

Abstract
The design and synthesis of a water-soluble 1,3-bis(diphenylene)-2-phenylallyl (BDPA) radical via the conjugate addition of a derivatized fluorene nucelophile is described. The compound is designed for use in dynamic nuclear polarization NMR. Its 9 GHz EPR spectrum in glycerol/water is reported.
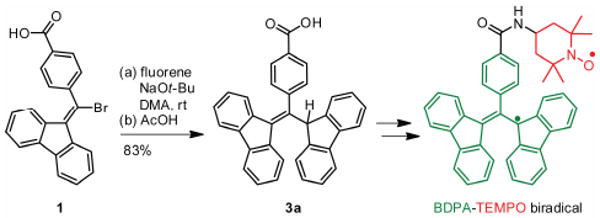
The 1,3-bis(diphenylene)-2-phenylallyl (BDPA) radical1 (Scheme 1, green) is an air-stable, carbon-centered radical that is unique among organic radicals in the extent of delocalization of its unpaired electron. The unpaired electron is predominantly located at the 1- and 3-positions of the compound's allyl core, but is further stabilized by delocalization into the two biphenyl ring systems attached at those positions.2 In addition, the propeller-like geometry of the compound has been suggested to sterically shield and further protect the radical from potential reaction partners.3
SCHEME 1.
The BDPA radical's resonance has a narrow line width in high-field EPR, presumably because it is highly delocalized, which makes it attractive for use in certain NMR experiments utilizing dynamic nuclear polarization (DNP).4 DNP can be used to increase the signal-to-noise ratio in NMR spectra by transferring the polarization of electrons to nuclei.5 Electrons are inherently easier to polarize because of their larger magnetic moment. Unfortunately, BDPA cannot be used in experiments that require an aqueous cosolvent, such as studies using DNP to improve NMR protein structure determination, because of its hydrophobicity.5b
We report the synthesis of a water-soluble BDPA (WS-BDPA) radical. Previous syntheses of BDPA derivatives have not focused on imparting water solubility.1b,3a,6 However, there are several reports of water-soluble derivatives of the triarylmethyl (trityl) radical.7 In addition to their use in DNP-NMR, some water-soluble trityl derivatives have been used as EPR probes for oxygen concentration and pH, which is a possible application of water-soluble BDPA derivatives.8
Building on our previously reported synthesis of a BDPA-TEMPO biradical9 (Scheme 1), the conjugate addition of a fluorene anion to compound 1 became the crucial transformation in our route to WS-BDPA. Compound 1 contains a carboxylic acid at the para-position of its phenyl ring that can aid in aqueous solubility. We reasoned that the addition of two additional carboxylic acids, masked as esters, at the 2- and 7-positions of the fluorene nucleophile would greatly improve the solubility of the resultant BDPA radical in polar solvents. However, we observed that adding electron-withdrawing groups to fluorene slows the conjugate addition. For example, when 2,7-dibromofluorene (pKa = 17.9 in DMSO) is used as the nucleophile instead of fluorene (pKa = 22.6 in DMSO)10 the required reaction time increases from 1 hour to 24 hours. The stabilization of the fluorene carbanion results in decreased nucleophilicity and less driving force to form the more stabilized BDPA carbanion (pKa = 14)11. As a result, a fluorene with esters directly attached would likely not be a good reaction partner with 1. A fluorene derivative with a saturated carbon between the aromatic ring system and the ester would likely have the desired nucleophilicity at the 9-position, but its benzylic protons located alpha to an ester could be deprotonated under the reaction conditions. The simplest route to separate the esters from the conjugated system and still maintain selectivity for deprotonation at the 9-position of the fluorene ring system was to introduce an ethylene linker, as in 2 (protons alpha to the ester in 2 have a pKa ≈ 30 in DMSO12).
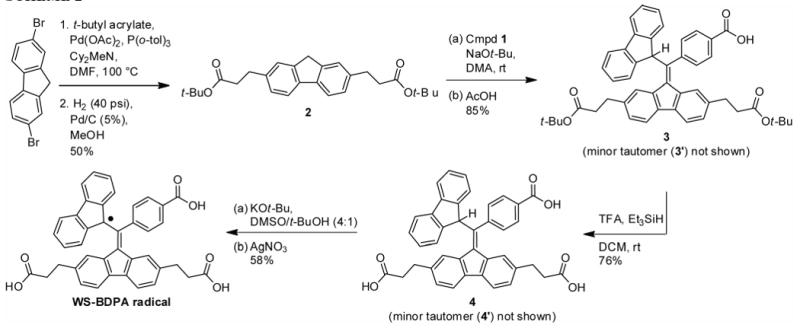
Diester 2 was synthesized in moderate yield from 2,7-dibromofluorene and t-butyl acrylate via a Heck reaction13 followed by hydrogenation (Scheme 2). The reaction conditions for the addition of 2 to 1 required adjustment from those of the previous procedure.9 We found that the slow addition of 2 to a stirring solution of 1 and excess sodium t-butoxide in dimethylacetamide (DMA) improved yields, as compared to the previous procedure in which 1 was added to a solution of the fluorene anion. Maintaining a low concentration of fluorene anion inhibits side reactions. In addition, we found that freshly prepared sodium t-butoxide14 was superior to the commercially available material, likely because it contained less sodium hydroxide and therefore caused less unwanted ester cleavage. In this regard, the use of dry DMA was also important. After protonation and purification, the product was isolated as a 2:1 mixture of tautomers.
SCHEME 2.
The tautomers have distinct 1H-NMR spectra, as shown in Figure 1. Previously, we reported NMR experiments and an X-ray crystal structure of 3a, which revealed the origin of the signals outside the normal range of aromatic protons, specifically those above 8.0 ppm and below 6.0 ppm.9 The upfield shift in signals HC and HC′ is caused by an interaction with the magnetic field of the nearby phenyl ring. The downfield signals (HB, HB′) result from a steric interaction with HA and HA′.15 An NOE of 20% was measured between the analagous protons in 3a.9
FIGURE 1.
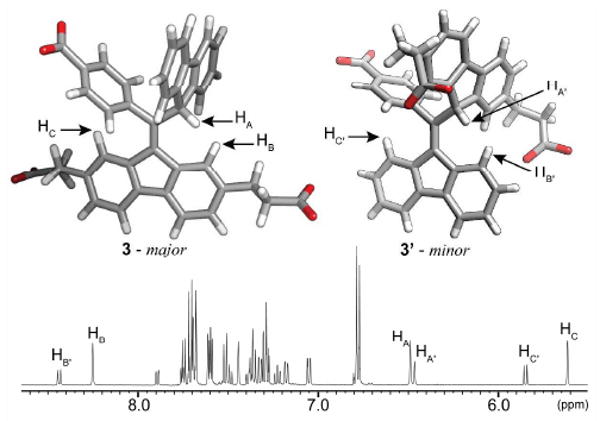
Molecular models of 3 and 3′ (t-butyl groups have been omitted for clarity) and the aromatic region of the 1H-NMR spectrum.
To prepare triacid 4, removal of the t-butyl groups is achieved with trifluoroacetic acid in dichloromethane with the addition of triethylsilane as a carbocation trap.16 The acidic conditions do not cause a change in the ratio of tautomers in 4 as compared to 3.
In order to generate the radical by one-electron oxidation, compound 4 was deprotonated to form the tetra-anion (see Figure 2, blue solution). We found that a large excess (20 equiv) of potassium t-butoxide in a 4:1 solution of DMSO/t-butanol provided the best results. Because excess base can cause reduction of the radical back to the carbanion, the amount of oxidant had to be increased to ensure complete oxidation. Addition of the oxidant was quickly followed by a dilution of the reaction mixture with acidic water (pH = 2), and subsequent extraction of the triacid into diethyl ether. The isolated red-brown powder showed no 1H-NMR signals at a concentration of 20 mM in d4-methanol beyond those of the solvent and water, as would be expected for a paramagnetic compound. HRMS and FT-IR agreed with the proposed structure. As shown in Figure 2, in a glycerol and water solution (3:2) the radical has a strong absorption in the visible region (λmax = 496 nm) and a weak absorption in the near-infrared region (λmax = 867 nm). BDPA in dichloromethane has similar absorbance maxima, which are 485 nm and 859 nm, respectively.1b,3a
FIGURE 2.
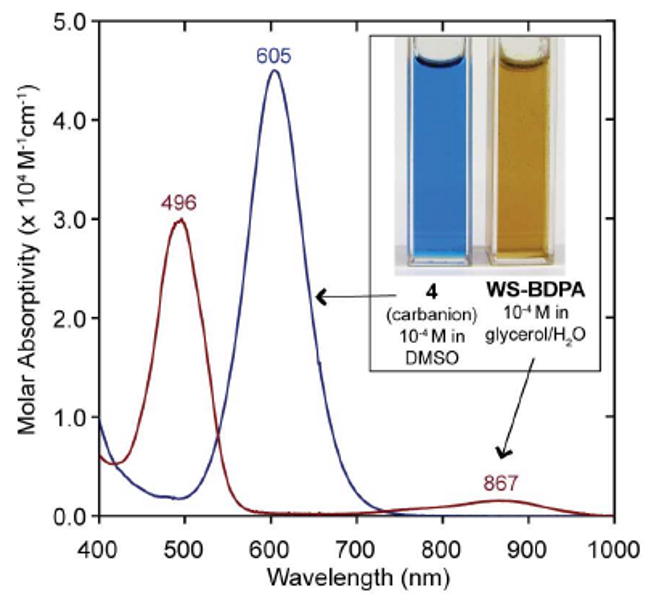
Absorption spectra of carbanion 4 in dimethylsulfoxide and WS-BDPA in 3:2 glycerol and water.
The solubility of the radical in aqueous solution (PBS buffer, pH = 8.0) was approximately 1.0 mM. Because the material was designed for use in DNP-NMR experiments dealing with biomolecules, we also tested the solubility of the radical in a 3:2 solution of glycerol and water, a solvent mixture favored for those experiments.5b The radical was soluble at 1 mM at pH =7, but required the addition of sodium bicarbonate to be soluble at 10 mM (pH = 8). Figure 3 shows the 9 GHz EPR spectrum of WS-BDPA in glycerol/water beneath the spectrum of BDPA in toluene. The radicals have the same g-value (g = 2.003) and a similar coupling pattern (see Supporting Information for simulated spectrum).1b,2,17 In addition, the radical was persistent at room temperature both in the solid-state and in solution, but detailed studies of its stability as compared to BDPA have not yet been performed.
FIGURE 3.
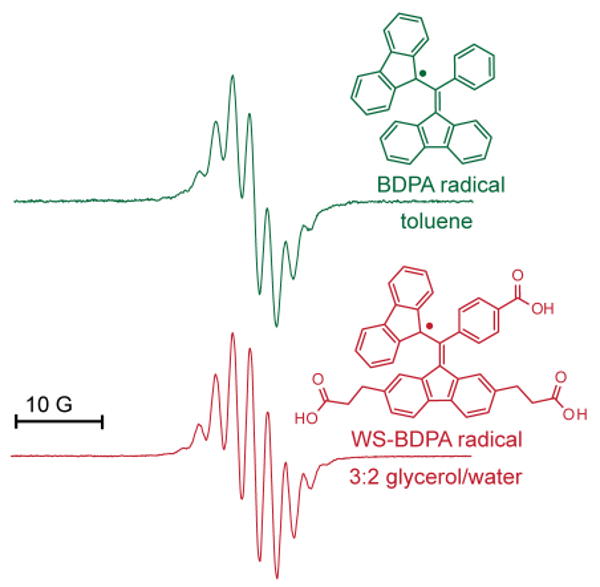
9 GHz EPR of BDPA (0.1 mM, 4 mm diameter tube) in toluene and WS-BDPA (10.0 mM, 150 μL flat cell) in glycerol/water. Experimental parameters: rt, 2 mW, modulation amplitude 10 mT.
In conclusion, the design and synthesis of a water-soluble derivative of the BDPA radical is reported. We characterize the pair of tautomers that are a consequence of constructing the BDPA skeleton with non-identical fluorene rings. Finally, the generation of the stable radical and its characterization by EPR spectroscopy in aqueous solution are reported.
Experimental Section
di-t-butyl 3,3′-(9H-fluorene-2,7-diyl)dipropanoate (2)
To a 250 mL flask were added 5.00 g (15.4 mmol, 1.0 equiv) of 2,7-dibromofluorene, 0.087 g (0.390 mmol, 2.5 mol %) palladium acetate, 0.470 g (0.780 mmol, 5 mol %) tri(o-tol)phosphine, 5 mL of dicyclohexylmethylamine, and 100 mL of anhydrous dimethylformamide. The solution was degassed with bubbling argon and 7.00 mL (6.13 g, 47.8 mmol, 3.1 equiv) of t-butyl acrylate was added. After heating to 80 °C for 4 hours and cooling overnight, excess water was added and the resulting solid was filtered, washed with water, and dried on the filter pad. The material was suspended in 150 mL of methanol and 1.00 g of 5% Pd/C was added. Hydrogenation was performed overnight in a Parr Hydrogenator at 40 PSI with shaking. After removal of the methanol, the crude material was chromatographed using silica gel and eluted with increasingly polar mixtures of hexane and ethyl acetate to yield 3.25 g (50%) of a white powder. 1H NMR (500 MHz, CD2Cl2, δ) (all coupling constants (J) in Hz): 7.64 (d, J = 7.5, 2H), 7.36 (s, 2H), 7.18 (d, J = 8.0, 2H), 3.82 (s, 2H), 2.94 (t, J = 7.5, 4H), 2.54 (t, J = 7.5, 4H), 1.39 (s, 18 H); 13C NMR (126 MHz, CD2Cl2, δ) 172.6, 144.1, 140.2, 140.0, 127.4, 125.6, 119.9, 80.6, 37.8, 37.2, 31.8, 28.4; HRMS (ESI): calcd for C27H34O4Na [M + Na]+: 445.2349; found, 445.2329. FT-IR (KBr, thin film) νmax (cm-1): 2978, 1729(s), 1454, 1365, 1257, 1133, 1009, 956, 817, 741; mp 99–101 °C (methanol).
4-((2,7-bis(3-(t-butoxy)-3-oxopropyl)-9H-fluoren-9-ylidene)(9H-fluoren-9-yl)methyl)benzoic acid (3) and 4-((2,7-bis(3-(tert-butoxy)-3-oxopropyl)-9H-fluoren-9-yl)(9H-fluoren-9-ylidene)methyl)benzoic acid (3′)
To a 100 mL flask were added 0.100 g (0.265 mmol, 1.0 equiv) of bromide 2, 0.255 g (2.65 mmol, 10 equiv) of sodium t-butoxide, and 6.0 mL of degassed, anhydrous dimethylacetamide (DMA). The solution was cooled to 0 °C. In a separate flask, 0.168 g (0.398 mmol, 1.5 equiv) of ester 1 was dissolved in 10 mL of DMA and this solution was slowly added to the first flask over a period of 20 minutes. The reaction became deep blue. After stirring at room temperature for 2 hours, excess (3 mL) of acetic acid was added, turning the reaction from deep blue to light yellow. Additional water was added and the solution was extracted with diethyl ether (3 × 30 mL). The ether was washed twice with water and once with brine and dried over sodium sulfate. After removal of the solvent, the crude material was purified by silica gel column chromatography using increasingly polar mixtures of ethyl acetate in dichloromethane, after which 0.163 g (85%) of a light yellow powder was isolated. 1H NMR (500 MHz, CD2 Cl2, δ) (all coupling constants (J) in Hz): Compound 3: 8.25 (s,1H), 7.75 (d, J = 8.0, 1H), 7.71 (d, J = 8.0, 2H), 7.69 (d, J = 8.0, 2H), 7.60 (ddd, J = 8.0, 1.0, 0.5, 2H), 7.59 (d, J = 8.0, 1H), 7.36 (td, J = 8.0, 1.0, 2H), 7.32 (dd, J = 8.0, 1.0, 1H), 7.29 (td, J = 8.0, 1.0, 2H), 7.05 (dd, J = 8.0, 1.0, 1H), 6.78 (d, J = 8.0, 2H,), 6.49 (s, 1H), 5.62 (s, 1H), 2.95 (t, J = 7.5, 2H), 2.54 (t, J = 7.5, 2H), 2.45 (t, J = 7.5, 2H), 2.07 (t, J = 7.5, 2H), 1.35 (s, 9H), 1.30 (s, 9H); Compound 3′: 8.44 (ddd, J = 8.0, 1.0, 0.5, 1H), 7.89 (ddd, J = 8.0, 1.0, 0.5, 1H), 7.75 (ddd, J = 8.0, 1.0, 0.5, 1H), 7.69 (d, J = 8.0, 2H), 7.52 (d, J = 8.0, 2H), 7.49 (td, J = 8.0, 1.0, 1H), 7.44 (s, 2H), 7.38 (td, J = 8.0, 1.0, 1H), 7.23 (td, J = 8.0, 1.0, 1H), 7.18 (d, J = 8.0, 2H), 6.79 (td, J = 8.0, 1.0, 1H), 6.78 (d, J = 8.0, 2H), 6.47 (s, 1H), 5.85 (d, J = 8.0, 1H), 2.90 (t, J = 7.5, 4H), 2.49 (t, J = 7.5, 4H), 1.35 (s, 18H); 13C NMR (126 MHz, CD2Cl2, δ): 172.5, 172.4, 172.3, 171.6, 171.5, 145.2, 144.9, 144.6, 144.4, 144.0, 143.5, 142.5, 141.8, 140.7, 140.6, 140.5, 140.4, 139.4 (2), 139.3, 139.0, 138.5, 136.7, 136.6, 130.0, 129.5, 129.2, 129.0, 128.6, 128.5 (3), 128.3, 128.2, 128.0, 127.7, 127.1, 126.5, 126.3, 125.9, 125.8 (2), 125.7, 120.6, 120.5, 120.2, 119.7, 119.3, 80.7 (2), 80.4, 52.8, 52.7, 37.8 (2), 36.7, 32.1, 31.7, 31.6, 28.3, 28.2; HRMS (ESI): calcd for C48H45O6 [M-H]-: 717.3222; found, 717.3206. FT-IR (KBr, thin film) νmax (cm- 1): 2977, 2930, 1727(s), 1694(s), 1607, 1447, 1419, 1367, 1260, 1148, 1018, 846, 822, 736.
3,3′-(9-((4-carboxyphenyl)(9H-fluoren-9-yl)methylene)-9H-fluorene-2,7-diyl)dipropanoic acid (4) and 3,3′-(9-((4-carboxyphenyl)(9H-fluoren-9-ylidene)methyl)-9H-fluorene-2,7-diyl)dipropanoic acid (4′)
To a 50 mL flask were added 0.150 g (0.209 mmol, 1.0 equiv) of 3, 9.0 mL of anhyd CH2Cl2, 1 drop triethylsilane, and 4.5 mL of TFA. The reaction mixture was stirred under an argon atmosphere for 30 minutes, after which excess sodium bicarbonate solution was added. The aqueous layer was washed with diethyl ether (2 × 10 mL), and acidified with 2M HCl to a pH of 1. The aqueous layer was extracted with diethyl ether (3 × 10 mL). The combined ether layers were washed with brine and dried over sodium sulfate. Removal of the solvent followed by drying under high vacuum yielded 0.0960 g (0.158 mmol, 76 %) of an off-white powder. 1H NMR (500 MHz, d4-methanol, δ) (all coupling constants(J) in Hz): Compound 4: 8.25 (s, 2H), 7.73 (d, J = 8.0, 1H), 7.64 (d, J = 8.0, 2H), 7.63 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0, 4H), 7.32 (m, 3H), 7.25 (t, J = 8.0, 2H), 7.04 (d, J = 16.0, 1H), 6.71 (d, J = 8.5, 2H), 6.45 (s, 1H), 5.64 (s, 1H), 2.98 (t, J = 8.0, 2H), 2.62 (t, J = 8.0, 2H), 2.48 (t, J = 8.0, 2H), 2.17 (t, J = 8.0, 2H). Compound 4′: 8.41 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 7.5, 1H), 7.72 (d, J = 7.5, 1H), 7.64 (d, J = 8.0, 2H), 7.48 (d, J = 8.0, 2H), 7.46 (t, J = 8.0, 1H), 7.43 (s, 2H), 7.36 (t, J = 8.0, 1H), 7.19 (t, J = 8.0, 1H), 7.16 (d, J = 8.0, 2H), 6.74 (t, J = 8.0, 1H), 6.71 (d, J = 8.0, 2H), 6.42 (s, 1H), 5.85 (d, J = 8.0, 1H), 2.92 (t, J = 8.0, 4H), 2.57 (t, J = 8.0, 4H). 13C NMR (126 MHz, d4-methanol and CDCl3, δ): 176.3, 176.1, 175.8, 169.7, 144.9, 144.6, 144.3, 144.2, 144.0, 142.8, 142.1, 140.9, 140.4 (2), 139.7, 139.4, 139.2, 138.9, 136.8, 136.7, 129.8 (2), 129.6, 129.5, 129.3, 129.2, 128.4, 127.8, 127.2, 126.5, 126.0, 125.9, 125.7, 120.7 (2), 120.5, 119.9, 119.6, 53.1, 53.0, 36.9, 36.8, 35.8, 32.2, 31.8, 31.6. HRMS (ESI): calcd for C40H29O6 [M-H]-: 605.1970; found, 605.1968. FT-IR (KBr, thin film) νmax (cm-1): 3600-2800 (br), 1700 (s), 1606, 1446, 1417, 1274, 1178, 1017, 824, 734.
WS-BDPA radical
To a 200 mL flask were added 0.040 g (0.066 mmol, 1.0 equiv) of compound 4, 0.150 g (1.34 mmol, 20 equiv) of sublimed potassium t-butoxide, and 50 mL of a 4:1 mixture of DMSO/t-BuOH. After 30 minutes, 0.180 g (1.06 mmol, 16.0 equiv) of silver nitrate was added. The solution immediately became brown and was diluted after 1 minute with 400 mL of 0.01 M HCl, and extracted with diethyl ether (3 × 10 mL). The combined organic layers were washed with water (3 × 25 mL) and brine. After drying with sodium sulfate, the solvent was removed. The resulting solid was washed with hexane, and 0.023 g (58%) of a dark red-brown powder was isolated. HRMS (ESI): calcd for C40H29O6 M-: 605.1970; found, 605.1968. FT-IR (KBr, thin film) νmax (cm-1): 3600-2800 (br), 1708 (s), 1604, 1446, 1417, 1282, 1177, 1018, 824, 730. EPR (9 GHz) [WS-BDPA] = 10 mM in 3:2 glycerol/H2O, quartz flat cell, g-value = 2.003.
Supplementary Material
Acknowledgments
We thank Dr. Thorsten Maly, Galia Debelouchina, and Professor Robert G. Griffin for helpful discussions. Funding for this project was provided by the National Science Foundation and the National Institute of Biomedical Imaging and Bioengineering (EB-002804).
Footnotes
Supporting Information: Additional experimental details, copies of 1H-, gCOSY, and 13C-NMR spectra for 2, 3, and 4. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Koelsch CF. J Amer Chem Soc. 1957;79:4439. [Google Scholar]; (b) Kuhn R, Neugebauer FA. Monatsh Chem. 1964;95:3. [Google Scholar]
- 2.Azuma N, Ozawa T, Yamauchi J. Bull Chem Soc Jpn. 1994;67:31. [Google Scholar]
- 3.(a) Breslin DT, Fox MA. J Phys Chem. 1993;97:13341. [Google Scholar]; (b) Griller D, Ingold KU. Acc Chem Res. 1976;9:13. [Google Scholar]
- 4.(a) Wind RA, Duijvestijn MJ, van der Luat C, Manenschijn A, Vriend J. Prog Nucl Magn Reson Spectrosc. 1985;17:33. [Google Scholar]; (b) Afeworki M, Schaefer J. Macromolecules. 1992;25:4092. [Google Scholar]; (c) Becerra LR, Gerfen GJ, Temkin RJ, Singel DJ, Griffin RG. Phys Rev Lett. 1993;71:3561. doi: 10.1103/PhysRevLett.71.3561. [DOI] [PubMed] [Google Scholar]; (d) Weis V, Bennati M, Rosay M, Griffin RG. J Chem Phys. 2000;113:6795. [Google Scholar]
- 5.(a) Barnes AB, Paëpe GD, Wel PCAvd, Hu KN, Joo CG, Bajaj VS, Mak-Jurkauskas ML, Sirigiri JR, Herzfeld J, Temkin RJ, Griffin RG. Appl Magn Reson. 2008;34:237. doi: 10.1007/s00723-008-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maly T, Debelouchina GT, Bajaj VS, Hu KN, Joo CG, Mak-Jurkauskas ML, Sigirhi JR, van der Wel PCA, Herzfeld J, Temkin RJ, Griffin RG. J Chem Phys. 2008;128:052211. doi: 10.1063/1.2833582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Plater MJ, Kemp S, Lattmann E. J Chem Soc, Perkin Trans. 2000;1:971. [Google Scholar]; (b) Nishide H, Yoshioka N, Saitoh Y, Gotoh R, Miyakawa T, Tsuchida E. J Macromol Sci Pure Appl Chem. 1992;A29:775. [Google Scholar]; (c) Solar SL. J Org Chem. 1963;28:2911. [Google Scholar]
- 7.(a) Reddy TJ, Iwama T, Halpern HJ, Rawal VH. J Org Chem. 2002;67:4635. doi: 10.1021/jo011068f. [DOI] [PubMed] [Google Scholar]; (b) Bowman MK, Mailer C, Halpern HJ. J Magn Reson. 2005;172:254. doi: 10.1016/j.jmr.2004.10.010. [DOI] [PubMed] [Google Scholar]; (c) Roques N, Maspoch D, Wurst K, Ruiz-Molina D, Rovira C, Veciana J. Chem Eur J. 2007;12:9238. doi: 10.1002/chem.200600447. [DOI] [PubMed] [Google Scholar]; (d) Liu Y, Villamena FA, Sun J, Xu Y, Dhimitruka I, Zweier JL. J Org Chem. 2008;73:1490. doi: 10.1021/jo7022747. [DOI] [PubMed] [Google Scholar]; (e) Anderson S, Golman K, Rise F, Wikstrom H, Wistrand LG. U.S. Patent No.: 5,530,140. 1996
- 8.Bobko AA, Dhimitruka I, Zweier JL, Khramtsov VV. J Amer Chem Soc. 2007;129:7240. doi: 10.1021/ja071515u. [DOI] [PubMed] [Google Scholar]
- 9.Dane EL, Maly T, Debelouchina GT, Griffin RG, Swager TM. Org Lett. 2009;11:1871. doi: 10.1021/ol9001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bordwell FG, McCollum GJ. J Org Chem. 1976;41:2391. [Google Scholar]
- 11.Kuhn R, Rewicki D. Liebigs Ann Chem. 1967;706:250. [Google Scholar]
- 12.Zhang XM, Bordwell FG, Van Der Puy M, Fried HE. J Org Chem. 1993;58:3060. [Google Scholar]
- 13.Patel BA, Ziegler CB, Cortese NA, Plevyak JE, Zebovitz TC, Terpko M, Heck RF. J Org Chem. 1977;42:3903. [Google Scholar]
- 14.Sodium t-butoxide was prepared by refluxing sodium with excess freshly distilled t-butanol followed by evaporation of excess solvent under high vacuum. The material was stored and weighed in a nitrogen glove box.
- 15.Cheney BV. J Am Chem Soc. 1968;90:5386. [Google Scholar]
- 16.Mehta A, Jaouhari R, Benson TJ, Douglas KT. Tett Lett. 1992;33:5441. [Google Scholar]
- 17.Watanabe K, Yamauchi J, Ohya-Nishiguchi H, Deguchi Y, Ishizu K. Bull Inst Chem Res Kyoto Univ. 1975;53:161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.