 5), linking this novel mode of DAT function to anomalous DA signaling associated with the A559V variant.
The exact mechanism by which phosphorylation increases DAT channel-like activity remains unclear. Based on the crystal structure of LeuT, a bacterial homolog of DAT (Yamashita et al., 2005), A559V faces out in TM12, far from the pore domain. Nonetheless, this relatively subtle variant, which is associated with a human disease, alters the phosphorylation state of DAT. N-terminal phosphorylation may alter the conformation of DAT in a manner that stabilizes a channel mode. Alternatively, phosphorylation could promote interaction of the N terminus with another part of DAT or with an associated protein to alter DAT function. The DAT N terminus has been shown to interact with soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein syntaxin 1A (SYN1A) (Lee et al., 2004) in a CaMKII-mediated manner to regulate DA efflux (Binda et al., 2008). Thus, the hDAT A559V/SYN1A interaction could possibly be responsible for stabilizing the channel-like state of DAT to sustain ADE. Conceivably, phosphorylation of the DAT N terminus could disrupt its interaction with a DAT intracellular domain to allow association with other intracellular proteins. For ion channels, such interactions among intracellular domains are responsible for regulating substrate permeation in many different channel families, including Shaker K+ channels, in which amino acids in the N-terminal tail bind near, and occlude, the channel pore (Hoshi et al., 1990). Importantly, this kind of interaction has been proposed to regulate the GABA transporter cycle (Quick et al., 2004). Thus, a common mechanism for ion channel and transporter regulation may be through a series of protein/protein interactions.
The A559V human DAT coding variant has been identified in two male siblings diagnosed with ADHD, as well as one individual diagnosed with bipolar disorder (Grünhage et al., 2000; Mazei-Robison et al., 2005). Prominent theories posit an alteration of DA neurotransmission in both disorders based, in part, upon the importance of DA signaling in circuits involved in motor function, mood, reward, and attention (Carlsson, 1987). Although A559V is not a common polymorphism, the identification of rare, functional variants has proven critical to the elucidation of molecular mechanisms underlying complex diseases such as Parkinson's disease and Alzheimer's disease (St George-Hyslop, 2000; Hardy et al., 2003). Thus, it is intriguing to speculate that anomalous transporter-mediated neurotransmitter efflux may be an unappreciated source of risk for mental illness, especially disorders associated with altered DA signaling. Our data point to the possibility that mutations in other genes upstream of DAT-mediated ADE, such as CaMKII or D2R, could potentially trigger similar anomalous efflux.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant MH081423 to E.B., The Peter F. McManus Charitable Trust to C.S., and NIH Grants DA020306 to K.E., MH63232 to R.J.C., DA22413, DA12408, and MH54137 to J.A.J., MH58921 to R.D.B., and DA13975 and MH58921 to A.G.
References
- Bannon MJ. 2004. Dopamine. Nature Encyclopedia of Life Sciences, Nature Publishing Group. Retrieved May 15, 2008 from www.els.net. [Google Scholar]
- Bannon MJ, Sacchetti P, Granneman JG. The dopamine transporter: potential involvement in neuropsychiatric disorders. In: Watson SJ, editor. Psychopharmacology: the fourth generation of progress. Nashville, TN: The American College of Neuropsychopharmacology; 2000. [Google Scholar]
- Binda F, Dipace C, Bowton E, Robertson SD, Lute BJ, Fog JU, Zhang M, Sen N, Colbran RJ, Gnegy ME, Gether U, Javitch JA, Erreger K, Galli A. Syntaxin 1A interaction with the dopamine transporter promotes amphetamine-induced dopamine efflux. Mol Pharmacol. 2008;74:1101–1108. doi: 10.1124/mol.108.048447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolan EA, Kivell B, Jaligam V, Oz M, Jayanthi LD, Han Y, Sen N, Urizar E, Gomes I, Devi LA, Ramamoorthy S, Javitch JA, Zapata A, Shippenberg TS. D2 receptors regulate dopamine transporter function via an extracellular signal-regulated kinases 1 and 2-dependent and phosphoinositide 3 kinase-independent mechanism. Mol Pharmacol. 2007;71:1222–1232. doi: 10.1124/mol.106.027763. [DOI] [PubMed] [Google Scholar]
- Carlsson A. Perspectives on the discovery of central monoaminergic neurotransmission. Annu Rev Neurosci. 1987;10:19–40. doi: 10.1146/annurev.ne.10.030187.000315. [DOI] [PubMed] [Google Scholar]
- Carvelli L, McDonald PW, Blakely RD, Defelice LJ. Dopamine transporters depolarize neurons by a channel mechanism. Proc Natl Acad Sci U S A. 2004;101:16046–16051. doi: 10.1073/pnas.0403299101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervinski MA, Foster JD, Vaughan RA. Psychoactive substrates stimulate dopamine transporter phosphorylation and down-regulation by cocaine-sensitive and protein kinase C-dependent mechanisms. J Biol Chem. 2005;280:40442–40449. doi: 10.1074/jbc.M501969200. [DOI] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci U S A. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N, Nikandrova Y, Bowton E, McMahon DG, Colbran RJ, Daws LC, Sitte HH, Javitch JA, Galli A, Gether U. Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron. 2006;51:417–429. doi: 10.1016/j.neuron.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Foster JD, Pananusorn B, Vaughan RA. Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J Biol Chem. 2002;277:25178–25186. doi: 10.1074/jbc.M200294200. [DOI] [PubMed] [Google Scholar]
- Galli A, Blakely RD, DeFelice LJ. Norepinephrine transporters have channel modes of conduction. Proc Natl Acad Sci U S A. 1996;93:8671–8676. doi: 10.1073/pnas.93.16.8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambalvo CT. Protein kinase C and dopamine transport—2. Effects of amphetamine in vitro. Neuropharmacology. 1992;31:1211–1222. doi: 10.1016/0028-3908(92)90049-u. [DOI] [PubMed] [Google Scholar]
- Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14:43–49. doi: 10.1016/0165-6147(93)90029-j. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Grünhage F, Schulze TG, Müller DJ, Lanczik M, Franzek E, Albus M, Borrmann-Hassenbach M, Knapp M, Cichon S, Maier W, Rietschel M, Propping P, Nöthen MM. Systematic screening for DNA sequence variation in the coding region of the human dopamine transporter gene (DAT1) Mol Psychiatry. 2000;5:275–282. doi: 10.1038/sj.mp.4000711. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cookson MR, Singleton A. Genes and parkinsonism. Lancet Neurol. 2003;2:221–228. doi: 10.1016/s1474-4422(03)00350-8. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlig KM, Binda F, Khoshbouei H, Blakely RD, McMahon DG, Javitch JA, Galli A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc Natl Acad Sci U S A. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor L, Gnegy ME. Protein kinase C inhibitors block amphetamine-mediated dopamine release in rat striatal slices. J Pharmacol Exp Ther. 1998;284:592–598. [PubMed] [Google Scholar]
- Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem. 2003;278:12070–12077. doi: 10.1074/jbc.M212815200. [DOI] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:E78. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Pei L, Moszczynska A, Vukusic B, Fletcher PJ, Liu F. Dopamine transporter cell surface localization facilitated by a direct interaction with the dopamine D2 receptor. EMBO J. 2007;26:2127–2136. doi: 10.1038/sj.emboj.7601656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Kim MY, Kim DH, Lee YS. Syntaxin 1A and receptor for activated C kinase interact with the N-terminal region of human dopamine transporter. Neurochem Res. 2004;29:1405–1409. doi: 10.1023/b:nere.0000026404.08779.43. [DOI] [PubMed] [Google Scholar]
- Lute BJ, Khoshbouei H, Saunders C, Sen N, Lin RZ, Javitch JA, Galli A. PI3K signaling supports amphetamine-induced dopamine efflux. Biochem Biophys Res Commun. 2008;372:656–661. doi: 10.1016/j.bbrc.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazei-Robison MS, Couch RS, Shelton RC, Stein MA, Blakely RD. Sequence variation in the human dopamine transporter gene in children with attention deficit hyperactivity disorder. Neuropharmacology. 2005;49:724–736. doi: 10.1016/j.neuropharm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Bowton E, Holy M, Schmudermaier M, Freissmuth M, Sitte HH, Galli A, Blakely RD. Anomalous dopamine release associated with a human dopamine transporter coding variant. J Neurosci. 2008;28:7040–7046. doi: 10.1523/JNEUROSCI.0473-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Morris AJ, Scarlata S. Regulation of effectors by G-protein alpha and beta-gamma subunits. Biochem Pharmacol. 1997;54:429–435. doi: 10.1016/s0006-2952(97)00032-4. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. Immunogold localization of the dopamine transporter: an ultrastructural study of the rat ventral tegmental area. J Neurosci. 1997;17:5255–5262. doi: 10.1523/JNEUROSCI.17-14-05255.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD. Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci. 2008;1129:35–46. doi: 10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlson GD, Wong DF, Tune LE, Ross CA, Chase GA, Links JM, Dannals RF, Wilson AA, Ravert HT, Wagner HN., Jr In vivo D2 dopamine receptor density in psychotic and nonpsychotic patients with bipolar disorder. Arch Gen Psychiatry. 1995;52:471–477. doi: 10.1001/archpsyc.1995.03950180057008. [DOI] [PubMed] [Google Scholar]
- Quick MW, Hu J, Wang D, Zhang HY. Regulation of a gamma-aminobutyric acid transporter by reciprocal tyrosine and serine phosphorylation. J Biol Chem. 2004;279:15961–15967. doi: 10.1074/jbc.M306924200. [DOI] [PubMed] [Google Scholar]
- Rayport S, Sulzer D, Shi WX, Sawasdikosol S, Monaco J, Batson D, Rajendran G. Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology. J Neurosci. 1992;12:4264–4280. doi: 10.1523/JNEUROSCI.12-11-04264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, McIntyre CC, Lisman JE. Reversal of synaptic memory by Ca2+/calmodulin-dependent protein kinase II inhibitor. J Neurosci. 2007;27:5190–5199. doi: 10.1523/JNEUROSCI.5049-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Niznik HB. Dopamine receptors and transporters in Parkinson's disease and schizophrenia. FASEB J. 1990;4:2737–2744. doi: 10.1096/fasebj.4.10.2197154. [DOI] [PubMed] [Google Scholar]
- St George-Hyslop PH. Molecular genetics of Alzheimer's disease. Biol Psychiatry. 2000;47:183–199. doi: 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Fukunaga K, Miyamoto E. Activation of nuclear Ca(2+)/calmodulin-dependent protein kinase II and brain-derived neurotrophic factor gene expression by stimulation of dopamine D2 receptor in transfected NG108–15 cells. J Neurochem. 2002;82:316–328. doi: 10.1046/j.1471-4159.2002.00967.x. [DOI] [PubMed] [Google Scholar]
- Vaughan RA, Huff RA, Uhl GR, Kuhar MJ. Protein kinase C-mediated phosphorylation and functional regulation of dopamine transporters in striatal synaptosomes. J Biol Chem. 1997;272:15541–15546. doi: 10.1074/jbc.272.24.15541. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Hitzemann R, Logan J, Schlyer DJ, Dewey SL, Wolf AP. Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse. 1993;14:169–177. doi: 10.1002/syn.890140210. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Newcorn J, Telang F, Solanto MV, Fowler JS, Logan J, Ma Y, Schulz K, Pradhan K, Wong C, Swanson JM. Depressed dopamine activity in caudate and preliminary evidence of limbic involvement in adults with attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2007;64:932–940. doi: 10.1001/archpsyc.64.8.932. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Fowler JS, Logan J, Abumrad NN, Hitzemann RJ, Pappas NS, Pascani K. Dopamine D2 receptor availability in opiate-dependent subjects before and after naloxone precipitated withdrawal. Neuropsychopharmacology. 1997;16:174–182. doi: 10.1016/S0893-133X(96)00184-4. [DOI] [PubMed] [Google Scholar]
- Wise RA. Drug-activation of brain reward pathways. Drug Alcohol Depend. 1998;51:13–22. doi: 10.1016/s0376-8716(98)00063-5. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na(+)/Cl(−)-dependent neurotransmitter transporters. Nature. 2005 doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zhang DQ, Stone JF, Zhou T, Ohta H, McMahon DG. Characterization of genetically labeled catecholamine neurons in the mouse retina. Neuroreport. 2004;15:1761–1765. doi: 10.1097/01.wnr.0000135699.75775.41. [DOI] [PubMed] [Google Scholar]