

Abstract
Background:
Behavioral variant frontotemporal dementia (bvFTD) is a common cause of younger onset dementia. Little is known about its rate of progression but a recently identified subgroup seems to have an excellent prognosis. Other determinants of survival are unclear.
Methods:
We analyzed survival in a large group of clinically diagnosed bvFTD patients (n = 91) with particular attention to demographic and clinical features at presentation. Of the 91 cases, 50 have died, with pathologic confirmation in 28.
Results:
Median survival in the whole group was 9.0 years from symptom onset, and 5.4 years from diagnosis. After the exclusion of 24 “phenocopy” cases, the analysis was repeated in a subgroup of 67 patients. The mean age at symptom onset of the pathologic group was 58.5 years and 16% had a positive family history. Their median survival was 7.6 years (95% confidence interval [CI] 6.6–8.6) from symptom onset and 4.2 years (95% CI 3.4–5.0) from diagnosis. The only factor associated with shorter survival was the presence of language impairment at diagnosis.
Conclusions:
Patients with definite frontotemporal dementia have a poor prognosis which is worse if language deficits are also present. This contrasts with the extremely good outcome in those with the phenocopy syndrome: of our 24 patients only 1 has died (of coincident pathology) despite, in some cases, many years of follow-up.
GLOSSARY
- ACE
= Addenbrooke’s Cognitive Examination;
- ADL
= activities of daily living;
- bvFTD
= behavioral variant frontotemporal dementia;
- CI
= confidence interval;
- FTD
= frontotemporal dementia;
- FTLD
= frontotemporal lobar degeneration;
- MMSE
= Mini-Mental State Examination.
Frontotemporal dementia (FTD) is an important cause of younger onset dementia.1 Of the clinical variants, the behavioral form (bvFTD) is the most common. Patients present with progressive changes in personality and social cognition, notably disinhibition, apathy, loss of empathy, altered eating patterns, stereotyped behaviors, and decline in manners and self-care. The diagnosis of bvFTD has a major impact on the family and is the main cause of considerable caregiver stress and burden.2
A frequently asked question, in clinical practice, is the likely progression and prognosis, but little published data are available to guide clinicians when advising patients and their families. Studies based on pathology as the gold standard for study inclusion indicate a very poor prognosis with survival of between 3 and 5 years.3,4 By contrast, a number of longitudinal clinic-based studies from Cambridge have identified a subgroup that fulfils clinical bvFTD criteria but with a benign course, suggesting a false-positive diagnosis, that are variously referred to as nonprogressors or phenocopy cases.5,6 Such cases present with identical clinical features to those with definite pathology7,8 but are distinguishable on the basis of normal brain imaging,5,9 better performance on cognitive measures, and relative preservation of activities of daily living.6,10 The aims of this study were to determine the prognosis of a large group of clinically diagnosed bvFTD patients with, and without, the inclusion of patients with a phenocopy syndrome, as well as to explore factors predictive of outcome in the progressive bvFTD cases.
METHODS
Case selection.
Review of the Cambridge Early Onset Dementia Clinic database identified 134 patients with a diagnosis of bvFTD presenting between 1990 and 2006, who met current consensus criteria for FTD11 with insidious onset, decline in social behavior and personal conduct, and emotional blunting as reported by family members. Forty-three patients were excluded for the following reasons: lost to follow-up (n = 11), inadequate clinical data (n = 9), and combined FTD with motor neuron disease (n = 23). The 91 patients included in the study were all 1) assessed by the same senior experienced behavioral neurologist (J.R.H.) and psychiatrist (Professor G.E. Berrios) to exclude major psychiatric illnesses (depression, alcohol or substance abuse, and schizophrenia); 2) investigated using standard protocols, which included a structured interview of caregivers for symptoms suggestive of FTD, the Cambridge Behavioural Inventory,12 the Mini-Mental State Examination (MMSE), and after 1997 the Addenbrooke’s Cognitive Examination (ACE),13 and neuroimaging by MRI scan; and 3) followed up for more than 3 years, with the exception of 8 patients who showed strong evidence of dementia on first assessment based on the finding of frontotemporal atrophy on MRI,5,9 impaired activities of daily living (ADL), and low score on the ACE (ACE <82). They all had English as a first language.
A retrospective review of the full medical records was conducted by an independent neurologist (B.G.), who had not been involved in diagnosis or care, using a study pro forma. Particular attention was paid to the first clinic attendance, diagnosis and symptom onset as reported by the family, performance on general cognitive screening tests: ACE13 and MMSE scores; and a positive family history, defined as having a first-degree relative with a history of younger onset dementia compatible with FTD. We extracted information on clinical features characteristic of FTD at presentation: 1) changes in personality and social behavior, apathy, disinhibition, stereotypic behavior, mental rigidity, alteration in food preference; 2) dysexecutive symptoms (poor planning, organization, and problem solving); 3) memory complaints; 4) psychotic symptoms (hallucinations and delusions); 5) disorders of language and communication noted by the examiner including paucity of speech (adynamism) or frank aphasia; and 6) motor features including parkinsonism, falls, and unsteadiness (those with motor neuron disease were excluded).
Standard protocol approvals, registrations, and patient consents.
This study has received approval from the ethical standards committee of Addenbrooke’s Hospital, Cambridge, UK. Received written consent was obtained from all patients in the study.
Statistics.
Data were analyzed using SPSS 15.0 (SPSS Inc., Chicago, IL). Survival analyses were conducted using Kaplan Meier methods (95% confidence limits). Survival curves across groups were examined with log rank tests. Endpoint to follow-up was date of death. The contribution of continuous variables (age at onset, MMSE and ACE at first visit) to risk of death was established with Cox proportional hazard regression models. The estimated hazard ratio, exp (b), measured the relationship between the intensities for individuals with and without a certain feature. Further analyses, to examine demographic and clinical features between groups at presentation, were conducted using independent t tests and χ2 tests where appropriate.
RESULTS
Demographic and clinical characteristics are presented in table 1. There were 59 men and 32 women. Eleven (12.1%) had a positive family history. The mean age at symptom onset was 57.2 ± 8.2 years and the mean duration from symptom onset to diagnosis was 3.6 ± 2.5 years. All of the patients presented with insidious changes in personality and social behavior as reported by their families. The most frequent feature was apathy (80%), followed by stereotypic behavior (77%), alteration in food preference (63%), and dysexecutive symptoms (62%). Memory complaints were present in 58%, disinhibition was present in 54%, and one third had a language disorder. A quarter had psychiatric symptoms (9% had hallucinations and 21% delusions). Motor symptoms such as parkinsonism, falls, and unsteadiness were present in 11%.
Table 1 Demographic and clinical characteristics of patients with frontotemporal dementia (FTD) and comparison of the pathologic and phenocopy subgroups
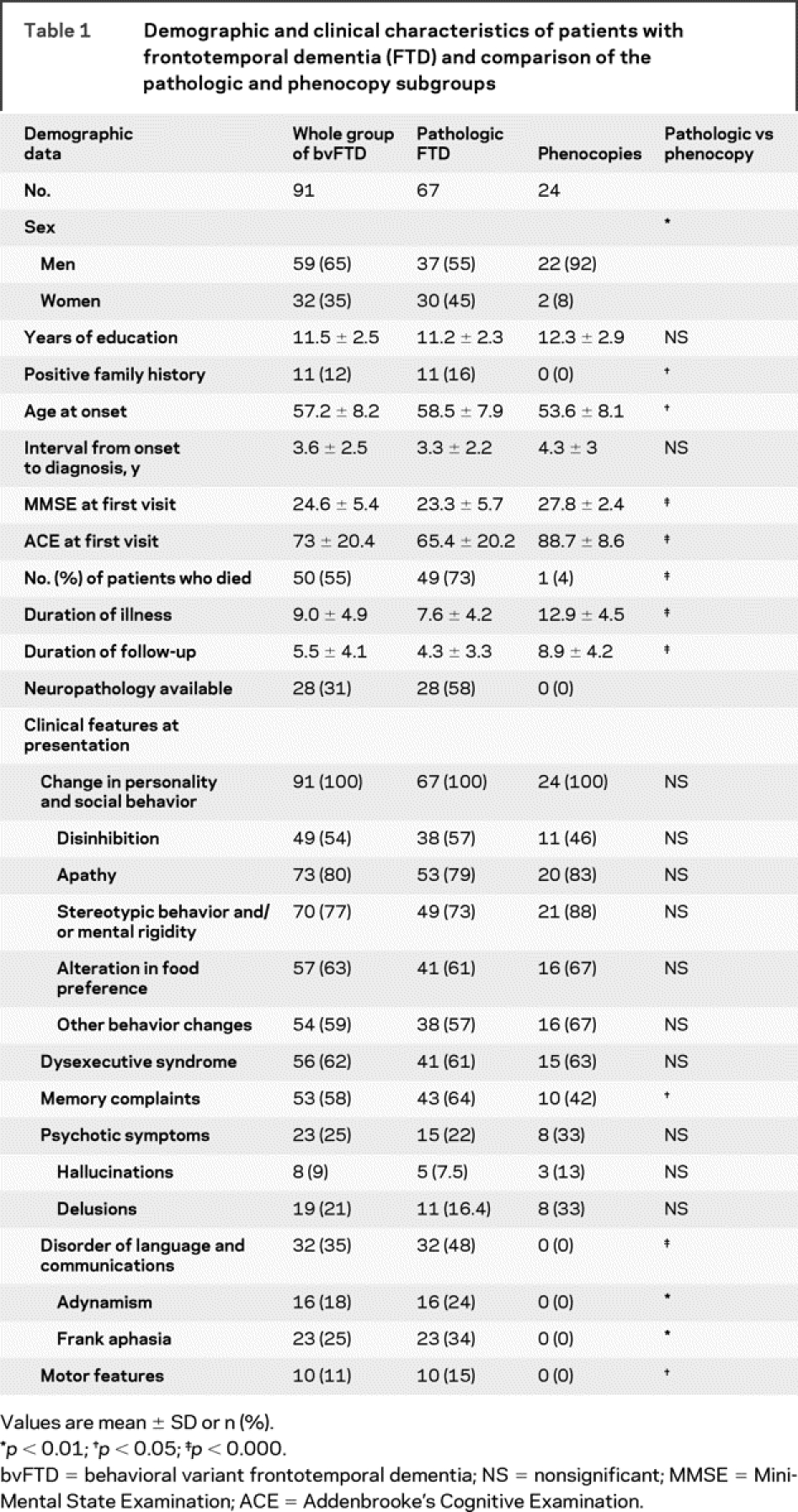
The average length of follow-up was 5.5 (±4.1) years. Overall, 50 (55%) died, of whom postmortem neuropathology was available for 28 (30.8%). The pathologic subtypes were tau positive pathology (frontotemporal lobar degeneration [FTLD]-tau) (n = 16; 57.1%), pathology with TDP-43 positive/tau-negative inclusions (FTLD-TDP/FTLD-UPS) (n = 11; 39.4%), and lacking distinctive histology (n = 1; 3.5%).14
The median survival for the whole group from symptom onset was 9.0 years (95% confidence interval [CI[ 8.0–10.0) and from diagnosis was 5.4 years (95% CI 4.6–6.2).
Pathologic vs phenocopy cases.
Close inspection of the clinical records indicated that disease progression was not evenly distributed. A subgroup showed no change over many years of follow-up. These patients have been previously described as nonprogressors or alternatively phenocopy cases.5,6,15 Inclusion of this subgroup in the survival analyses would undoubtedly have a major effect on overall survival in this cohort and would modify the contribution of predictive factors. We decided, therefore, to separate the patients into pathologic and phenocopy cases and explored factors affecting survival in the pathologic cases only. Nonprogression was defined as a lack of progression on the ACE and ADLs over a period of 3 years follow-up. In addition, all nonprogressors had a normal MRI at presentation as assessed using a previously described scale which rates frontal and temporal lobes,9,16 normal being a rating of 0 or 0.5.
Based upon these classification criteria, the total group of 91 patients comprised 67 pathologic and 24 phenocopy cases. Comparisons of demographic and clinical features between the 2 subgroups are shown in table 1. It is notable that among the 24 phenocopy cases, 22 (92%) were men and 2 (8%) were women. The pathologic group was significantly older (58.5 vs 53.6 years) than the phenocopy group and was also more impaired on the ACE (65.4 vs 88.7). In addition, a positive family history was seen in those with true FTD only (16% vs 0%). Disorders of language and communication were significantly more common in the pathologic subgroup (48% vs 0%). Motor features were also more common although present in a minority (15% vs 0%). Of the 67 pathologic cases 49 (73.1%) have died compared to only 1 (4.2%) from the phenocopy group who developed a mesothelioma. Unfortunately, consent for postmortem brain examination was not obtained from the family.
Survival analyses in progressive cases.
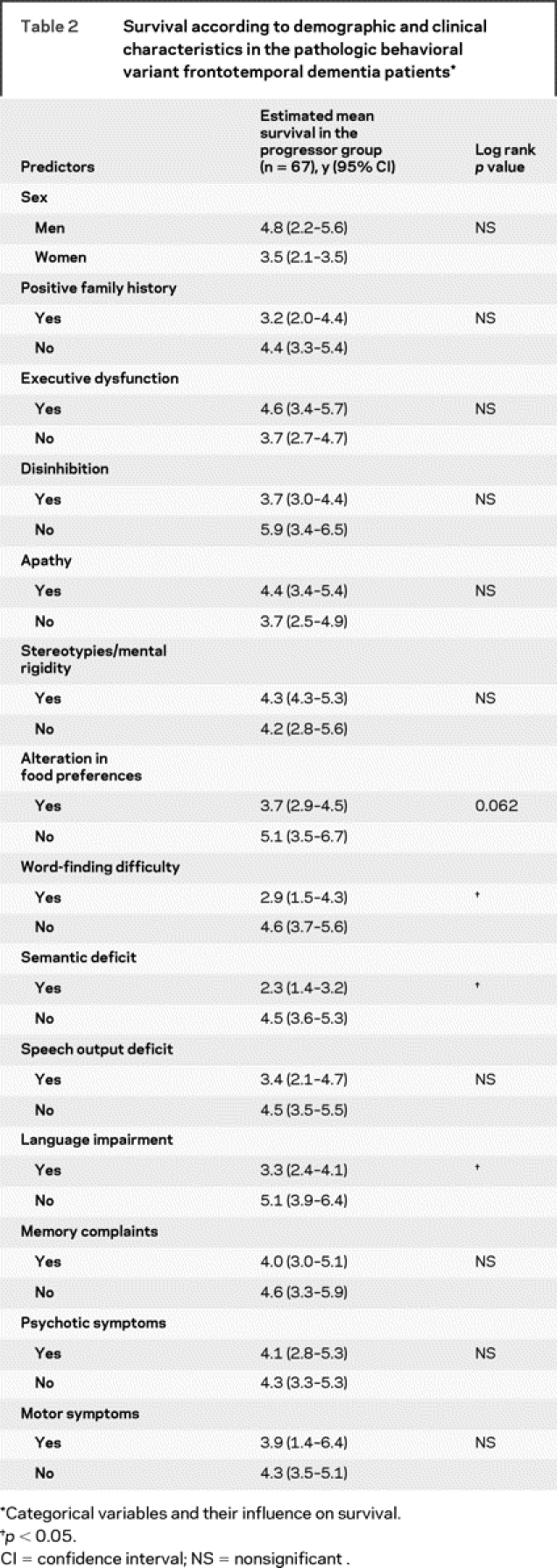
Kaplan-Meier survival analyses indicated a mean survival of 7.6 years (95% CI 6.6–8.6) from symptom onset and of 4.2 years (95% CI 3.4–5.0) from diagnosis of the progressors. The only factor impacting on survival was the presence of language disturbances at first assessment: semantic deficits, word-finding difficulty, and general language impairment were all associated with shorter survival (figure). In contrast, however, impaired speech output was not. One additional feature (alteration in food preference) approached significance. No other clinical features (executive dysfunctions, memory deficits, ACE or MMSE scores at first visit, psychotic symptoms, motor features) or demographic features (age at onset, sex, family history, behavioral features, memory performance) had an influence on survival (table 2).

Figure Survival plots: Pathologic behavioral variant frontotemporal dementia (bvFTD) patients
Kaplan-Meier curves for the pathologic bvFTD patients (A). The survival plots in B split the progressive patients into those with and without language impairment. I = cases not reaching death within the study period.
Table 2 Survival according to demographic and clinical characteristics in the pathologic behavioral variant frontotemporal dementia patients
DISCUSSION
The survival of this large group of clinically diagnosed bvFTD patients was not dissimilar to that previously reported3,4,17 with mean survival of 9.0 years (95% CI 8.0–10.0) from symptom onset and 5.4 years (95% CI 4.6–6.2) from diagnosis. These findings are, however, fallacious as the overall sample comprised 2 distinct subgroups. After removing the subgroup without evidence of progression from the analyses (i.e., phenocopy cases), survival in the pathologic subgroup (n = 67) was substantially shorter at 7.6 years (95% CI 6.6–8.6) from symptom onset and only 4.2 years (95% CI 3.4–5.0) from diagnosis compared to the whole sample. The latter figure is in line with studies examining survival that have used a pathologic diagnosis of FTD as the gold standard for entry selection criteria.3,4 This finding supports the view that previous clinical studies that have examined survival were biased due to the presence of phenocopy cases. When factors predictive of survival in the pathologic subgroup were examined, presence of language deficits, in particular word-finding difficulty and semantic deficit, was associated with shortened disease duration (see also17 for a similar finding reporting letter fluency performance as a predictor of survival). It is unclear why such deficits impact on survival in this subgroup. We suspect that combined behavioral and language deficits reflect conjoint frontal and temporal pathology and indicate, therefore, an extensive brain disease at presentation. Although the prevalence of other features (motor features, MMSE and ACE scores, age at disease onset, and positive family history for dementia) differed between progressors and phenocopies, none of these variables contributed to survival outcome.
These findings add to the growing literature that supports the existence of a benign group of patients that mimic clinical features suggestive of bvFTD without progression to frank dementia and prolonged survival. Such patients are typically men with normal performance on general cognitive tests such as the ACE,9,15 preserved executive function tasks6 and activities of daily living,10 as well as a normal MRI at presentation.9 A recent study has also demonstrated normal FDG-PET metabolism in all brain regions in this group.18 This study also indicates that a family history of dementia is much more common in the pathologic group than the phenocopy syndrome (16% vs 0%) and that patients with true FTD are significantly older.
A full discussion of the etiology of the nonprogressive syndrome is beyond the scope of this article. The finding of normal structural and metabolic imaging even after many years of follow-up19 makes a neurodegenerative disease extremely unlikely, although a pathologic confirmation of the phenocopy concept is still outstanding. A proportion may suffer a lifetime personality disorder which decompensates in midlife, while others may have a neuropsychiatric syndrome yet to be fully elucidated. Once patients with this phenocopy syndrome are excluded, then the survival for bvFTD from diagnosis is short. A parallel study has shown that the annual rate of decline on the ACE in pathologic patients is 10–12 points per year.15 We have also found evidence, in the present study, that mixed behavioral and language features at presentation is associated with poor prognosis.
The distinction between bvFTD patients likely to progress and those likely to remain stable over many years is critical. It has implications, both in terms of counseling individual families and for entry into trials of disease-modifying therapies. When faced with a patient with putative bvFTD, a prognosis can be established on the basis of sex, cognitive performance on the ACE, executive dysfunction, and the presence of language abnormalities and frontotemporal atrophy on MRI. More tentatively, motor abnormalities on examination may also be added to the list but this requires further confirmation. Once the diagnosis of probable pathologic bvFTD is established, then a prognosis can be given to the person with dementia and his or her family. The 50% survival for this group of patients is only 5 years and 80% are deceased by 8 years, making it a highly malignant disease and a high priority for the development of disease-modifying treatments.
DISCLOSURE
Dr. Garcin reports no disclosures. Dr. Lillo is supported by a CONYCIT scholarship, government of Chile, and Faculty of Medicine, University of Chile. Dr. Hornberger reports no disclosures. Dr. Piguet receives research support from the National Health and Medical Research Council of Australia and was awarded the Clinical Career Development Award Fellowship #510184. K. Dawson reports no disclosures. Dr. Nestor has served on a scientific advisory board for GlaxoSmithKline; received funding for travel from Eisai Inc.; and receives research support from the Medical Research Council UK. Dr. Hodges serves on the editorial board of Aphasiology, Cognitive Neuropsychiatry, and Cognitive Neuropsychology; receives royalties from publishing Cognitive Assessment for Clinicians (Oxford University Press, 2007) and Frontotemporal Dementia Syndromes (Cambridge University Press, 2007); and receives research fellowship from the Australian Research Council Federation (FF0776229).
Address correspondence and reprint requests to Prof. John R. Hodges, Prince of Wales Medical Research Institute, Cnr Barker St and Easy St, Randwick, Sydney, NSW 2031, Australia j.hodges@powmri.edu.au
Disclosure: Author disclosures are provided at the end of the article.
Received April 2, 2009. Accepted in final form August 12, 2009.
REFERENCES
- 1.Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology 2002;58:1615–1621. [DOI] [PubMed] [Google Scholar]
- 2.Mioshi E, Bristow M, Cook R, Hodges JR. Factors underlying caregiver stress in frontotemporal dementia and Alzheimer’s disease. Dement Geriatr Cogn Disord 2009;27:76–81. [DOI] [PubMed] [Google Scholar]
- 3.Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology 2003;61:349–354. [DOI] [PubMed] [Google Scholar]
- 4.Rascovsky K, Salmon DP, Lipton AM, et al. Rate of progression differs in frontotemporal dementia and Alzheimer disease. Neurology 2005;65:397–403. [DOI] [PubMed] [Google Scholar]
- 5.Davies RR, Kipps CM, Mitchell J, Kril JJ, Halliday GM, Hodges JR. Progression in frontotemporal dementia: identifying a benign behavioral variant by magnetic resonance imaging. Arch Neurol 2006;63:1627–1631. [DOI] [PubMed] [Google Scholar]
- 6.Hornberger M, Piguet O, Kipps C, Hodges JR. Executive function in progressive and nonprogressive behavioral variant frontotemporal dementia. Neurology 2008;71:1481–1488. [DOI] [PubMed] [Google Scholar]
- 7.Piguet O, Hornberger M, Shelley BP, Kipps CM, Hodges JR. Sensitivity of current criteria for the diagnosis of behavioral variant frontotemporal dementia. Neurology 2009;72:732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornberger M, Shelley BP, Kipps CM, Piguet O, Hodges JR. Can progressive and non-progressive behavioral variant frontotemporal dementia be distinguished at presentation? J Neurol Neurosurg Psychiatry (in press 2009). [DOI] [PubMed]
- 9.Kipps CM, Davies RR, Mitchell J, Kril JJ, Halliday GM, Hodges JR. Clinical significance of lobar atrophy in frontotemporal dementia: application of an MRI visual rating scale. Dement Geriatr Cogn Disord 2007;23:334–342. [DOI] [PubMed] [Google Scholar]
- 10.Mioshi E, Kipps CM, Hodges JR. Activities of daily living in behavioral variant frontotemporal dementia: differences in caregiver and performance-based assessments. Alzheimer Dis Assoc Disord 2009;23:70–76. [DOI] [PubMed] [Google Scholar]
- 11.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 12.Wedderburn C, Wear H, Brown J, et al. The utility of the Cambridge Behavioural Inventory in neurodegenerative disease. J Neurol Neurosurg Psychiatry 2008;79:500–503. [DOI] [PubMed] [Google Scholar]
- 13.Mathuranath PS, Nestor PJ, Berrios GE, Rakowicz W, Hodges JR. A brief cognitive test battery to differentiate Alzheimer’s disease and frontotemporal dementia. Neurology 2000;55:1613–1620. [DOI] [PubMed] [Google Scholar]
- 14.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 2009;117:15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kipps CM, Nestor PJ, Dawson CE, Mitchell J, Hodges JR. Measuring progression in frontotemporal dementia: implications for therapeutic interventions. Neurology 2008;70:2046–2052. [DOI] [PubMed] [Google Scholar]
- 16.Broe M, Hodges JR, Schofield E, Shepherd CE, Kril JJ, Halliday GM. Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 2003;60:1005–1011. [DOI] [PubMed] [Google Scholar]
- 17.Roberson ED, Hesse JH, Rose KD, et al. Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology 2005;65:719– 725. [DOI] [PubMed] [Google Scholar]
- 18.Kipps CM, Hodges JR, Fryer TD, Nestor PJ. Combined magnetic resonance imaging and positron emission to-mography brain imaging in behavioural variant frontotemporal degeneration: refining the clinical phenotype. Brain (in press 2009). [DOI] [PubMed]
- 19.Kipps CM, Nestor PJ, Fryer TD, Hodges JR. Behavioural variant frontotemporal dementia: not all it seems? Neurocase 2007;13:1–11. [DOI] [PubMed] [Google Scholar]