

Febrile seizures (FS) affect 3% of children aged 6 months to 6 years and have a strong genetic component with recurrence risk ratios of 3–5 in first-degree relatives.1 In rare families with autosomal dominant FS, mutations in the sodium channel alpha 1 subunit gene, SCN1A, have been identified.2,3 However, FS usually show complex inheritance with a polygenic basis. The search for susceptibility variants has been slow, so the recent report of a common splice site single nucleotide polymorphism (SNP) in SCN1A (IVS5N + 5 G>A, rs3812718) is notable. Two patient groups were studied: the first, adults with focal epilepsy with and without a history of FS (n = 90 and 486 respectively); the second, children with FS (n = 144). Patients homozygous for the A allele were reported to have a genotype relative risk of ∼3.0 for FS.4 The rs3812718 SNP is a plausible candidate for FS as it influences relative expression of neonatal and adult transcripts of SCN1A, which plays a key role in membrane excitability.5
To confirm the association between rs3812718 and FS, independent replication studies are vital.6 We tested the primary hypothesis of the original study, that an AA genotype at this SCN1A polymorphism confers increased risk of FS. For this, we recruited 558 unrelated Australian (predominantly Caucasian) patients from epilepsy clinics at 2 tertiary hospitals in Melbourne. The criteria for a diagnosis of FS were identical to that of the original study. Patients with an afebrile seizure history prior to the onset of FS were excluded. The genotyping methodology has been described previously.7 Approval was obtained from the Human Research Ethics Committees of Austin and Melbourne Health.
The cohort of 558 patients comprised 76 (14%) cases with and 482 (86%) without a history of FS. A total of 382 (68%) patients had idiopathic generalized epilepsies; 137 (25%) had focal epilepsies; 12 (2%) had both idiopathic generalized and focal epilepsies; 17 (3%) had unclassified epilepsy; and 10 (2%) had only ever experienced FS.
To ensure statistical consistency with the original findings,4 we utilized the Armitage trend test. No other SCN1A polymorphisms were examined.
Results.
Our Australian study did not replicate the association reported between the AA genotype of the rs3812718 SCN1A SNP and increased risk of FS despite using identical FS classification, statistical methodology, and a similar sample size4 (table). We tested the primary hypothesis of the original study in a cohort of mixed idiopathic generalized and focal epilepsy patients who had a confirmed history of FS. We then performed a secondary post hoc analysis to more closely replicate the population of the original study. We compared our focal epilepsy and FS group (including n = 10 pure FS cases) with our focal epilepsy control group (table). Finally, we compared the Australian FS group (n = 76) with the European control group (n = 701) as described in the original article.4 On all occasions the results were negative; however, we did not have the sample size required to test the hypothesis in a pure FS group.
Table IVS5N + 5 G>A (rs3812718) genotypic frequencies in an Australian population
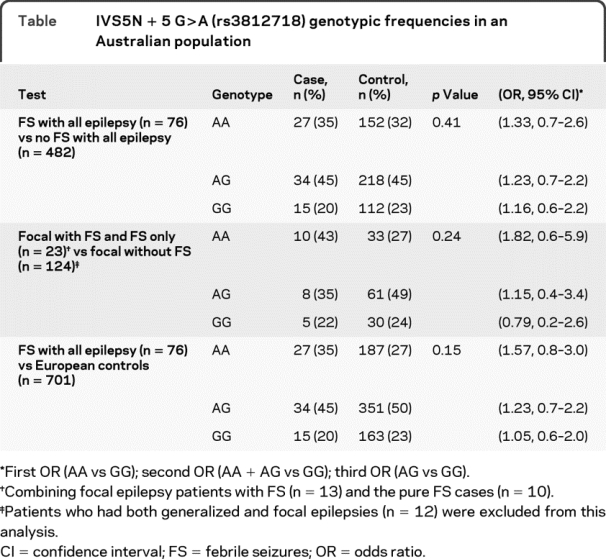
Because our Australian cohort consisted of predominantly patients with generalized epilepsy, we also evaluated a pure focal epilepsy cohort from the EPIGEN consortium.7 SNP SCN1A rs922224, which is in complete linkage disequilibrium with rs3812718 (r2 = 1) in Western European populations, was studied. Focal epilepsy patients (n = 1,589) from the United Kingdom, Ireland, Belgium, and the United States showed no difference between those with (n = 232) and those without a history of FS (n = 1,357) (p = 0.9, odds ratio AA vs GG homozygotes = 1.0, 95% confidence interval 0.7–1.5) or focal epilepsy with FS vs population controls (n = 1,806) (p = 0.3, odds ratio AA vs GG = 1.2, 95% confidence interval 0.8–1.8).
Our failure to replicate the original findings raises the possibility that the original observation was a false positive.
ACKNOWLEDGMENT
The authors thank the patients for their participation and the research assistants at the Epilepsy Research Centre, Department of Medicine (Austin Health), and the Epilepsy Program of The Royal Melbourne Hospital for their assistance. They also thank Leslie Sheffield (Department of Paediatrics, Murdoch Childrens Research Institute, the University of Melbourne, Australia) and Cassandra Szoeke (Department of Neurology, the Royal Melbourne Hospital, the University of Melbourne, Australia) for their help in establishing the cohort at Royal Melbourne Hospital.
APPENDIX
The EPIGEN Consortium contribution to this work was coordinated by Dr. S.M. Sisodiya. The Consortium additionally comprises the following: C. Depondt, M. Pandolfo (Department of Neurology, Hôpital Erasme, Université Libre de Bruxelles, Brussels, Belgium); G. Cavalleri, N. Delanty, S. Alhusaini (The Department of Clinical Neurological Sciences and Molecular and Cellular Therapeutics, RCSI Research Institute, Royal College of Surgeons in Ireland); C. Doherty (Division of Neurology, Beaumont Hospital, Dublin, Ireland); E.L. Heinzen, D.B. Goldstein, R. Radtke, T.J. Urban (Institute for Genome Sciences and Policy, Center for Human Genome Variation, Duke University, Durham, NC); C. Catarino, D. Kasperaviciute, S.K. Tate (Department of Clinical and Experimental Epilepsy, UCL Institute of Neurology, London, UK). C.D., M.P., N.D., S.A., C.D., R.R., C.C., S.M.S., and S.K.T. participated in the collection of the EPIGEN data. G.L.C., E.L.H., D.B.G., T.J.U., and D.K. analyzed the EPIGEN data. The Australian authors (S.P., I.E.S., T.J.O., and S.F.B.) were not part of the EPIGEN consortium.
*Members of the EPIGEN Consortium are listed in the appendix.
Disclosure: The EPIGEN Consortium was supported by grants from the UK Medical Research Council (G0400126), The Wellcome Trust (084730), UCLH CDRC grant (F136), and the National Society for Epilepsy, UK. The work was partly undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. The collection of the Belgian patients was supported by the Fonds National de la Recherche Scientifique and the Fondation Erasme, Université Libre de Bruxelles. The collection of the Irish patient cohort was supported as part of the Programme for Human Genomics and the Programme for Research in Third Level Institutions (PRTLI3) funded by the Irish Higher Education Authority. The authors report funding support of NHMRC, and the Faculty of Medicine, of the University of Melbourne. S. Petrovski reports no disclosures. Dr. Scheffer has served on scientific advisory boards for and received funding for travel from UCB and Janssen-Cilag EMEA; serves on the editorial boards of the Annals of Neurology and Epilepsia; may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic compound; has received speaker honoraria from UCB, Janssen-Cilag EMEA, and Eli Lilly and Company; and receives/has received research support from the National Health and Medical Research Council of Australia, Health Research Council of New Zealand, The University of Melbourne, the Jack Brockhoff Foundation, and the Perpetual Charitable Trustees. Dr. Sisodiya has served on a scientific advisory board and a speakers’ bureau for UCB; holds patent WO/2007/101991 (issued: 2007): Therapeutic target; has received speaker honoraria from Eisai Inc.; and has received research support from UCB and the Tuberous Sclerosis Association. Dr. O’Brien has chaired a scientific advisory board for Janssen-Cilag EMEA; serves on the editorial boards of Epilepsia, the Journal of Clinical Neuroscience, and Epilepsy and Behavior; has received speaker honoraria from Janssen-Cilag EMEA and Sanofi-Aventis; and has received research support from UCB, Abbott, National Health and Medical Research Council of Australia, and the Royal Melbourne Hospital Neuroscience Foundation. Dr. Berkovic has served on scientific advisory boards for UCB and Janssen-Cilag EMEA; has received funding for travel and honoraria from UCB; serves/has served on the editorial boards of Brain and Epileptic Disorders; and has received research support from UCB, the National Health and Medical Research Council of Australia, and the American Epilepsy Society.
Received June 23, 2009. Accepted in final form September 17, 2009.
Address correspondence and reprint requests to Professor Samuel F. Berkovic, Department of Medicine, University of Melbourne, Level 1, Neurosciences Building, Heidelberg Repatriation Hospital, Austin Health, 300 Waterdale Road, West Heidelberg. Vic. 3081, Australia; s.berkovic@unimelb.edu.au
&NA;
- 1.Helbig I, Scheffer IE, Mulley JC, Berkovic SF. Navigating the channels and beyond: unravelling the genetics of the epilepsies. Lancet Neurol 2008;7:231–245. [DOI] [PubMed] [Google Scholar]
- 2.Mantegazza M, Gambardella A, Rusconi R, et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc Natl Acad Sci USA 2005;102:18177–18182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheffer IE, Zhang Y, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain Dev 2009;31:394–400. [DOI] [PubMed] [Google Scholar]
- 4.Schlachter K, Gruber-Sedlymar U, Stogmann E, et al. A splice site variant in the sodium channel gene SCN1A confers risk of febrile seizures. Neurology 2009;72:974–978. [DOI] [PubMed] [Google Scholar]
- 5.Heinzen EL, Yoon W, Tate SK, et al. Nova2 Interacts with a Cis-Acting Polymorphism to Influence the Proportions of Drug-Responsive Splice Variants of SCN1A. Am J Hum Genet 2007;80:876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ott J. Association of genetic loci: replication or not, that is the question. Neurology 2004;63:955–958. [DOI] [PubMed] [Google Scholar]
- 7.Cavalleri GL, Weale ME, Shianna KV, et al. Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: a case-control study. Lancet Neurol 2007;6:970–980. [DOI] [PubMed] [Google Scholar]