

Abstract
Neurons in the central nervous system (CNS) lose their ability to regenerate early in development, but the underlying mechanisms are unknown. By screening genes developmentally regulated in retinal ganglion cells (RGCs), we identified Krüppel-like factor–4 (KLF4) as a transcriptional repressor of axon growth in RGCs and other CNS neurons. RGCs lacking KLF4 showed increased axon growth both in vitro and after optic nerve injury in vivo. Related KLF family members suppressed or enhanced axon growth to differing extents, and several growth-suppressive KLFs were up-regulated postnatally, whereas growth-enhancing KLFs were down-regulated. Thus, coordinated activities of different KLFs regulate the regenerative capacity of CNS neurons.
Adult mammalian central nervous system (CNS) axons are unable to regenerate after injury, but immature CNS neurons regenerate axons robustly (1–3). In addition to the development of an inhibitory CNS environment (4, 5), a developmental loss in neurons’ intrinsic capacity for axon growth is thought to contribute to regeneration failure. For example, after birth, axonal outgrowth from rat retinal ganglion cells (RGCs, a type of CNS neuron) slows substantially (6). Similar developmental declines in axon growth ability have been observed in mammalian tissue explants of brainstem (7), cerebellum (8, 9), entorhinal cortex (10), and retina (2). Various cell-autonomous factors such as cAMP (cyclic adenosine 3′,5′-monophosphate) and CREB (cAMP response element–binding protein) (11, 12), Bcl-2 (B cell lymphoma/leukemia 2) (13, 14), Rho/ROCK (Rho-associated kinase) (15), Cdh1-APC (anaphase-promoting complex) (16, 17), and PTEN (phosphatase and tensin homolog) (18) have been suggested to play roles in this process. However, manipulating these regulators of axon growth, even when simultaneously overcoming environmental inhibition, only partially restores regeneration, suggesting that additional intrinsic axon growth regulators remain to be identified.
To investigate the molecular basis for the developmental loss of axon growth ability in RGCs, we took advantage of the fact that coculture with amacrine cell membranes is sufficient to signal embryonic RGCs to decrease their rapid axon growth (6). Addition of the transcriptional inhibitor actinomycin D blocked this effect of amacrine membranes, and embryonic RGCs retained their capacity for axon growth (Fig. 1A). These data suggest that gene transcription is required for the developmental loss of intrinsic axon growth ability in RGCs.
Fig. 1.
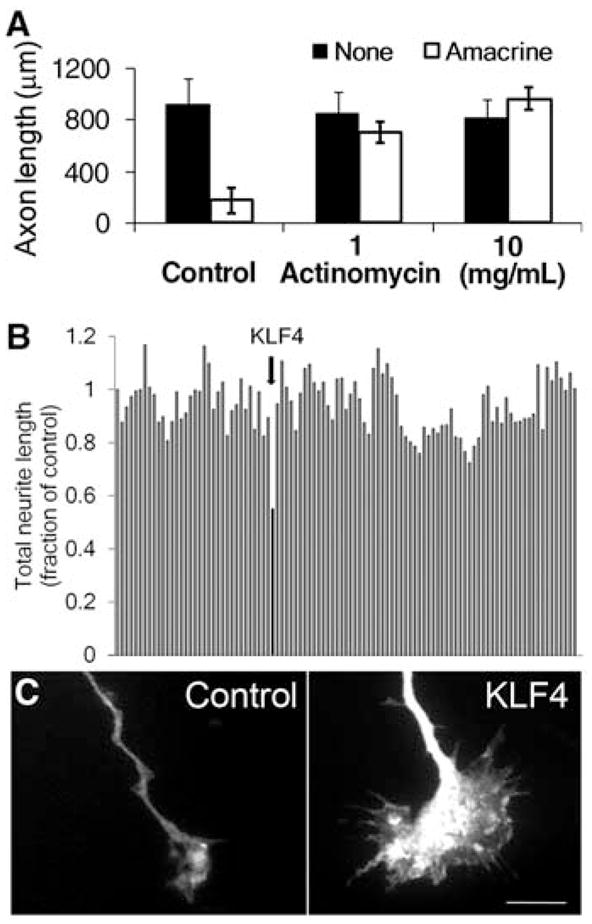
A screen of developmentally regulated genes identifies KLF4 as an inhibitor of neurite growth. (A) Purified embryonic RGCs were cultured in the presence (white bars) or absence (black bars) of amacrine cell membranes for 3 days, and replated away from amacrine cell membranes, after which RGC axon growth was measured. Actinomycin D blocked RGCs’ decrease in axon growth caused by amacrine cell membranes (mean ± SEM). (B and C) E18 hippocampal neurons were cotransfected with 111 candidate genes and enhanced green fluorescent protein (EGFP), cultured for 3 days on laminin, and immunostained for Tau to visualize neurites. (B) Neurite length of cotransfected (EGFP+) neurons. Bars represent average neurite length normalized to EGFP control (far left). KLF4 (arrow) decreased neurite growth by 50%. (C) EGFP+ growth cones of EGFP+/KLF4-transfected neurons (right) were enlarged compared to control-transfected neurons (left). Scale bar, 10 μm.
To identify candidate genes, we profiled gene expression from embryonic day 17 (E17) through postnatal day 21 (P21) RGCs (19), spanning the period when axon growth ability declines in vivo (6, 13). We screened 111 candidates whose expression changed more than threefold by over-expression in embryonic hippocampal neurons, and used automated image acquisition and neurite tracing (KSR instrument, Cellomics) for rapid, unbiased quantification of neurite length (20); the investigator (D.L.M.) was blinded to gene identity until the screen was complete. The zinc-finger transcription factor, Krüppel-like factor–4 (KLF4), was the most effective suppressor of neurite outgrowth, decreasing average length by 50% (Fig. 1B). In a separate, blinded screen examining growth cone morphologies, KLF4 again emerged as the most interesting candidate gene as growth cones in KLF4-overexpressing hippocampal neurons were consistently enlarged (e.g., Fig. 1C).
Although KLF4 regulates cell survival in other systems (21–23), we detected no differences in survival between KLF4- and control-transfected hippocampal neurons (fig. S1A). To determine if the growth-suppressive effect was specific either to axons or dendrites, we manually traced Tau+ and MAP2+ neurites (fig. S1B). Overexpression of KLF4 in embryonic hippocampal neurons significantly decreased the lengths of both axons (Tau+/MAP2−) and dendrites (Tau+/MAP2+) (figs. S1 and S2). We also observed a reduction in branching (fig. S3) and in the percentage of neurons that extended neurites (fig. S1C). Taken together, these findings suggest that KLF4 acts independently of cell survival to suppress axon and dendrite initiation and elongation by hippocampal neurons in vitro.
We next asked whether KLF4 regulates axon growth of RGCs. KLF4 expression increased postnatally, both by microarray analysis (19) (Fig. 2A) and by quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR; Fig. 2B) of acutely purified RGCs. We purified RGCs from E20 rats and transfected them with FLAG-tagged KLF4 (24) or a FLAG-only control. Overexpression of KLF4 in embryonic RGCs reduced the percentage of neurons extending neurites (Fig. 2E), reduced neurite branching (fig. S4), and reduced axon and, less so, dendrite lengths (Fig. 2, C and D). The average axon length of KLF4-transfected RGCs continued to increase over 3 days, but at a slower rate than control-transfected neurons (fig. S5), suggesting that KLF4 overexpression decreases elongation rate. Furthermore, truncated KLF4 that lacked a C-terminal DNA binding domain (fig. S2A) (24) had no effect on axon growth (Fig. 2, C and D). Thus, KLF4 suppresses axon growth in embryonic RGCs, and KLF4’s DNA binding domain is required for its growth-suppressive activity.
Fig. 2.

KLF4 is developmentally regulated in RGCs, and its overexpression decreases axon growth in a zinc-finger–dependent manner. (A and B) KLF4 expression in RGCs increases at birth, as measured in acutely purified rat RGCs by microarray [three probe sets (A); (19)] or in acutely purified mouse RGCs by qRT-PCR [(B); fold change from E18]. Two biological replicates are plotted with their average in (B). (C to E) FLAG-KLF4-WT, FLAG-KLF4-^C lacking the C-terminal zinc finger DNA binding domain, or FLAG or mCHERRY controls were transfected into E20 RGCs. (C) After 2 days, RGCs were immunostained for FLAG or GFP (green, transfected cells), and Tau or MAP2 (red) as marked [nuclear DAPI (4′,6-diamidino-2-phenylindole) is blue]. Scale bar, 50 μm (main panels), 10 μm (inset). (D) Hand-tracing revealed that FLAG-KLF4-WT overexpression decreased axon growth; overexpression of FLAG-KLF4-^C was similar to that of controls (**P < 0.001, *P < 0.02, unpaired t test, post-Bonferroni correction; mean ± SEM). (E) E20 RGCs were transfected with either mCherry-pIRES2-eGFP (control) or KLF4-pIRES2-EGFP and plated for 1, 2 or 3 days in vitro (DIV). At 1 to 3 DIV, more control-transfected RGCs extended at least one neurite >10 μm than KLF4-transfected RGCs (*P < 0.001, paired t test; mean ± SEM).
We next tested whether knocking out KLF4 in developing RGCs enhances axon growth ability. Because KLF4-null mice die perinatally (25), we used a Cre/lox strategy to target KLF4 knockout to RGCs. Floxed-KLF4 mice (26) were crossed to ROSA-EYFP (enhanced yellow fluorescent protein) reporter mice and Thy-1–promoter Cre recombinase mice. About 50% of RGCs purified from Thy-1-cre/ROSA-EYFP mice were EYFP+ (fig. S6). There was no effect of transgenic Cre expression on RGC neurite growth, neurite initiation, or survival in vitro (figs. S7 and S8). To examine axon growth from KLF4-deficient RGCs in vitro, we purified RGCs from P12 Thy1-cre+/−/KLF4fl/fl/ROSA-EYFP+ (“KO”) or Thy1-cre−/ −/KLF4fl/fl/ROSA-EYFP+ (“WT”) littermate mice and cultured them for 3 days (Fig. 3A). No effect of KLF4-KO was seen on survival (fig. S8). P12 KLF4-KO RGCs showed a statistically significant increase in neurite initiation compared to controls (Fig. 3B), mirroring our previous finding that overexpression of KLF4 decreases neurite initiation (Fig. 2E). We also observed a significant increase in neurite lengths in KLF4-KO RGCs (Fig. 3C). These data demonstrate that knocking out KLF4 enhances axon growth ability in P12 RGCs in vitro.
Fig. 3.

KLF4 knockout increases RGC neurite growth in vitro and regeneration of adult RGCs in vivo. (A to C) Purified P12 RGCs were cultured from Thy1-cre−/ −/KLF4fl/fl/Rosa+ (Cre− WT) and Thy1-cre+/−/KLF4fl/fl/Rosa+ (Cre+ KO) mice, and plated for 3 DIV before Tau immunostaining and automated tracing. (A) Immunostaining for Tau (red) demonstrated low levels of growth of Cre− WT RGCs (left) but increased levels of axon growth of Cre+ KLF4-KO RGCs (right; Rosa+ yellow cells, arrowheads). (B) KLF4-KO RGCs have a higher percentage of cells with neurites, compared to controls (N = 3; *P < 0.02, t test; mean ± SEM). (C) When all YFP+ RGCs were measured, KLF4 KO RGCs extended longer neurites than WT RGCs (representative experiment shown; *P < 0.001; mean ± SEM). (D and E) Two weeks after optic nerve crush of Thy1-cre+/KLF4+/+ (WT), Thy1-cre+/KLF4fl/+ (Het), and Thy1-cre+/KLF4fl/fl (KO) mice, regenerating fibers were anterogradely labeled by intravitreal injection of Alexa 594–labeled cholera toxin B. Regenerating fibers were counted at specified distances from the lesion site. (D) More fibers regenerate in KO mice compared to WT or Het (n = 10 WT, 4 Het, and 7 KO mice; P < 0.001 for KO versus WT or Het; no difference between WT and Het by mixed-model analysis of covariance; mean ± SEM). (E) Partial projections of sectioned optic nerve from WT and KO mice show regenerating axons more than 1 mm distal to the lesion site in KO nerve. Scale bar, 200 μm.
We next asked if knocking out KLF4 during development enhances regeneration from adult RGCs in vivo. Thy1-cre+/KLF4fl/fl (KO), Thy1-cre+/KLF4fl/+ (Het), or Thy1-cre+/KLF4+/+ (WT) littermate mice were subjected to optic nerve crush, and after 2 weeks we assessed regeneration of RGC axons in the optic nerve. By adulthood, there were no differences in RGC number between KO, Het, and WT animals (fig. S9A). Compared to controls, however, KLF4 KO mice showed an increased number of regenerating axons at multiple distances from the injury site (Fig. 3, D and E). KLF4 KO did not affect RGC survival after injury (fig. S9B), showing that this increase in regenerating axons was not secondary to an increased RGC number. Thus, knocking out KLF4 expression during development increases the regenerative potential of adult RGCs.
Although knocking out KLF4 enhanced axon growth and regeneration, the size of the effect led us to speculate that other KLF family members might compensate for the loss of KLF4. The KLF family comprises 17 related transcription factors with homologous DNA binding domains and divergent activation and repression domains (27). KLFs often regulate gene expression interactively, with both cooperative and competitive relationships among family members (28–30). Our microarray data suggested that many KLFs are expressed by RGCs (19) and that some are developmentally regulated (fig. S10). We reprofiled the expression of all 17 KLF family members in developing RGCs by RT-PCR and detected transcripts for 15 (Fig. 4E). Furthermore, qRT-PCR revealed that KLF6 and KLF7 transcripts decrease more than 10-fold, whereas KLF9 increases more than 250-fold (Fig. 4, A to C). Thus, expression of multiple KLFs is regulated in developing RGCs.
Fig. 4.

Multiple KLF family members are developmentally regulated in RGCs and differentially affect CNS neurite growth. (A to C) RGCs from multiple ages were purified by immunopanning and analyzed by qRT-PCR. Transcript abundance is normalized to E19. KLF6 (A) and KLF7 (B) decrease more than 5-fold postnatally, whereas KLF9 (C) increases 250-fold. Each marker type is a separate experiment, and the line shown is the average; N = 2 to 3. (D) P4 RGCs were cotransfected with KLFs and EGFP and plated for 2 days on laminin. Bars represent average total neurite length of transfected (EGFP+) neurons [n > 700; *P < 0.05, **P < 0.01; analysis of variance (ANOVA) with post hoc Dunnett’s test; mean ± SEM; pooled data from two replicate experiments]. (E) P5 cortical neurons were cotransfected with individual KLFs and mCherry, plated for 3 days on laminin, and immunostained for β-III tubulin. (Top) KLF family members are grouped according to defined structural domains (27) and clustered by amino acid similarity (Clustal analysis, Vector NTI). (Middle) Bars represent average total neurite length of transfected (mCherry+) neurons and are colored by the presence of known motifs (above). Nine KLFs significantly decreased neurite length, and two increased neurite length (N > 3, n > 100; *P < 0.05, **P < 0.01, ANOVA with post hoc Dunnett’s test; mean ± SEM). (Bottom) Purified RGCs from different ages were analyzed by RT-PCR with KLF-specific primers, ordered according to the overlying bar graph. Transcripts for all KLFs except KLF1 and -17 were detected in developing RGCs. (F) P5 cortical neurons were cotransfected with combinations of KLFs with IRES-mCherry (red) or IRES-EGFP (green) reporters and cultured as above (DNA loading controls, fig. S13). Bars represent average neurite length of dually transfected neurons (mCherry+, EGFP+). Coexpression of KLF4 or -9 blocked the growth-promoting effects of KLF6 or -7 (N = 3, n > 25; *P < 0.05, **P < 0.01, ANOVA with post hoc Dunnett’s test; mean ± SEM).
Do other KLF family members also regulate neurite growth? Other KLFs can affect neurite branching in response to thyroid hormone (KLF9) (31) or neurite outgrowth in zebrafish retinal explants (KLF6 and -7) (32). In RGCs, overexpression of KLF9 significantly decreased growth, similar to KLF4, and KLF6 and -7 increased neurite growth 13 and 23%, respectively (Fig. 4D). We comprehensively surveyed all 17 KLF family members’ on neurite growth in cortical neurons in vitro, and found that although no KLFs affected cell survival (fig. S11), eight KLFs, including KLF4 and -9, suppressed neurite growth, and KLF6 and -7 again significantly increased neurite growth, 35 and 60%, respectively (Fig. 4E). As with KLF4, effects on neurite growth depended on the DNA binding domain (figs. S12 and S13). Clustering KLFs by sequence similarity revealed an association between functional domains (27) and effects on neurite outgrowth (Fig. 4E). For instance, overexpression of the BTEB cluster and the cluster containing KLF4 (orange and pink bars, respectively, Fig. 4E) decreased neurite growth. The TIEG and PVALS/T (33)–containing clusters (blue and green bars, Fig. 4E) had no effect on neurite length. KLF6 and KLF7, with 85% homologous activation domains, both increased neurite length (yellow bars, Fig. 4E). To explore coordinate regulation of neurite growth by KLFs, we coexpressed all two-way combinations of KLFs -4, -6, -7, and -9 in cortical neurons. The negative effects of KLF4 on neurite growth were dominant over the otherwise positive effects of KLF6 or -7; the negative effects of KLF9 summed with those of KLF6 or -7 to no net effect (Fig. 4F), suggesting a complexity to KLF-KLF interactions in regulating neurite growth. Thus, during development, RGCs down-regulate at least two growth-enhancing KLFs (KLF6 and -7), and up-regulate at least two growth-suppressive KLFs (KLF4 and -9), which may be dominant in their effect over KLF6 and -7.
These findings that the KLF family of transcription factors regulates axon growth in a number of CNS neurons have important implications. First, although KLF4 has been implicated in a wide variety of cellular events including differentiation (34, 35), cancer progression (36–38), and stem cell reprogramming (39), this function for KLF4 in postmitotic neurons advances our knowledge of the transcriptional regulation of axon regeneration. KLF4 targets relevant for regeneration may include genes selectively expressed in neurons or important in growth cone function. Second, the clustering of KLF gene function according to domain homology may provide a key for understanding how KLFs cooperate and compete to determine cellular phenotype, whether for axon regeneration or for other systems (27). Third, the decrease in RGCs’ intrinsic axon growth ability (6) parallels changes in expression within the KLF family: Postnatal RGCs express higher levels of axon growth-suppressing KLFs and lower levels of axon growth-enhancing KLFs; similar changes can be found in published corticospinal motor neuron data (40). Thus, manipulating multiple KLF genes may be a useful strategy to add to existing approaches to increase the intrinsic regenerative capacity of mature CNS neurons damaged by injury or disease.
Supplementary Material
Acknowledgments
This work was funded by a National Eye Institute (NEI) R03 grant (EY016790, J.L.G.) and subsequently by a National Institute of Neurological Disorders and Stroke (NINDS) R01 grant (NS061348, J.L.G.) and critical funding from The Glaucoma Foundation and the Seigal Foundation (J.L.G.), the C. H. Neilsen Foundation (V.P.L. and J.L.G.), the Ralph Wilson Medical Research Foundation and the W.G. Ross Foundation (V.P.L.), The Buoniconti Fund to Cure Paralysis (V.P.L. and J.L.B.), an NEI P30 grant (EY014801), and an unrestricted grant from Research to Prevent Blindness to the University of Miami. D.L.M. is a Lois Pope LIFE Fellow, with support from NINDS training grants T32 NS07492 and T32 NS007459. We are indebted for generous gifts of FLAG-tagged KLF4 (C. Liu, University of Texas), mCherry (R. Tsien, University of California, San Diego), and KLF2 constructs (J. Lingrel, University of Cincinnati). We are grateful to W. Feuer (University of Miami) for statistical contributions and to R. Corredor (University of Miami) for optimizing RGC electroporation. We thank the following lab members and collaborators for their meaningful contributions: Ne. Patel, Ni. Patel, J. Gallardo, N. Sharifai, E. Walford, M. Velarde, A. Sloan, M. Benny, A. Oliva, Y. Shi, E. Hernandez, G. Lambert, and G. Gaidosh. The authors declare they have no conflicts of interest.
Footnotes
References and Notes
- 1.Nicholls J, Saunders N. Trends Neurosci. 1996;19:229. doi: 10.1016/0166-2236(96)10021-7. [DOI] [PubMed] [Google Scholar]
- 2.Chen DF, Jhaveri S, Schneider GE. Proc Natl Acad Sci USA. 1995;92:7287. doi: 10.1073/pnas.92.16.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bregman BS, Kunkel-Bagden E, McAtee M, O’Neill A. J Comp Neurol. 1989;282:355. doi: 10.1002/cne.902820304. [DOI] [PubMed] [Google Scholar]
- 4.Yiu G, He Z. Nat Rev Neurosci. 2006;7:617. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Case LC, Tessier-Lavigne M. Curr Biol. 2005;15:R749. doi: 10.1016/j.cub.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Goldberg JL, Klassen MP, Hua Y, Barres BA. Science. 2002;296:1860. doi: 10.1126/science.1068428. [DOI] [PubMed] [Google Scholar]
- 7.Blackmore M, Letourneau PC. J Neurobiol. 2006;66:348. doi: 10.1002/neu.20224. [DOI] [PubMed] [Google Scholar]
- 8.Dusart I, Airaksinen MS, Sotelo C. J Neurosci. 1997;17:3710. doi: 10.1523/JNEUROSCI.17-10-03710.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouslama-Oueghlani L, Wehrle R, Sotelo C, Dusart I. J Neurosci. 2003;23:8318. doi: 10.1523/JNEUROSCI.23-23-08318.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li D, Field PM, Raisman G. Eur J Neurosci. 1995;7:1164. doi: 10.1111/j.1460-9568.1995.tb01106.x. [DOI] [PubMed] [Google Scholar]
- 11.Cai D, et al. J Neurosci. 2001;21:4731. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao Y, et al. Neuron. 2004;44:609. doi: 10.1016/j.neuron.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 13.Chen DF, Schneider GE, Martinou JC, Tonegawa S. Nature. 1997;385:434. doi: 10.1038/385434a0. [DOI] [PubMed] [Google Scholar]
- 14.Cho KS, et al. J Cell Sci. 2005;118:863. doi: 10.1242/jcs.01658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehmann M, et al. J Neurosci. 1999;19:7537. doi: 10.1523/JNEUROSCI.19-17-07537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A. Science. 2004;303:1026. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- 17.Lasorella A, et al. Nature. 2006;442:471. doi: 10.1038/nature04895. [DOI] [PubMed] [Google Scholar]
- 18.Park KK, et al. Science. 2008;322:963. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang JT, et al. J Neurosci. 2007;27:8593. doi: 10.1523/JNEUROSCI.4488-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchser WJ, Pardinas JR, Shi Y, Bixby JL, Lemmon VP. Biotechniques. 2006;41:619. doi: 10.2144/000112279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu S, Tai C, MacVicar BA, Jia W, Cynader MS. Brain Res. 2009;1250:49. doi: 10.1016/j.brainres.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 22.Ghaleb AM, Katz JP, Kaestner KH, Du JX, Yang VW. Oncogene. 2007;26:2365. doi: 10.1038/sj.onc.1210022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagos EG, Ghaleb AM, Dalton WB, Bialkowska AB, Yang VW. Oncogene. 2009;28:1197. doi: 10.1038/onc.2008.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang W, et al. Mol Cell Biol. 2006;26:2055. doi: 10.1128/MCB.26.6.2055-2064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segre JA, Bauer C, Fuchs E. Nat Genet. 1999;22:356. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 26.Katz JP, et al. Development. 2002;129:2619. doi: 10.1242/dev.129.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaczynski J, Cook T, Urrutia R. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang J, et al. Nat Cell Biol. 2008;10:353. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 29.Eaton SA, et al. J Biol Chem. 2008;283:26937. doi: 10.1074/jbc.M804831200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang DT, Zhao W, Mahatan CS, Geiman DE, Yang VW. Nucleic Acids Res. 2002;30:2736. doi: 10.1093/nar/gkf400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cayrou C, Denver RJ, Puymirat J. Endocrinology. 2002;143:2242. doi: 10.1210/endo.143.6.8856. [DOI] [PubMed] [Google Scholar]
- 32.Veldman MB, Bemben MA, Thompson RC, Goldman D. Dev Biol. 2007;312:596. doi: 10.1016/j.ydbio.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 33.Abbreviations for the amino acid residues are as follows: A, Ala; E, Glu; G, Gly; I, Ile; L, Leu; P, Pro; S, Ser; T, Thr; and V, Val.
- 34.Ghaleb AM, et al. Cell Res. 2005;15:92. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dai X, Segre JA. Curr Opin Genet Dev. 2004;14:485. doi: 10.1016/j.gde.2004.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rowland BD, Peeper DS. Nat Rev Cancer. 2006;6:11. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 37.Safe S, Abdelrahim M. Eur J Cancer. 2005;41:2438. doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 38.Black AR, Black JD, Azizkhan-Clifford J. J Cell Physiol. 2001;188:143. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 39.Zhao R, Daley GQ. J Cell Biochem. 2008;105:949. doi: 10.1002/jcb.21871. [DOI] [PubMed] [Google Scholar]
- 40.Arlotta P, et al. Neuron. 2005;45:207. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.