

Abstract
Background:
COL4A1 mutations cause familial porencephaly, infantile hemiplegia, cerebral small vessel disease (CSVD), and hemorrhagic stroke. We recently described hereditary angiopathy with nephropathy, aneurysm, and muscle cramps (HANAC) syndrome in 3 families with closely localized COL4A1 mutations. The aim of this study was to describe the cerebrovascular phenotype of HANAC.
Methods:
Detailed clinical data were collected in 14 affected subjects from the 3 families. MRI and magnetic resonance angiography (MRA) were performed in 9 of them. Skin biopsies were analyzed by electron microscopy in affected subjects in the 3 families.
Results:
Only 2 of 14 subjects had clinical cerebrovascular symptoms: a minor ischemic stroke at age 47 years and a small posttraumatic hemorrhage under anticoagulants at age 48 years. MRI-MRA showed cerebrovascular lesions in 8 of 9 studied subjects (mean age 39.4 years, 21–57 years), asymptomatic in 6 of them. Unique or multiple intracranial aneurysms, all on the carotid siphon, were observed in 5 patients. Seven patients had a CSVD characterized by white matter changes (7/7) affecting subcortical, periventricular, or pontine regions, dilated perivascular spaces (5/7), and lacunar infarcts (4/7). Infantile hemiplegia, major stroke, and porencephaly were not observed. Skin biopsies showed alterations of basement membranes at the dermoepidermal junction associated with expansion of extracellular matrix between smooth vascular cells in the arteriolar wall.
Conclusion:
The cerebrovascular phenotype in hereditary angiopathy with nephropathy, aneurysm, and muscle cramps syndrome associates a cerebral small vessel disease and a large vessel disease with aneurysms of the carotid siphon. It is consistent with a lower susceptibility to hemorrhagic stroke than in familial porencephaly, suggesting an important clinical heterogeneity in the phenotypic expression of disorders related to COL4A1 mutations.
GLOSSARY
- CADASIL
= cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy;
- CPK
= creatine phosphokinase;
- CSVD
= cerebral small vessel disease;
- DPS
= dilated perivascular spaces;
- HANAC
= hereditary angiopathy with nephropathy, aneurysm, and muscle cramps;
- HERNS
= hereditary endotheliopathy with retinopathy, nephropathy, and stroke;
- ICA
= intracranial aneurysm;
- MRA
= magnetic resonance angiography;
- OMIM
= Online Mendelian Inheritance in Man;
- RVCL
= retinal vasculopathy with cerebral leukodystrophy;
- WMC
= white matter changes.
Type IV collagen is a major component of basement membranes. Type IV collagen α1 and α2 chains form the widely expressed α1α1α2(IV) heterotrimers.1–3 Mutations in the COL4A1 gene, which encodes the α1(IV) chain, have been reported in 6 families with autosomal dominant forms of porencephaly, in which a porencephalic cavity is caused by a cerebral hemorrhage that occurred during the intrauterine or neonatal period.4–9 A broad range of neurologic features have been described in families with hereditary porencephaly related to COL4A1 mutations, including infantile hemiplegia, mental retardation, and hemorrhagic strokes in childhood or in adults often triggered by head trauma. The cerebrovascular phenotype also includes a cerebral small vessel disease (CSVD) with leukoencephalopathy, lacunar infarcts, microbleeds, macrobleeds, and dilated perivascular spaces (DPS), sometimes without porencephaly. Three additional families with CSVD and COL4A1 mutations have been reported, 1 with hemorrhagic stroke and retinal arteriolar tortuosity, 1 with hemorrhagic stroke and cataract, and the other with ischemic stroke and anterior segment dysgenesis of the eye (Axenfeld-Rieger anomaly).10–13 One sporadic case with recurrent intracerebral hemorrhage was also reported.14 No manifestations suggestive of a systemic disease were observed in these patients. Consequently, COL4A1 mutations are a new cause of CSVD, in addition to lipohyalinosis secondary to hypertension, and to other hereditary conditions such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), retinal vasculopathy with cerebral leukodystrophy (RVCL), and amyloid angiopathy.
We have recently described an autosomal dominant syndrome in 3 unrelated French families that we named HANAC syndrome for hereditary angiopathy with nephropathy, aneurysm, and muscle cramps.15,16 HANAC syndrome is due to COL4A1 mutations affecting glycine residues that are in close proximity in exons 24 and 25 within the triple-helix domain of the protein. HANAC syndrome has a broad phenotypical spectrum with systemic as well as cerebrovascular features. The systemic phenotype includes a nephropathy with either hematuria or bilateral renal cysts, a muscular disease with cramps and/or elevated creatine phosphokinase (CPK), and retinal arterial tortuosity. Because ultrastructural examination of the kidney and the skin shows abnormal thickening, multilamination, and/or focal disruption of the basement membranes, HANAC is considered as a systemic “basalopathy.” The objective of the present study is to give a detailed description of neurologic manifestations and brain imaging in the 3 recently reported families with HANAC syndrome.
METHODS
A total of 14 affected members have been identified in the 3 families (A, B, and C) with HANAC syndrome (figure e-1 on the Neurology® Web site at www.neurology.org). As previously reported, genetic analysis showed COL4A1 mutations localized in exon 24 in family A (c.1493G>T) and in exon 25 in family B (c.1555G>A) and family C (c.1583G>A).16
Standard protocol approvals, registration, and patient consents.
The study received approval from the ethical regional committee on human experimentation (“Comité de Protection des Personnes-Ile de France VI number P061017–IDRCB 2007-A01441-52). Written informed consent was obtained from all patients participating in the study.
Clinical data.
Nine of the 14 patients with COL4A1 mutations were directly interviewed and examined for neurologic evaluation: 3 affected brothers and their 3 children in family A, 2 patients in family B (the father and her daughter), and 1 patient in family C. Clinical information on the 4 deceased patients and on the affected subjects who declined to participate in this study was obtained through their relatives. The neurologic interview recorded mental retardation, infantile hemiparesis, strokes and their types, cranial traumas and their consequences, headaches, epilepsy, dementia, and sudden death. Risk factors for cerebral vascular disorders were recorded.
Clinical evaluation of systemic involvement, including renal, muscle, eye, and cardiovascular symptoms, has been reported in detail.16 All 14 patients and 3 relatives without COL4A1 mutation (B.III-2, B.II-1, and C.III-2) had fundus examination and retinal angiography.
Cerebral imaging.
Brain MRI was performed in 9 patients and in 1 unaffected relative (C.III-2) using a 1.5-T magnetic resonance scanner (contiguous slices of 5 mm thickness) and included T1-weighted images, T2-weighted images, fluid-attenuated inversion recovery sequences, and magnetic resonance angiography (MRA). T2 gradient-echo (T2*) weighted images were recorded in 7 patients. When MRA was inconclusive about the presence of an intracranial aneurysm (ICA), 16-row multislice CT angiography was performed. Four-axis conventional angiography was available in 1 patient.
All imaging data were analyzed by a consensus of 2 neurologists (S.A. and P.F.) and 1 neuroradiologist (B.M.). The following lesions were assessed: white matter changes (WMC) defining leukoencephalopathy, lacunar infarcts, DPS, microbleeds, macrobleeds, and porencephaly. MRI lesions were categorized by anatomic cerebral region (appendix e-1). For lobar white matter, it was specified when lesions affected the arcuate fibers and the center semiovale. WMC were rated according to the Scheltens visual scale, taking into account size, number, and anatomic distribution of the lesions17 (appendix e-1). The numbers of lacunar infarcts and microbleeds were recorded.
The size of each ICA was defined by its maximal diameter. The carotid siphon was divided according to Fisher classification into 5 segments from its beginning to its end with 3 extradural (C5 to C3) and 2 intradural (C2 and C1) segments. Other intracranial arteries abnormalities, such as dolichoectasia, were also examined.
Skin biopsy.
A punch biopsy was performed in healthy skin of subjects A.III.3, A.IV.4, B.II.1, and C.II.3 (with the COL4A1 mutation) and C.III.2 (without the mutation). Electron microscopy was performed as previously described.16
RESULTS
Clinical data.
None of the 14 subjects had infantile hemiparesis or mental retardation. There was no history of stroke during the neonatal period and childhood, or history of spontaneous subarachnoid or intracerebral hemorrhage or dementia in the 3 families. None of the 14 subjects had diabetes or hypertension, 1 patient had hypercholesterolemia (A.III.1), 2 subjects were past smokers (A.III.1 and A.III.3), and 1 was an active smoker (A.III.5). Neurologic examination was normal for CNS functions in the 9 subjects whom we directly assessed. For the 4 deceased subjects, the age of death ranged from 60 to 69 years. The causes of death were not neurologic. Subject A.II.1, whose genotype is unknown, died suddenly at age 6 months from an unknown cause. No other sudden death was recorded.
One patient (A.IV.4) had a unique seizure at age 19 years without recurrence in the absence of any therapy. Three subjects (A.III.1, A.IV.1, and C.II.3) had migraine without aura. Only 2 of the 14 subjects with COL4A1 mutations had a history of cerebrovascular injury. Patient A.III.3 had an acute cerebellar ataxia without vertigo lasting 2 days at age 47 years; MRI with diffusion-weighted imaging, performed only 8 days later, did not show any recent infarct. The diagnosis of brainstem minor ischemic stroke was retained, and investigations revealed neither cardiac nor large artery cause. Patient A.III.1 presented a posttraumatic cerebromeningeal hemorrhage at age 48 years, while the patient was receiving anticoagulants for venous thrombosis of the lower limb. A cranial trauma was indeed responsible for confusion, aphasia, and headaches related to a left temporal hemorrhagic contusion measuring 23 mm with adjacent subarachnoid hemorrhage. His mental status improved within 24 hours, and his language totally recovered within 4 weeks. Notably, his brother (A.III.5) had a serious brain trauma due to a car crash at age 30 years, with initial loss of consciousness and transient confusion lasting 1 day. No CT scan was performed. He denied any persistent neurologic deficit.
All patients showed retinal arteriolar tortuosity and had muscle cramps, with elevation of CPK, whereas 3 relatives without COL4A1 mutation did not have any retinal or muscular abnormalities.16 Renal involvement was observed in 10 patients, consisting of renal cysts in 4, hematuria in 7 (family A), and renal insufficiency in 5. Raynaud phenomenon was recorded in 6 patients, and supraventricular arrhythmia was recorded in 5.16
Radiologic data.
Brain MRI was performed in 9 subjects (mean age 39.4 years, 21–57 years), during the third decade of life in 4 of them and during the fifth and sixth decades in the other 5 patients. All but 1 affected subject (A.III.5, aged 44 years) showed at least 1 abnormality. Four patients had a combination of ICAs and CSVD, 3 had only CSVD, and 1 had an isolated ICA (tables 1 and e-1). Interestingly, no porencephaly was observed. Brain imaging performed in 1 unaffected relative (C.III.2) was normal.
Table 1 White matter changes and other components of cerebral small vessel disease on brain MRI
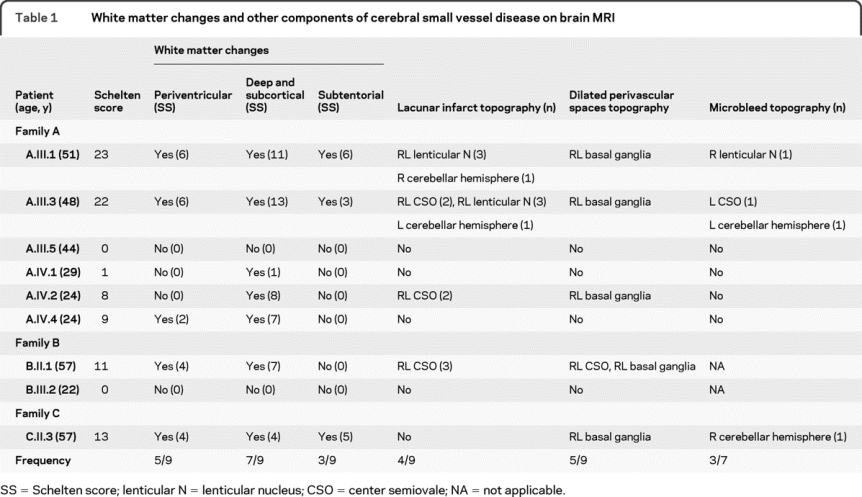
A total of 12 ICAs were present in the 5 subjects with at least 1 affected member in each family: 3 of 6 in family A (A.III.3, AIV.2, and A.IV4), 1 of 2 in family B (B.III.2), and 1 of 1 in family C (C.II.3) (figure 1 and table e-1). Multiple ICAs were observed in 3 patients. All ICAs were asymptomatic and discovered on systematic radiologic examinations at ages ranging from 21 to 57 years. The majority of ICAs were small; only 1 was larger than 7 mm. The ICAs presented a large base of implantation without identifiable neck. All were localized on the intracranial carotid, at different levels of the carotid siphon. Six ICAs were extradural (1 on C4 and 5 on C3), and 6 were intradural (4 on C2 and 2 on C1). Eleven of the 12 aneurysms were localized on the right carotid, and 1 was localized on the left carotid. Additionally, 2 patients (A.III.3 and C.II.3) had dolichosiphons of both carotids.

Figure 1 Intracranial aneurysms in HANAC syndrome
Intracranial aneurysms (arrows) or dolichoectasia (star) are shown in the 5 patients of the 3 families. Conventional angiography demonstrates multiple aneurysms on the right distal carotid artery in patient A.III.3 (A). Note an aspect of dolichoarteries of the carotid siphon. Magnetic resonance angiography (MRA) on axial slice (B) shows an aneurysm of the right distal carotid artery in patient A.IV.2. CT angiography (C) demonstrates 2 aneurysms of the right distal carotid artery with the form of glove finger in patient A.IV.4. (D) shows a right distal carotid aneurysm on axial slice of MRA in patient B.III.2. CT angiography shows multiple intracranial aneurysms of the right carotid arteries (E) and dolichoectasia (F) of the left distal carotid artery in patient C.II.3. HANAC = hereditary angiopathy with nephropathy, aneurysm, and muscle cramps.
Seven patients had MRI abnormalities consistent with a CSVD, including 5 of 6 in family A (A.III.1, A.III.3, A.IV.3, A.IV2, and A.IV.4), 1 of 2 in family B (B.II.1), and 1 of 1 in family C (C.III.3). All these 7 patients had WMC with a periventricular (5/7), subcortical (7/7), and/or subtentorial (3/7) distribution (table 1). Periventricular WMC showed an equal distribution between anterior, posterior, and lateral horns (figure 2). Subcortical WMC especially affected the frontal and parietal lobes, with the largest and most confluent lesions being observed in the inferoposterior areas of these lobes. Temporal and occipital lobes as well as U fibers were globally spared. WMC were predominantly located in the centrum semiovale and sometimes involved the external capsule and the posterior part of the posterior limb of the internal capsule (figure 2). Subtentorial WMC affected the core of the pons respecting the surface (figure 2). No cortical involvement was noted. In addition, 15 lacunar infarcts were observed in 4 patients (A.III.1, A.III.3, A.IV.2, and B.II.1), including 9 located in white matter (7 in the centrum semiovale and 2 in cerebellum hemisphere) and 6 in gray matter, all in the lenticular nucleus (table 1 and figure e-2). Five patients (A.III.1, A.III.3, A.IV.2, B.II.1, and C.II.3) had DPS in the basal ganglia and/or centrum semiovale (table 1 and figure e-2). Interestingly, only 3 (A.III.1, A.III.3, and C.II.3) of the 7 subjects investigated by T2* sequences had microbleeds, with 1 or 2 microbleeds per patient (table 1 and figure e-2). Patient A.III.1 had a 10-mm posttraumatic hemorrhagic lesion of the left external temporal region, but no other patient had a macrobleed.

Figure 2 White matter changes
Fluid-attenuated inversion recovery (FLAIR) images show multiple periventricular and subcortical cerebral white matter changes in patients A.III.1 at age 51 years (A), A.III.3 at age 48 years (B), A.IV.2 at age 24 years (C), A.IV.4 at age 24 years (D), B.II.1 at age 57 years (E), and C.II.3 at age 57 years (F). FLAIR images denote subtentorial white matter changes of the rostral pons in patients A.III.1 (G), A.III.3 (H), and C.II.3 (I).
Globally, CSVD seemed more severe in older than in younger patients; the Scheltens score was generally higher in the oldest generation (table 1). However, the severity of WMC was not homogeneous among patients of the same age. Indeed, the 44-year-old patient A.III.5 did not have any MRI abnormalities. Moreover, the 29-year-old patient A.IV.1 had very mild WMC in comparison with his 24-year-old brother (A.IV.2).
Ultrastructural skin arterial lesions.
Significant ultrastructural anomalies were found in affected subjects although their skin had a completely normal appearance on physical examination and light microscopy. Replication of the lamina densa was observed at the dermoepidermal junction in the affected subjects of the 3 families (A.III.3, A.IV.4, B.II.1, and C.II.3; figure 3, A–C). The wall of dermal arterioles was also markedly altered. Vascular smooth muscle cells were dissociated, with abnormal spreading of the basement membrane (figure 3, D–F). The vessel wall had a normal appearance in the skin of a control sample, unaffected subject C.III.2 (data not shown).

Figure 3 Skin basement membrane abnormalities
Ultrastructural examination of skin biopsies in affected patients of the 3 families (A.IV.4 [A and D], B.II.1 [B and E], and C.II.3 [C and F]) shows segmental basement membrane (BM) replications at the dermoepidermal junction (arrow, A–C). In dermal vessels, vascular smooth muscle cells are dissociated by abnormal expansion and thickening of the BM (open arrow and asterisk, D–F).
DISCUSSION
Since the initial description of 3 distinct phenotypes associated with COL4A1 mutations that include autosomal dominant type I porencephaly (Online Mendelian Inheritance in Man [OMIM] 175780), cerebral small vessel disease with hemorrhages (OMIM 607595), and HANAC syndrome (OMIM 611773), identification of additional families now clearly indicates that autosomal dominant porencephaly and CSVD are part of a continuum with overlapping neurologic clinical and imaging features (table 2). This study focused on cerebrovascular disease in HANAC syndrome and revealed 4 distinctive features. First, although cerebrovascular lesions are common, they are usually asymptomatic in contrast with patients with prominent brain disease related to COL4A1 mutations, in whom various degrees of infantile hemiplegia, mental retardation, and stroke were commonly observed. Second, in addition to CSVD dominated by leukoencephalopathy, MRI analysis revealed unique or multiple aneurysms electively localized on the carotid siphon, which indicates that mutations in COL4A1 are not only involved in CSVD but also in large artery disease. Third, no patient with HANAC syndrome displayed porencephalic cavities, macrobleed, or a large number of microbleeds. Fourth, cerebrovascular disease in HANAC is part of a multiorgan disease with systemic vasculopathy as attested to by the results of skin biopsy.
Table 2 Phenotypic and genotypic characteristics of families with COL4A1-related disorders

In human and mice mutants with porencephaly related to COL4A1 mutation, the phenotype includes a high susceptibility to hemorrhagic strokes frequently triggered by birth trauma, brain trauma, or anticoagulant treatment.4,11 By contrast, in HANAC syndrome, only 1 of the 14 affected members had a small hemorrhage after a serious head trauma despite anticoagulant therapy. Another subject had a severe head trauma without any trace of cerebral hemorrhage on follow-up MRI. These findings strongly suggest that COL4A1 mutations underlying HANAC syndrome are associated with a lower risk of hemorrhagic strokes than COL4A1 mutations underlying familial porencephaly and CVSD. The absence of severe brain hemorrhages may be due to environmental and/or genetic modifiers. Mice with COL4A1 mutations also show important phenotypic heterogeneity,4,18 with a significant influence of the genetic background on the ocular phenotype.19 Because of the small number of families and patients with COL4A1 mutations described so far, a precise genotype-phenotype correlation with neurologic as well as systemic manifestations remains hazardous. Nevertheless, our data raise the hypothesis of a mutation effect on the phenotypic expression, because COL4A1 mutations responsible for HANAC syndrome are closely localized in a very narrow stretch of the gene, whereas other mutations usually affect more C-terminal glycine residues (table 2).
CSVD in HANAC syndrome is dominated by leukoencephalopathy affecting periventricular, deep regions and the pons. WMC were noted despite the young age of the patients and in the absence of vascular risk factors, such as hypertension, usually associated with WMC.20 The highest lesion load was observed in frontal and parietal white matter predominantly in posterior regions, especially in the centrum semiovale. The temporal lobe and arcuate fibers are spared in contrast with the abnormalities observed in CADASIL.21,22 Most of the WMC and lacunar infarcts were located in the territory of lenticulostriate small perforating arteries, leptomeningeal long penetrating arteries, and pontine perforating arteries with a pattern recovering partially those of WMC associated with hypertension and arteriolosclerosis.22,23
The severity of WMC seems to be variable between and within families with HANAC, suggesting the presence of genetic and/or environmental modifiers or subtle differences in the consequences of the different mutations. In a previously reported family of 6 affected members with a COL4A1 mutation but without HANAC disease, 2 subjects died of intracranial hemorrhage, whereas MRI signal abnormalities did not change after a follow-up period of 7 years in the other patients.24 These findings correspond with our data showing that COL4A1 mutation carriers may have a great diversity in clinical and radiologic expression of cerebrovascular injury.
COL4A1 mutations in HANAC syndrome constitute a new monogenic cause of familial ICAs with 55% of the affected patients. ICAs were previously described in only 3 patients from 3 different families with COL4A1 mutation but without HANAC syndrome5,7,12,13 (table 2). The other hereditary diseases associated with ICAs are rare and consist mainly of diseases of connective tissue and extracellular matrix such as autosomal dominant polycystic kidney disease and Ehlers-Danlos syndrome type IV with mutations in COL3A1.24–26 The extracellular matrix of the arterial wall plays an important role in strength and elasticity of intracranial arteries. Collagen types III and IV and elastin fibers are decreased in the wall of ICAs as well as in skin biopsies of patients with ICA.27 Moreover, genes encoding extracellular matrix protein, including COL4A1, have been associated with a susceptibility to ICA formation in the general population.28 In vascular basement membrane, COL4A1 forms a sheetlike network beneath the endothelium and surrounding smooth muscle cells.1,2 Our findings suggest that COL4A1 mutations observed in HANAC syndrome lead to a disruption of extracellular matrix and a remodeling of the vascular wall with the formation of ICA.
A surprising fact about ICAs in HANAC syndrome was their elective localization on the carotid siphon both on intradural and extradural segments. The collagen type IV network is essential for the cohesiveness of basement membranes under conditions of increasing mechanical demands.29 The carotid siphon constitutes a peculiar zone due to the conjunction of a high blood flow and a tortuous curving form. In HANAC syndrome, the occurrence of basement membrane defects in this area, which is subject to high mechanical stress, may predispose to ICA formation in the carotid siphon. Intriguingly, no patient had a spontaneous subarachnoid hemorrhage, which may be due to the small size of these ICAs or to a low risk of rupture of aneurysms associated with COL4A1 mutations.
In addition to CSVD and ICA, COL4A1 mutations in HANAC syndrome are responsible for a multiorgan disease affecting the kidney, the musculature, and the retinal small arterioles, which seems to be the hallmark of the syndrome. The ultrastructural alterations observed in the skin, particularly in dermal arterioles, demonstrate a systemic vasculopathy. Interestingly, skin biopsy also shows changes in blood vessel morphology in 2 autosomal dominant cerebral angiopathies: CADASIL (OMIM 125310) due to mutations in the NOTCH3 receptor and hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS), a distinctive subtype of RVCL caused by mutations in TREX1 transcription factor.30,31 Patients with CADASIL and transgenic mice expressing mutant Notch3 show an enlargement of the inter–smooth muscle cell space, in skin vessels and tail artery sections. These anomalies precede the accumulation of granular osmiophilic material in transgenic mice.32 In HERNS, the brain disease is associated with renal abnormalities, Raynaud phenomenon, and retinal vasculopathy with ultrastructural alterations affecting capillaries in the brain and other tissues,33 but with a distinct feature from HANAC syndrome. Skin biopsy may provide a cost-effective guide before practicing an expensive and time-consuming genetic screening of COL4A1 mutations.
AUTHOR CONTRIBUTIONS
Sonia Alamowitch and Emmanuelle Plaisier designed the study, retrieved the data, and drafted the report. Pascal Favrole and Béatrice Marro retrieved the clinical and radiologic data. Catherine Prost and Zhiyong Chen retrieved the data of skin biopsy. Tom Van Agtmael helped with the interpretation of the data. Pierre Ronco initiated the project and contributed to design and revision of the report.
ACKNOWLEDGMENT
The authors thank Safa Benhassine and Marie-Chrisine Verpont for technical assistance.
DISCLOSURE
Dr. Alamowitch serves on the editorial committee of the Revue Neurologique. Dr. Plaisier, Dr. Favrole, Dr. Prost, and Mrs. Chen report no disclosures. Dr. Van Agtmael receives research support from the Medical Research Council UK [G0601268 (PI)]. Dr. Marro reports no disclosures. Dr. Ronco serves as a Section Editor for Nephrology Dialysis Transplantation and serves on the editorial boards of the Journal of American Society of Nephrology, Nature Clinical Practice Nephrology, and Kidney International; and has received research support from Amgen SAS.
Supplementary Material
Address correspondence and reprint requests to Dr. Sonia Alamowitch, Stroke Unit, Department of Neurology, Tenon University Hospital, AP-HP, 4 Rue de la Chine, Paris, France sonia.alamowitch@tnn.aphp.fr
Supplemental data at www.neurology.org
*These authors contributed equally to this work.
Supported by grants from INSERM, Université Pierre et Marie Curie–Paris 6 (BQR and legs Tessier), Assistance Publique–Hôpitaux de Paris (Délégation à la Recherche Clinique, Contrat CIRC), Association pour l’Utilisation du Rein Artificiel (AURA), Agence Nationale de le Recherche (ANR-08-Genopath-018-02), and Délégation Générale à la Santé, and through Coordination Theme 1 (Health) of the European Community’s 7th Framework Programme (grant agreement no. HEALTH-F2-2007- 201590). T.V.A. is supported by an MRC New Investigator Research Grant.
Disclosure: Author disclosures are provided at the end of the article.
Received April 19, 2009. Accepted in final form September 9, 2009.
REFERENCES
- 1.Mayne R. Collagenous proteins of blood vessels. Arteriosclerosis 1986;6:585–593. [DOI] [PubMed] [Google Scholar]
- 2.Shekhonin BV, Domogatsky SP, Muzykantov VR, et al. distribution of type I, III, IV and V collagen in normal and atherosclerotic human arterial wall: immunomorphological characteristics. Coll Relat Res 1985;5:355–368. [DOI] [PubMed] [Google Scholar]
- 3.Urabe N, Naito I, Saito K, et al. Basement membrane type IV collagen molecules in the choroids plexus, pia mater and capillaries in the mouse brain. Arch Histol Cytol 2002;65:133–143. [DOI] [PubMed] [Google Scholar]
- 4.Gould DB, Phalan FC, Breedveld GJ, et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 2005;308:1167–1171. [DOI] [PubMed] [Google Scholar]
- 5.Mancini GMS, De Coo IFM, Lequin MH, Arts WF. Hereditary porencephaly: clinical and MRI findings in two Dutch families. Eur J Paediatr Neurol 2004;8:45–54. [DOI] [PubMed] [Google Scholar]
- 6.Aguglia U, Gambardella A, Breedveld GJ, et al. Suggestive evidence for linkage to chromosome 13qter for autosomal dominant type 1 porencephaly. Neurology 2004;62:1613–1615. [DOI] [PubMed] [Google Scholar]
- 7.Breedveld G, de Coo RF, Lequin MH, et al. Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. J Med Genet 2006;43:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van der Knaap MS, Smit LM, Barkhof F, et al. Neonatal porencephaly and adult stroke related to mutations in collagen IV A1. Ann Neurol 2006;59:504–511. [DOI] [PubMed] [Google Scholar]
- 9.De Vries LS, Koopman C, Groenendaal F, et al. COL4A1 mutation in two preterm siblings with antenatal onset of parenchymal hemorrhage. Ann Neurol 2009;65:12–18. [DOI] [PubMed] [Google Scholar]
- 10.Vahedi K, Massin P, Guichard JP, et al. Hereditary infantile hemiparesis, retinal arteriolar tortuosity, and leukoencephalopathy. Neurology 2003;60:57–63. [DOI] [PubMed] [Google Scholar]
- 11.Gould DB, Phalan FC, van Mil SE, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med 2006;354:1489–1496. [DOI] [PubMed] [Google Scholar]
- 12.Sibon I, Coupry I, Menegon P, et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann Neurol 2007;62:177–184. [DOI] [PubMed] [Google Scholar]
- 13.Shah S, Kumar Y, McLean B, et al. A dominant inherited mutation in collagen IV A1 (COL4A1) causing childhood onset stroke without porencephaly. Eur J Paediatr Neurol Epub 2009 May 28. [DOI] [PubMed]
- 14.Vahedi K, Kubis N, Boukobza M, et al. COL4A1 Mutation in a patient with sporadic, recurrent intracerebral hemorrhage. Stroke 2007;38:1461–1464. [DOI] [PubMed] [Google Scholar]
- 15.Plaisier E, Alamowitch S, Gribouval O, et al. Autosomal-dominant familial hematuria with retinal arteriolar tortuosity and contractures: a novel syndrome. Kidney Int 2005;67:2354–2360. [DOI] [PubMed] [Google Scholar]
- 16.Plaisier E, Gribouval O, Alamowitch S, et al. COL4A1 mutations and hereditary angiopathy with nephropathy, aneurysm and cramps (HANAC) syndrome. N Engl J Med 2007;357:2687–2695. [DOI] [PubMed] [Google Scholar]
- 17.Scheltens P, Barkhof F, Leys D, et al. A semiquantitative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. J Neurol Sci 1993;114:7–12. [DOI] [PubMed] [Google Scholar]
- 18.Van Agtmael T, Schötzer-Schrehardt U, McKie L, et al. Dominant mutations of COL4A1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum Mol Genet 2005;14:3161–3168. [DOI] [PubMed] [Google Scholar]
- 19.Gould DB, Marchant JK, Savinova OV, et al. COL4A1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet 2007;16:798–807. [DOI] [PubMed] [Google Scholar]
- 20.Van Swieten JC, Van den Hout JH, Van Ketel BA, et al. Periventricular lesions in the white matter on magnetic resonance imaging in the elderly. Brain 1991;114:761–774. [DOI] [PubMed] [Google Scholar]
- 21.Chabriat H, Levy C, Taillia H, et al. Patterns of MRI lesions in CADASIL. Neurology 1998;51:452–457. [DOI] [PubMed] [Google Scholar]
- 22.Auer DP, Pütz B, Gôssl C, et al. Differential lesion patterns in CADASIL and sporadic subcortical arteriosclerotic encephalopathy: MR imaging study with statistical parametric comparison. Radiology 2001;218:443–451. [DOI] [PubMed] [Google Scholar]
- 23.Lammie GA. Hypertensive cerebral small vessel disease and stroke. Brain Pathol 2002;12:358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vahedi K, Boukobza M, Massin P, et al. Clinical and brain MRI follow-up study of a family with COL4A1 mutation. Neurology 2007;69:1564–1568. [DOI] [PubMed] [Google Scholar]
- 25.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000;342:673–680. [DOI] [PubMed] [Google Scholar]
- 26.Rossetti S, Chauveau D, Kubly V, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet 2003;361:2196–2201. [DOI] [PubMed] [Google Scholar]
- 27.Ruigrok YM, Rinkel GJ, Wijmenga C. Genetics of intracranial aneurysms. Lancet Neurol 2005;4:179–189. [DOI] [PubMed] [Google Scholar]
- 28.Ruigrok YM, Rinkel GJ, Van’t Slot R, et al. Evidence in favor of the contribution of genes involved in the maintenance of the extracellular matrix of the artery wall to the development of intracranial aneurysms. Hum Mol Genet 2006;15:3361–3368. [DOI] [PubMed] [Google Scholar]
- 29.Poschl E, Schlotzer-Schrehardt U, Brachvogel B, et al. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004;131:1619–1628. [DOI] [PubMed] [Google Scholar]
- 30.Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 1996;383:707–710. [DOI] [PubMed] [Google Scholar]
- 31.Richards A, Van Den Maagdenberg AM, Jen JC, et al. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophie. Nat Genet 2007;39:1068–1070. [DOI] [PubMed] [Google Scholar]
- 32.Ruchoux MM, Domenga V, Brulin P, et al. Transgenic mice expressing mutant Notch3 develop vascular alter-ations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am J Pathol 2003;162:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jen J, Cohen AH, Yue Q, et al. Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS). Neurology 1997;49:1322–1330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.