

Abstract
Mesenchymal stem cell (MSC) therapy is poised to establish a new clinical paradigm; however, recent trials have produced mixed results. Although MSC were originally considered to treat connective tissue defects, preclinical studies revealed potent immunomodulatory properties that prompted the use of MSC to treat numerous inflammatory conditions. Unfortunately, although clinical trials have met safety endpoints, efficacy has not been demonstrated. We believe the challenge to demonstrate efficacy can be attributed in part to an incomplete understanding of the fate of MSC following infusion. Here, we highlight the clinical status of MSC therapy and discuss the importance of cell-tracking techniques, which have advanced our understanding of the fate and function of systemically infused MSC and might improve clinical application.
Introduction to mesenchymal stem cell (MSC) therapy
Imagine a simple intravenous cell therapy that can restore function to damaged or diseased tissue, avoid host rejection and reduce inflammation throughout the body without the use of immunosuppressive drugs. Such a breakthrough would revolutionize medicine. Fortunately, pending regulatory approval, this approach might not be far off. Specifically, cell therapy utilizing adult mesenchymal stem cells (MSC, Box 1), multipotent cells with the capacity to promote angiogenesis, differentiate to produce multiple types of connective tissue and downregulate an inflammatory response, are the focus of a multitude of clinical studies currently underway. MSC are being explored to regenerate damaged tissue and treat inflammation, resulting from cardiovascular disease and myocardial infarction (MI), brain and spinal cord injury, stroke, diabetes, cartilage and bone injury, Crohn’s disease and graft versus host disease (GvHD) [1]. The problems, however, are that some recent late stage clinical trials have failed to meet primary endpoints, and the fate of MSC following systemic infusion as well as the mechanisms through which they impact host biology are largely unknown [2].
Box 1. MSC phenotype.
Although they have donned many names, i.e. mesenchymal stem cells, mesenchymal stromal cells, multipotent stromal cells, marrow stromal cells and colony-forming unit-fibroblastic, MSCs were originally described as adherent cells from bone marrow that form colonies [42]. Later these cells were found to have multilineage differentiation potential because they could form connective tissue cell types capable of producing bone, adipose and cartilage [43].The International Society for Cellular Therapy (ISCT) defines human MSCs as tissue- culture plastic adherent cells capable of osteogenesis, adipogenesis and chondrogenesis that are positive for CD73, CD90 and CD105 but negative for CD11b, CD14, CD34, CD45, CD79a and HLA-DR surface markers [44]. Despite these guidelines, characterizing and defining the MSC phenotype represents an ongoing challenge [2,45,46]. Bone marrow-derived MSCs are a heterogeneous population of cells and MSC characteristics such as surface marker expression, proliferation rate and differentiation potential are dependent on passage, cell density and the cell culture media [46]. The discovery of MSCs in fat and virtually all other mature tissues [47] has introduced additional nuances in that MSC properties seem to depend on the tissue from which they are isolated [46]. Although MSCs were initially considered for therapy based on their multilineage differentiation capacity, their ability to secrete cytokines and growth factors that are antiapoptotic, proangiogenic and have the potential to reduce scarring and inflammation have positioned MSCs for a broader spectrum of clinical applications [48]. In particular, the use of MSCs to downregulate inflammation offers significant therapeutic potential for treating inflammatory diseases. Specifically, MSCs possess the ability to reduce B-cell proliferation, monocyte maturation and secretion of interferon-γ and TNF-α while promoting T-regulatory cell induction and secretion of IL-10 [39,40]. Table I presents a summary of MSC traits and properties.
Table 1.
Reported MSC characteristics
| Surface markers |
Differentiation potential |
Secreted factors |
|---|---|---|
| CD44+ | Osteogenic | VEGF, Ang-1, SDF-1, |
| CD73+ | Adipogenic | PDGF, TSG-6, bFGF, |
| CD90+ | Chondrogenic | FGF-7, IL-1, IL-6, IL-10, |
| CD105+ | Myogenic | PIGF, MCP-1, TGFβ, |
| CD11b− | Endothelial | PGE-2, IDO, M-CSF, HGF, |
| CD14− | Epithelial | MMP-9, Sfrp, Thymosin β4, |
| CD34− | Neuronal | Plasminogen, Tenacin C, |
| CD45− | [1,43,46] | Thrombospondin 1 [4,39,40,48] |
| CD79a− | ||
| HLA-DR− | ||
| [21,44] |
In this article, we highlight the recent paradigm shift that has occurred in therapeutic use of MSC based on their immunomodulatory properties as opposed to their multilineage differentiation capacity. We discuss the clinical state of MSC therapy in addition to cell-tracking techniques that have been developed with in vivo models to elucidate the mechanisms through which MSC provide a therapeutic effect.
Paradigm shift in the use of MSC for therapy
Although the initial applications conceived for MSC therapy focused on their multilineage differentiation capacity, and more specifically on the potential of MSC to differentiate into osteogenic cells that produce bone tissue as a treatment for fractures, osteogenesis imperfecta or spinal fusion, recent clinical trials have focused almost entirely on the ability of MSC to exert their biological function through trophic mechanisms, including the secretion of cytokines that might serve both paracrine and endocrine functions [3–6]. This shift stemmed from observations that MSC therapy resulted in reduction of inflammation, apoptosis and fibrosis in numerous disease models despite a lack of MSC differentiation and engraftment in the injured tissue. Thus, it was hypothesized that regeneration must be due to trophic factors rather than differentiation (reviewed in Ref. [7]). This paradigm shift towards utilizing trophic properties of MSC for therapy also included a shift from local delivery of MSC to systemic administration, which is less invasive and more convenient, particularly for multiple dosing regimens. However, similar to bone marrow transplantation, where a small percentage of the total hematopoietic stem cells that are infused reach the bone marrow [8,9], only a small percentage of the infused MSC (often <1%) reach the target tissue with cell entrapment commonly observed in capillaries within the liver, spleen and lung [1].
Clinical state of MSC therapy
Mixed results from recent clinical trials have evoked promise and discouragement from both the scientific and clinical communities. Early studies demonstrating that MSC modulate immune function in human [10] and mouse [11] in vitro cultures and within rodent models generated optimism for the prospect of treating some of the most chronic and elusive inflammatory conditions in the developed world. For example, numerous groups have shown reduced scarring and increased cardiac output following MSC therapy in animal models of MI [12–14]. A recently completed phase I trial, using a single infusion of allogeneic MSC (see Glossary; Osiris Therapeutics, Inc., Columbia, MD, Prochymal™ product) in patients within 10 days of acute MI corroborates these findings [15]. In the randomized placebo-controlled dose-escalating trial, patients receiving MSC experienced a 4-fold decrease in arrhythmias and premature ventricular contractions (PVCs), and showed improved overall health compared to patients receiving placebo. Magnetic resonance imaging (MRI) of a subset of patients 1-year post-treatment revealed a significant increase in left ventricular ejection fraction (LVEF). Interestingly, an increase in the dose of MSC reduced the rate of PVCs but not any of the other metrics. Importantly, there were no significant adverse events, and thus this trial validated the safety of allogeneic MSC; however, the viability of MSC post-treatment and the role of MSC in the recovery of cardiac function remain to be elucidated. These results should be considered with cautious optimism; the BOOST trial, which assessed intra-coronary delivery of MSC, initially showed significant improvement in LVEF over control, but this difference was not significant after 18 months [16], thus long-term followup of intravenous MSC therapy is needed. A phase II trial using MSC to treat GvHD reported a reduced 2-year mortality rate [17]. These promising results provided significant motivation for large-scale, placebo-controlled clinical trials. Although phase I and phase II safety trials progressed without severe adverse events, the phase III randomized, placebo-controlled trials failed to reach their primary endpoints. These trials utilized MSC as a firstand second-line therapy to treat GvHD and steroid–refractory GvHD, respectively [18]. Interestingly, these trials illuminated the significant placebo effect that is common with stem cell-based therapies. It is important to consider that the placebo effect has the potential to mask modest therapeutic efficacy. Treatment resulted in a statistically significant improvement over those receiving placebo in patients with steroid–refractory liver or gastrointestinal GvHD and a clinically significant improvement over controls among pediatric patients [18]. Further analysis of the data is ongoing. A trial targeting chronic obstructive pulmonary disease (COPD) with Prochymal™ is underway and preliminary data (gathered 6 months after treatment) showed reduced systemic inflammation compared to controls as measured by C-reactive protein, but there was no significant improvement in pulmonary function [19]. Although the mixed clinical data could be considered a major setback to the entire MSC field, these trials extended initial phase I safety data to thousands of patients, and we believe this should be considered a critical milestone, particularly given that typical doses include hundreds of millions of allogenic MSC. It is also important to consider that it took several decades to optimize bone marrow transplantation before it became a standard of care. Thus, we need to focus on reaching the challenges that were highlighted by these clinical trials, which probably stem from our lack of understanding of the fate of MSC following systemic infusion. Enhanced understanding of fundamental MSC biology should allow more systematic engineering approaches to reduce variability and achieve higher efficacy.
It is possible that the inability to meet primary clinical endpoints in phase III trials resulted from a low efficiency of engrafted cells, which is often described in animal models [2], that reduces the potential for long-term availability of immunomodulatory cytokines. Intriguingly, positive data have emerged from clinical trials despite the lack of data supporting long-term survival and engraftment of systemically delivered MSC. This could result from the dominant use of allogenic MSC in animal studies and human trials (see Table 1 for the source of MSC used in clinical trials), which can be recognized and quickly disposed of by the host immune system. Hare and colleagues (University of Miami, Miami, FL, USA) are currently recruiting patients to determine if autologous MSC (see Glossary) exhibit enhanced therapeutic efficacy compared to allogeneic MSC in a National Institutes of Health-funded study for heart failure. In addition to these considerations, it is also possible that once introduced into the body, MSC do not secrete the same repertoire or concentration of cytokines that have been observed in vitro. The lack of data supporting long-term engraftment and the limited knowledge of cell fate for systemically administered MSC could be due to a lack of sufficient technologies to monitor MSC fate in vivo, an area we believe deserves attention.
Table 1.
State of clinical trials using exogenous MSCs.
| Condition by Organ System | Trialsa(Patients) | Allogeneic | Autogeneic | Trophicb | Differentiateb | IVc | Localc | IAc | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Multiple Systems | 18 (1067) | ||||||||||
| GvHD | 16 (1027) | 15 | 1 | 16 | 16 | ||||||
| Sjgren’s Syndrome | 1 (20) | 1 | 1 | 1 | |||||||
| SLE(Lupus) | 1 (20) | 1 | 1 | 1 | |||||||
| Bone/Cartilage | 26 (1487) | ||||||||||
| Arthritis-Foot Fusion | 1 (100) | 1 | 1 | 1 | |||||||
| Bone Fracture | 2 (210) | 2 | 2 | 2 | |||||||
| Bone Neoplasms | 1 (50) | 1 | 1 | 1 | |||||||
| Cartilage Defects | 4 (185) | 1 | 3 | 4 | 4 | ||||||
| Meniscectomy | 2 (110) | 2 | 2 | ||||||||
| Osteodysplasia | 2 (58) | 1 | 1 | 2 | 1 | 1 | |||||
| Osteogenesis Imperfecta | 3 (35) | 3 | 3 | ||||||||
| Osteonecrosis | 2 (51) | 1 | 1 | 2 | 1 | 1 | |||||
| Periodontitis | 1 (10) | 1 | 1 | 1 | |||||||
| Spinal Fusion | 8 (678) | 8 | 8 | 8 | |||||||
| Cardiovascular | 19 (951) | ||||||||||
| Dilated Cardiomyopathy | 2 (80) | 2 | 2 | 2 | |||||||
| Heart Failure | 3 (200) | 2 | 1 | 2 | 1 | 3 | |||||
| Ischemic Heart Disease | 3 (160) | 3 | 2 | 1 | 3 | ||||||
| Myocardial Infarction | 7 (428) | 4 | 3 | 6 | 1 | 3 | 4 | ||||
| Limb Ischemia | 4 (83) | 3 | 1 | 4 | 3 | 1 | |||||
| Gastrointestinal | 3 (480) | ||||||||||
| Crohn’s | 3 (480) | 3 | 3 | 3 | |||||||
| Kidney | 6 (136) | ||||||||||
| Acute Kidney Injury | 1 (15) | 1 | 1 | 1 | |||||||
| Kidney Transplant | 4 (101) | 2 | 2 | 4 | 4 | ||||||
| Lupus Nephritis | 1 (20) | 1 | 1 | 1 | |||||||
| Liver | 7 (204) | ||||||||||
| Cirrhosis | 6 (203) | 6 | 3 | 3 | 2 | 4 | |||||
| Fam. Hypercholesterolemia | 1 (1) | 1 | 1 | 1 | |||||||
| Lung | 1 (60) | ||||||||||
| COPD | 1 (60) | 1 | 1 | 1 | |||||||
| Nervous | 12 (294) | ||||||||||
| Multiple System Atrophy | 1 (NA) | 1 | 1 | 1 | |||||||
| Neuroblastoma | 1 (15) | 1 | 1 | 1 | |||||||
| Spinal Cord Injury | 2 (103) | 2 | 2 | 2 | |||||||
| Multiple Sclerosis | 4 (84) | 1 | 3 | 4 | 3 | 1 | |||||
| Parkinson’s Disease | 1 (5) | 1 | 1 | 1 | |||||||
| ALS | 1 (24) | 1 | 1 | 1 | |||||||
| Stroke | 2 (63) | 2 | 1 | 1 | 2 | ||||||
| Pancreas | 4 (210) | ||||||||||
| Type 1 Diabetes | 3 (110) | 2 | 1 | 3 | 3 | ||||||
| Type 2 Diabetes | 1 (100) | 1 | 1 | 1 | |||||||
| Skin | 5 (455) | ||||||||||
| Diabetic Wounds | 3 (360) | 3 | 2 | 1 | 3 | 1 | |||||
| Systemic Sclerosis | 1 (20) | 1 | 1 | 1 | |||||||
| Epidermolysis Bullosa | 1 (75) | 1 | 1 | 1 | |||||||
| Total | 101 (5,344) | 59 (3,385) | 42 (1,959) | 65 (3,588) | 36 (1,756) | 48 (2,495) | 49 (2,683) | 5 (166) | |||
| Completed trials (n=21) | Scheduled for completion (n = 63) | ||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | Not specified/other | |
| 1 | 1 | 2 | 8 | 8 | 21 | 20 | 13 | 5 | 1 | 17/4 | |
Data collected from ClinicalTrials.gov registry on 13 March, 2010. Searches for ‘Mesenchymal Stem Cells’, ‘Mesenchymal Stromal Cells’, ‘Multipotent stromal cells’, ‘bone marrow stromal cells’, ‘Stem cells for Spinal Fusion’, ‘Prochymal’, and ‘connective tissue progenitor’ returned 142 unique results, and of those the 101 reported here used exogenous delivery of MSCs. Based on information provided in the trial summary, it is estimated that approximately 85% of trials utilize culture expanded cells. Excluded trials involved expanded access to existing trials, recruitment of endogenous MSCs to sites of injury, and others that did not pertain to MSC therapy.
Trials were categorized as Trophic, if the rationale for the study was dependent on MSC’s pro-angiogenic, anti-apoptotic, or immune modulating properties. Trials were categorized as Differentiate if the rationale depended on the differentiation of delivered MSCs.
IV, intravenous; IA, intra-arterial; Local, delivered in scaffold or injected directly into target tissue.
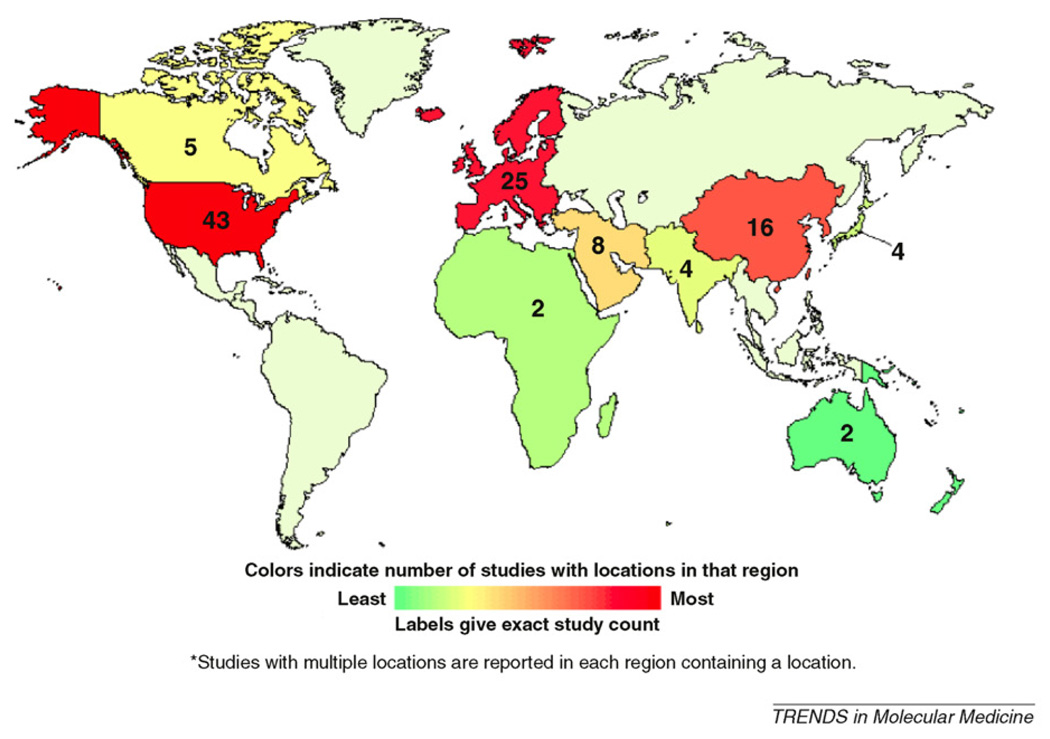
Supplemental figure displays trial distribution on a global map.
Monitoring MSC fate in vivo
A large fraction of systemically infused MSC typically become trapped within the lungs as emboli owing to their large size and their repertoire of cell-surface adhesion receptors [20–23]. Alternatively, they arrest and interrupt blood flow during the first pass through the precapillary level [24]. Such passive arrest prevents the majority of infused MSC from homing to damaged or diseased tissues. Despite these complications, numerous animal studies and some clinical trials have reported favorable outcomes following systemic infusion of MSC [12,17,25–27].The lack of specific homing is perhaps why high dosing is used in clinical trials; 150–300 million MSC are typically administered with each infusion [28]. This prompts the questions: Can entrapped MSC transmigrate through the endothelium?; How long do the entrapped MSC survive?; and Can they provide benefit to distant organs? Several recent publications have attempted to address these questions.
Lee et al. used a cross-species experimental design and real-time PCR (rtPCR) to track the fate of systemically administered human MSC in a mouse model [4]. rtPCR analysis for human-specific Alu sequences (see Glossary)in blood samples showed that within 5 min of MSC infusion through the tail vein, 99% of MSC were cleared from the circulation. Within 10–30 min, a resurgence of ~2–3% of the infused MSC was observed within the blood stream. Tissue samples from various organs revealed that the majority of cells were initially found in the lung, which is consistent with previous studies [20,22]. Then, 15 min after infusion, 83% of the human DNA was detected in the lung, whereas only trace amounts were detected in other tissues. The authors attempted to reduce lung entrapment by decreasing the number of infused cells, blocking key adhesion integrins and pretreating the MSC with rat white-blood cells (to sensitize them to Stromal Cell-Derived Factor-1); however, the fraction of trapped MSC remained unchanged. Histological analysis revealed that the MSC formed emboli in the afferent blood vessels of the lung, a common finding for systemic infusion of other cell types including hematopoietic stem cells and endothelial progenitor cells [9,29]. No MSC were found in the bone marrow, which contradicted other studies [21,30] and highlighted a potential shortcoming of PCR-based techniques, which could be approximately 10-fold less sensitive than radiolabeling techniques [31,32].
In addition to PCR-based techniques for tracking the fate of systemically administered MSC, alternative approaches leverage the advantages of light and fluorescent microscopy that are well suited for small animal models. The Lin group has characterized tumor–cell, hematopoietic stem cell and MSC trafficking in the skull of living mice using in vivo confocal and two-photon microscopy, which provides highresolution spatial delineation of the location of a cell [21,33,34]. Similarly, Toma et al. utilized intravital microscopy, which permits detailed real-time and serial imaging of in vivo phenomenon, to examine the entrapment of MSC within a microvascular bed [24]. In this model, the cremaster muscle of the rats was exposed and fluorescently labeled MSC were injected into the iliac artery. The density of MSC in varying depths of the vasculature was monitored over time using differential interference contrast and fluorescence imaging. All MSC arrested within 5 min of injection with 92% of the injected MSC entrapped during the first pass within the cremaster muscle. However, MSC were only trapped at the precapillary level, resulting in blockage of blood flow to the capillary bed. The number of viable MSC in the cremaster muscle decreased drastically over the next 72 h; only 14% of those originally entrapped survived, as determined by preserved nuclear morphology. As intravital microcopy is best suited for monitoring cells within a preselected location, redistribution of the MSC to other tissues is challenging to evaluate.
One method that can address this is bioluminescence imaging, which lacks single-cell resolution but enables whole-organism tracking of cell distribution. For example, Wang et al. used MSC expressing a firefly–luciferase reporter gene in combination with bioluminescence imaging in a metastatic breast cancer model [35]. This allowed noninvasive whole-animal tracking of intravenously injected MSC and their progeny over the course of several days. In healthy controls, MSC were initially found in the pulmonary capillaries but quickly dispersed after 1 day. The reduction of signal in the lungs can be attributed both to redistribution of MSC to other tissues as well as to cell death. Bioluminescence can be extremely valuable in characterizing MSC affinity and tropism for inflammatory and tumor microenvironments as has been reviewed by Spaeth et al. [36].
Recent cell tracking studies have provided valuable insight into the distribution of MSC following systemic infusion and have begun to help elucidate the process of cell localization within specific tissues. However, it is critical to note that whole-animal imaging techniques such as bioluminescence lack the resolution to determine if cells remain in the vasculature or have undergone transendothelial migration. Aside from passive cell entrapment, which appears to be a dominant mechanism through which infused MSC reach their final destination, characterization of the host vasculature is required to better understand active homing mechanisms. The vascular expression of adhesion molecules and endothelial presentation of cytokines can vary substantially within a vascular bed [34]. Thus, future studies should employ multiple methods, summarized in Table 2, to assess the final destination of the infused cells through both macroscopic distribution and microscopic spatial localization analysis.
Table 2.
In Vivo Cell monitoring techniques
| Technique | Detected agent | Detection limit | Temporal resolution | Whole animal |
|---|---|---|---|---|
| PCR [32] | Sequence unique to donor | 50,000 cells | Requires sacrifice | Yes |
| Radiolabeling (SPECT) [32] | Isotope | 5000 cells | 30 s/projection | Yes |
| Intravital microscopy [49,55] | Fluorescent marker, i.e. reporter gene, antibody, quantum dots |
Single cell | <1 s | No |
| In vivo confocal [50] | Fluorescent marker | Single cell | <1 s | No |
| Bioluminescence microscopy [51] | Luciferase gene+substrate | 10 cells | <1 min | Yes |
| MRI [52] | Magnetic nanoparticles | 10–20 cells/voxel | >10 min depending on size of region |
Yes |
Therapeutic implications and concluding remarks
The results from multiple clinical trials using systemically administered MSC illuminate critical challenges that must be addressed, yet provide the young field of MSC therapy with rationale for additional ‘steps’ forward. Importantly, research has already begun to identify the fate and function of MSC following systemic infusion. With evidence for massive cell entrapment in the lungs and in capillary beds of other tissues, approaches are being developed to enhance cell homing to target tissues through genetic and chemical engineering approaches [2,37]. It is possible that targeted delivery of cells is unnecessary for certain applications, as the therapeutic effects of MSC are systemic; however, enhanced delivery to specific tissues could increase the efficiency of cell therapy and reduce the number of infused cells, potentially reducing the cost of developing a therapeutic product. Conventional wisdom suggests that promoting transmigration and longevity of MSC, perhaps even non-specifically, could increase therapeutically relevant systemic effects (i.e. where engrafted cells continue to secrete cytokines that are released into the circulation). More research is needed to determine if the few MSC that engraft in target tissues [38] mediate regeneration through differentiation into more mature connective tissue cell types and whether or not the engrafted cells integrate and coordinate with the native tissue to restore function. With the discovery of secreted TSG-6 by MSC entrapped within the lungs (Box 2) and knowledge of several other MSC-secreted immunomodulatory factors (Box 1), there is now evidence that the therapeutic effects could in part result from systemic (endocrine) effects in addition to previously described (local) paracrine signaling and direct cell–cell interactions. For example, Nemeth et al. demonstrated that MSC in direct contact with macrophages secrete prostaglandin E2, which reprograms macrophages to increase production of the potent anti-inflammatory cytokine interleukin-10 (IL-10) [39,40].
Box 2. MSC fate sheds light on function.
Despite mass entrapment of systemically administered MSCs within the lung and other tissues, tail vein injection in rodent models of MI still provides a functional improvement that is typically evidenced by decreased scar size and increased cardiac output. In the seminal paper by Lee et al., a paracrine factor that is released by embolized MSCs was identified; this factor promotes tissue regeneration through a systemic effect, similar to the action of a conventionally administered drug [4].
A transcriptome analysis of embolized MSCs from the lungs generated a list of 451 upregulated transcripts with rtPCR analysis showing that TSG-6, a known anti-inflammatory protein, had the largest increase in mRNA levels [53]. TSG-6, which was originally discovered by secretome analysis of skin fibroblasts following incubation with tumor necrosis factor (TNF)-α [54], is a 30- kDa protein that inhibits neutrophil migration and the production and activity of both plasmin and matrix metalloproteinases (MMPs) [53]. Interestingly, MSC secretion of TSG-6 was 120-fold greater than that of fibroblasts obtained from the same human donor [4]. Two infusions of recombinant TSG-6 following MI (without administration of MSCs) decreased infarct size, reduced scaring and improved cardiac function, yet not to the same extent as MSCs. MSCs with TSG-6 knock down by RNA interference did not impact infarct size. The authors hypothesized that the embolism of the MSCs in the lung creates a local injury that activates the MSCs to secrete TSG-6, which enters circulation and downregulates the inflammatory process at the site of MI.
MI is characterized by invasion of neutrophils, monocytes and macrophages that secrete MMPs, breaking down the dead myocardium to replace it with a fibrous scar [12]. MSC secretion of TSG-6 and infusion of recombinant TSG-6 interrupted this process during the initial 48 h of wound healing, resulting in a reduced inflammatory process and improved regeneration of the infarcted tissue. This study utilized xenografts (see Glossary); human MSCs were injected into a murine model. Xenografts have different distribution kinetics than allogeneic MSCs in murine models [51] (allogenic MSCs are the standard for human clinical trials). Because the Lee et al. proposed mechanism for enhanced therapeutic efficacy depends on entrapment and activation of xenogenic MSCs in the lungs, allogeneic MSCs, which have been shown in mouse models to disperse from the lungs within hours of infusion, might produce substantially different results.
The heterogeneity of the MSC population presents a challenge for generalizing findings from different groups, as it is known that differences in culture conditions, source, passage and cell density all impact MSC phenotype [41]. Moving forward, it is important to characterize the conditions needed to develop therapeutically relevant cells; and in tandem, cell-tracking techniques that can be performed in large animal models and in humans, which would enhance understanding of MSC engraftment, allow long-term assessment of cell phenotype and ultimately increase therapeutic potential (Box 3). Furthermore, development of such tracking technologies for animal models could make it possible to monitor cells following systemic infusion into patients. Unlike conventional drugs, which are designed to act through a known pathway, cell therapies are living therapeutics, which can multiply, senesce, undergo necrosis or apoptosis, or alter their phenotype, and thereby drastically change their therapeutic potential. The ability to track the location of cells and monitor viability and functional characteristics (e.g. differentiation state) could provide feedback for potential clinical interventions and for the development of a consistently efficacious treatment. Despite an incomplete explanation of their role in regeneration, there are multiple clinical trials being performed. As shown in Table 1, the ClinicalTrials.-gov registry currently lists 101 trials that are using exogenous MSC to treat a wide range of damaged, diseased or inflamed tissues. Because only 21 of these trials have been completed, we can anticipate an abundance of new human data in the near future for a wide range of therapeutic applications (21 trials are scheduled to be completed in 2010 and 20 trials in 2011). Through investigation of MSC biology, discovery of their therapeutic mechanisms within animal models and testing their therapeutic potential within human trials, we will hopefully achieve many more steps forward to make MSC therapy a new clinical paradigm.
Box 3. Outstanding questions.
MSC homing
Which adhesion molecules mediate MSC homing?
MSC engraftment
How should MSC engraftment be defined?
Do MSC persist long term and how can this time frame be extended?
Which tissue microenvironments provide favorable sites for MSC engraftment?
MSC monitoring
What are the best approaches to monitor MSC therapy and how might these approaches be connected to clinical interventions to improve the therapeutic outcome?
How should MSC distribution and phenotype be monitored in animals and in humans?
MSC function
What is the kinetics of cytokine secretion and how does this change as MSCs differentiate into more mature progeny?
In addition to TSG-6, which MSC-secreted cytokines have systemic effects?
Therapy optimization
What are the optimal conditions to develop therapeutically relevant cells (with increased homing potential and/or increased cytokine production)?
Can MSCs be replaced by MSC supernatant and how might the supernatant be standardized?
Can MSC therapy be improved by shifting the balance between systemic (endocrine) and local (paracrine or cell–cell contact) activity? How might this change for treatment of different diseases?
How can patients be stratified to select those who would be most responsive?
What is the optimal dosing regimen?
Acknowledgment
This work was supported by National Institute of Health grant DE019191 and by the American Heart Association grant #0970178N to JMK.
Glossary
- Allogeneic
cells originate from a donor of the same species as the recipient.
- Alu sequences
a repetitive sequence of several hundred base pairs that occur frequently in primate genomes.
- Autologous
donor cells originate from the recipient.
- Endocrine signaling
secreted factors exert effects on distant cells.
- Paracrine signaling
secreted factors exert effects on neighboring cells.
- Xenograft
cells originate from a donor of a different species than the recipient.
References
- 1.Phinney DG, Prockop DJ. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair – current views. Stem Cells. 2007;25:2896–2902. doi: 10.1634/stemcells.2007-0637. [DOI] [PubMed] [Google Scholar]
- 2.Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4:206–216. doi: 10.1016/j.stem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Iso Y, et al. Multipotent human stromal cells improve cardiac function after myocardial infarction in mice without long-term engraftment. Biochem. Biophys. Res. Commun. 2007;354:700–706. doi: 10.1016/j.bbrc.2007.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee RH, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ortiz LA, et al. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc. Natl. Acad. Sci. U. S. A. 2007;104:11002–11007. doi: 10.1073/pnas.0704421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tögel F, et al. Vasculotropic, paracrine actions of infused mesenchymal stem cells are important to the recovery from acute kidney injury. Am. J. Physiol. Renal Physiol. 2007;292:F1626–F1635. doi: 10.1152/ajprenal.00339.2006. [DOI] [PubMed] [Google Scholar]
- 7.van Poll D, et al. Mesenchymal stem cell therapy for protection and repair of injured vital organs. Cell. Mol. Bioeng. 2008;1(1):42–50. (doi:10.1007/s12195-008-0001-2) [Google Scholar]
- 8.van Hennik PB, et al. Seeding efficiency of primitive human hematopoietic cells in nonobese diabetic/severe combined immune deficiency mice: implications for stem cell frequency assessment. Blood. 1999;94:3055–3061. [PubMed] [Google Scholar]
- 9.Cui J, et al. Bone marrow cell trafficking following intravenous administration. Br. J. Haematol. 1999;107:895–902. doi: 10.1046/j.1365-2141.1999.01779.x. [DOI] [PubMed] [Google Scholar]
- 10.Di Nicola M, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 11.Krampera M, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–3729. doi: 10.1182/blood-2002-07-2104. [DOI] [PubMed] [Google Scholar]
- 12.Laflamme M, Murry C. Regenerating the heart. Nat. Biotechnol. 2005;23:845–856. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 13.Mangi AA, et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat. Med. 2003;9:1195–1201. doi: 10.1038/nm912. [DOI] [PubMed] [Google Scholar]
- 14.Schuleri K, et al. Autologous mesenchymal stem cells produce reverse remodelling in chronic ischaemic cardiomyopathy. Eur. Heart J. 2009;30:2722–2732. doi: 10.1093/eurheartj/ehp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hare JM, et al. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J. Am. Coll. Cardiol. 2009;54:2277–2286. doi: 10.1016/j.jacc.2009.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer GP, et al. Intracoronary bone marrow cell transfer after myocardial infarction: eighteen months’ follow-up data from the randomized, controlled BOOST (BOne marrOw transfer to enhance ST-elevation infarct regeneration) trial. Circulation. 2006;113:1287–1294. doi: 10.1161/CIRCULATIONAHA.105.575118. [DOI] [PubMed] [Google Scholar]
- 17.Le Blanc K, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 18.Mills CR. Osiris Therapeutics Announces Preliminary Results for Prochymal Phase III GvHD Trials. Press Release; 2009. [cited 1 November, 2009]. available from: http://investor.osiris.com/releasedetail.cfm?ReleaseID=407404. [Google Scholar]
- 19.Mills CR. Osiris Therapeutics Reports Interim Data for COPD Stem Cell Study. Press Release; 2009. [cited 1 November, 2009]. available from: http://investor.osiris.com/releasedetail.cfm?ReleaseID=391580. [Google Scholar]
- 20.Schrepfer S, et al. Stem cell transplantation: the lung barrier. Transplant Proc. 2007;39:573–576. doi: 10.1016/j.transproceed.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 21.Sackstein R, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 22.Gao J, et al. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs (Print) 2001;169:12–20. doi: 10.1159/000047856. [DOI] [PubMed] [Google Scholar]
- 23.Barbash IM, et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation. 2003;108:863–868. doi: 10.1161/01.CIR.0000084828.50310.6A. [DOI] [PubMed] [Google Scholar]
- 24.Toma C, et al. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ. Res. 2009;104:398–402. doi: 10.1161/CIRCRESAHA.108.187724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Blanc K, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 26.Giordano A, et al. From the laboratory bench to the patient’s bedside: an update on clinical trials with mesenchymal stem cells. J. Cell. Physiol. 2007;211:27–35. doi: 10.1002/jcp.20959. [DOI] [PubMed] [Google Scholar]
- 27.Chen SL, et al. Improvement of cardiac function after transplantation of autologous bone marrow mesenchymal stem cells in patients with acute myocardial infarction. Chin. Med. J. 2004;117:1443–1448. [PubMed] [Google Scholar]
- 28.NIH. Evaluation of PROCHYMAL (Adult Human Stem Cells for Treatment-Resistant Moderate-to-Severe Crohn’s Disease. 2008 27 June, 2008 (cited 29 June, 2008); available from: http://clinicaltrials.gov/ct2/show/NCT00482092?term=osiris&rank=3.
- 29.Smits PA, et al. Distribution of circulation-derived endothelial progenitors following systemic delivery. Endothelium. 2007;14:1–5. doi: 10.1080/10623320601177254. [DOI] [PubMed] [Google Scholar]
- 30.Rombouts WJ, Ploemacher RE. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia. 2003;17:160–170. doi: 10.1038/sj.leu.2402763. [DOI] [PubMed] [Google Scholar]
- 31.Kraitchman D. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005;112:1451–1461. doi: 10.1161/CIRCULATIONAHA.105.537480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ. Res. 2004;95:9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- 33.Lo Celso C, et al. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–96. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sipkins DA, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang H, et al. Trafficking mesenchymal stem cell engraftment and differentiation in tumor-bearing mice by bioluminescence imaging. Stem Cells. 2009;27:1548–1558. doi: 10.1002/stem.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spaeth E, et al. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–738. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 37.Wagner J, et al. Optimizing mesenchymal stem cell-based therapeutics. Curr. Opin. Biotechnol. 2009;20(5):531–536. doi: 10.1016/j.copbio.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Quevedo HC, et al. Allogeneic mesenchymal stem cells restore cardiac function in chronic ischemic cardiomyopathy via trilineage differentiating capacity. Proc. Natl Acad. Sci. U. S. A. 2009;106:14022–14027. doi: 10.1073/pnas.0903201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–3506. doi: 10.1182/blood-2007-02-069716. [DOI] [PubMed] [Google Scholar]
- 40.Németh K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee RH, et al. The CD34-like protein PODXL and alpha6-integrin (CD49f) identify early progenitor MSCs with increased clonogenicity and migration to infarcted heart in mice. Blood. 2009;113:816–826. doi: 10.1182/blood-2007-12-128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friedenstein A, et al. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp. Hematol. 1976;4(5):267–274. [PubMed] [Google Scholar]
- 43.Caplan AI. Why are MSCs therapeutic? New data: new insight. J. Pathol. 2009;217:318–324. doi: 10.1002/path.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 45.Bianco P, et al. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–319. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prockop D. Repair of tissues by adult stem/progenitor cells (MSCs): controversies, myths, and changing paradigms. Mol. Ther. 2009;17(6):939–946. doi: 10.1038/mt.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.da Silva Meirelles L, Chagastelles P. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006;119(11):2204–2213. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 48.Burchfield JS, Dimmeler S. Role of paracrine factors in stem and progenitor cell mediated cardiac repair and tissue fibrosis. Fibrogenesis Tissue Repair. 2008;1(1):1–11. doi: 10.1186/1755-1536-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rüster B, et al. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood. 2006;108:3938–3944. doi: 10.1182/blood-2006-05-025098. [DOI] [PubMed] [Google Scholar]
- 50.Runnels JM, et al. Imaging molecular expression on vascular endothelial cells by in vivo immunofluorescence microscopy. Mol. Imaging. 2006;5:31–40. [PMC free article] [PubMed] [Google Scholar]
- 51.Kidd S, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescence imaging. Stem Cells. 2009;27(10):2614–2623. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kraitchman DL, et al. In vivo magnetic resonance imaging of mesenchymal stem cells in myocardial infarction. Circulation. 2003;107:2290–2293. doi: 10.1161/01.CIR.0000070931.62772.4E. [DOI] [PubMed] [Google Scholar]
- 53.Milner CM, et al. TSG-6: a pluripotent inflammatory mediator? Biochem. Soc. Trans. 2006;34:446–450. doi: 10.1042/BST0340446. [DOI] [PubMed] [Google Scholar]
- 54.Lee TH, et al. A novel secretory tumor necrosis factor-inducible protein (TSG-6) is a member of the family of hyaluronate binding proteins, closely related to the adhesion receptor CD44. J. Cell Biol. 1992;116:545–557. doi: 10.1083/jcb.116.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shah, Bhranti S, et al. Labeling of mesenchymal stem cells by bioconjugated quantum dots. Nano Letters. 2007;7(10):3071–3079. doi: 10.1021/nl071547f. [DOI] [PMC free article] [PubMed] [Google Scholar]