

Abstract
Methamphetamine (METH) is an illicit and potent psychostimulant, which acts as an indirect dopamine agonist. In the striatum, METH has been shown to cause long lasting neurotoxic damage to dopaminergic nerve terminals and recently, the degeneration and death of striatal cells. The present study was undertaken to identify the type of striatal neurons that undergo apoptosis after METH. Male mice received a single high dose of METH (30 mg/kg, i.p.) and were killed 24 h later. To demonstrate that METH induces apoptosis in neurons, we combined terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining with immunohistofluorescence for the neuronal marker neuron-specific nuclear protein (NeuN). Staining for TUNEL and NeuN was colocalized throughout the striatum. METH induces apoptosis in approximately 25% of striatal neurons. Cell counts of TUNEL-positive neurons in the dorsomedial, ventromedial, dorsolateral and ventrolateral quadrants of the striatum did not reveal anatomical preference. The type of striatal neuron undergoing cell death was determined by combining TUNEL with immunohistofluorescence for selective markers of striatal neurons: dopamine- and cAMP-regulated phosphoprotein, of apparent Mr 32,000, parvalbumin, choline acetyltransferase and somatostatin (SST). METH induces apoptosis in approximately 21% of dopamine- and cAMP-regulated phosphoprotein, of apparent Mr 32,000-positive neurons (projection neurons), 45% of GABA-parvalbumin-positive neurons in the dorsal striatum, and 29% of cholinergic neurons in the dorsal–medial striatum. In contrast, the SST-positive interneurons were refractory to METH-induced apoptosis. Finally, the amount of cell loss determined with Nissl staining correlated with the amount of TUNEL staining in the striatum of METH-treated animals. In conclusion, some of the striatal projection neurons and the GABA-parvalbumin and cholinergic interneurons were removed by apoptosis in the aftermath of METH. This imbalance in the populations of striatal neurons may lead to functional abnormalities in the output and processing of neural information in this part of the brain.
Keywords: methamphetamine, apoptosis, striatum, projection neurons, interneurons
Methamphetamine (METH) is a potent and addictive psychostimulant. The neurotoxic effects of this substituted amphetamine are associated with its ability to induce an overflow of dopamine in the synapse by displacing vesicular dopamine stores (Raiteri et al., 1979; Liang and Rutledge, 1982a,b; Schmidt et al., 1985; Sulzer et al., 1995). Displaced dopamine molecules and its metabolites can be readily oxidized to reactive quinones and semiquinones further generating reactive oxygen (Cadet and Brannock, 1998) and nitrogen radicals (Lipton and Rosenberg, 1994; Imam et al., 1999) that affect a wide range of modifications of proteins, sugars, and lipids. Augmented levels of dopamine in the synapse induced by METH have been shown in humans (Wilson et al., 1996; McCann et al., 1998), non-human primates (Seiden et al., 1976; Villemagne et al., 1998; Harvey et al., 2000) and rodents (Wagner et al., 1980; Yu et al., 2002). These studies also show that METH causes toxicity of the striatal nerve terminals. Administration of amphetamines and its analogs like METH results in long-lasting neurochemical depletions in dopamine and serotonin levels, inhibition of tyrosine hydroxylase and tryptophan hydroxylase activity, and dopamine reuptake sites (Davidson et al., 2001; Hanson et al., 2004; McCann and Ricaurte, 2004). In addition to the damage at the dopamine terminals, there is emerging evidence demonstrating that METH causes injury to cell bodies in various brain regions. For example, significant cortical gray matter and hippocampal deficits were seen in the brains of humans who used METH (Thompson et al., 2004). Rodents treated with METH displayed Fluoro-Jade B-positive degenerating neurons in the striatum (Yu et al., 2004). Repeated exposure to METH resulted in cell body injury in the parietal cortex of rats (Eisch and Marshall, 1998). In addition, a number of studies demonstrated the induction by METH of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)-positive apoptotic cells in the cortex and striatum of rodents (Stumm et al., 1999; Deng and Cadet, 2000; Deng et al., 2001; Zhu et al., 2002; Cadet et al., 2003; Loonam et al., 2003). In view of the neurotoxic effects of METH and the selective vulnerability of some striatal neurons toward dopaminergic manipulation, it is important to determine the phenotype of striatal neuron killed by an acute administration of METH.
The striatum is the largest nucleus of the basal ganglia and is associated with disorders of movement (Hickey and Chesselet, 2003; Singer and Minzer, 2003). There is increasing evidence that it is also involved in the control of attention, executive function, motivated behaviors (Alexander et al., 1986) as well as neuropsychiatric conditions such as obsessive compulsive disorders, psychoses and addictive behaviors (Calabresi et al., 1997). This structure is heterogeneous in its internal organization as well as in its afferent and efferent connections. Approximately 90% of the neurons of the striatum are GABAergic medium spiny projection neurons that are further subdivided into striatonigral projections containing substance P and dynorphin and striatopallidal projections containing enkephalin. The remaining 10% make up various striatal interneurons, of which the GABA-parvalbumin, somatostatin (SST), and cholinergic interneurons are considered the most prevalent (Gerfen, 1992). The selective loss of some striatal neurons has been associated with some neurodegenerative disorders. For example, striatal atrophy and choreiform movements seen in Huntington's disease are attributed to the progressive degeneration of striatal medium spiny projection neurons (Reiner et al., 1988; Albin et al., 1990). Considerable evidence now shows that it is the enkephalin-positive striatopallidal neurons that are the most vulnerable (Albin et al., 1992; Richfield et al., 1995, 2002; Mitchell et al., 1999; Deng et al., 2004) while striatal aspiny interneurons are spared in this disease (Ferrante et al., 1985; Reiner et al., 1988; Cicchetti et al., 1996; Hickey and Chesselet, 2003). Excitotoxic lesions by quinolinic acid are associated with increased concentrations of SST and neuropeptide Y as a result of preferential sparing of striatal interneurons (Beal et al., 1986, 1991). Damage to the striatum by the mitochondrial inhibitor, 3-nitopropionic acid, is specific to projection neurons while dopaminergic axons and nicotinamide adenine dinucleotide phosphate (NADPH)-diaphorase positive interneurons are spared (Guyot et al., 1997). Interneurons that contain NADPH-diaphorase, SST and neuropeptide Y are also less vulnerable in models of ischemia (Uemura et al., 1990). In the light of these findings, excessive METH use poses a serious health concern because the neuronal deficits in the aftermath of METH may lead to the eventual impairment of normal striatal function.
In the present study we employed immunohistofluorescence to assess the impact of METH on selective neuronal populations of the striatum. We combined TUNEL staining with immunohistofluorescence for SST, choline acetyltransferase (ChAT), and parvalbumin to assess the interneurons, and dopamine- and cAMP-regulated phosphoprotein, of apparent Mr 32,000 (DARPP-32) for the projection neurons. Our results demonstrate that the projection neurons, the cholinergic, and the GABA-parvalbumin neurons are decreased in number in the striatum after METH.
EXPERIMENTAL PROCEDURES
Animals and drug administration
A single i.p. injection of METH (10, 20, 30 or 40 mg/kg of body weight; Sigma, St. Louis, MO, USA) was given to male ICR mice 10–11 weeks of age (Taconic, Germantown, NY). All animals were housed individually with food and water available ad libitum on a 12-h light/dark cycle. Animals were habituated for two weeks prior to any drug treatment. Twenty-four hours post-treatment, animals are either killed by decapitation or first anesthetized with ketamine/acepromazine (100 mg/kg, 3 mg/kg of body weight) and then perfused transcardially with phosphate-buffered saline, pH 7.5 (PBS), containing 5000 U/ml of heparin followed by 4% paraformaldehyde in PBS. The brains were then either dissected out and immediately placed on dry ice or post-fixed in 4% paraformaldehyde in PBS overnight and cryoprotected in 30% sucrose in PBS at 4 °C. The tissue was then frozen and stored at −80 °C until use. Procedures of animal use were according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Hunter College of the City University of New York. All efforts were made to minimize the number of animals used and their suffering.
TUNEL histochemistry
The method was as described by Xu et al. (2005) with minor modifications. In brief, fresh frozen 20 μm coronal sections were collected between bregma 0.38 mm±0.1 mm and fixed in 4% paraformaldehyde for 30 min. After a wash with PBS, pH 7.6, the sections were immersed in 0.4% Triton-X-100 in PBS for 5–10 min at 70 °C. Sections were washed and TUNEL reactions (Roche Applied Science, Indianapolis, IN, USA) were applied directly onto sections and incubated for 1 h in a humidified chamber. After TUNEL staining, sections were counterstained with DAPI. Stained sections were washed in PBS and overlaid with a glass coverslip with Vectashield (Vector Laboratories, Burlingame, CA, USA). Images were taken using Molecular Dynamics CLSM Multiprobe 2001 scanning confocal system (Molecular Dynamics, Sunnyvale, CA, USA) or with a Nikon Eclipse E400 epifluorescent scope (Nikon, Melville, NY, USA) attached to a Hamamatsu digital camera C4742-95 (Hamamatsu, Bridgewater, NJ, USA) using FITC filters.
Double-labeling immunohistofluorescence
Free-floating serial coronal sections 20 μm in thickness were collected between bregma 0.38 mm±0.1 mm. The tissue was washed in PBS and then immersed in heated 0.4% Triton X-100 in PBS for 5 min and immediately washed in PBS. For neuron-specific nuclear protein (NeuN) or parvalbumin immunohistofluorescence, nonspecific binding sites were blocked with M.O.M.® Mouse Ig Blocking Reagent (Vector Laboratories) for 1 h. Sections were washed and incubated with M.O.M.® diluent for 15 min. Sections were then incubated in mouse anti-NeuN (1:500, Chemicon, Temecula, CA, USA) or mouse anti-parvalbumin (1:2000, Chemicon) in M.O.M® diluent in 0.1% Triton-X-100 in PBS overnight at 4 °C. Sections were washed in PBS and blocked again with 5% normal goat serum in 0.1% Triton X-100 in PBS for 30 min. After PBS washes, sections were incubated with Cy3-conjugated goat anti-mouse (1:1000, Chemicon) in 1% normal goat serum/0.1% Triton X-100 in PBS for 1 h and washed with PBS.
For SST or DARPP-32 immunohistofluorescence, nonspecific binding sites were blocked with 10% normal goat serum and 0.2% Triton X-100 in PBS for 1 h. Sections were washed in PBS and incubated with rabbit anti-SST (1:500, Chemicon) in 2% normal goat serum/0.1% Triton X-100 in PBS or rabbit anti-DARPP-32 (1:200, Cell Signal) overnight at 4 °C. After a wash with PBS, sections were incubated with Cy3-conjugated goat anti-rabbit (1: 1000, Chemicon) in 1% normal goat serum/0.2% Triton X-100 in PBS for 1 h and rinsed with PBS.
For ChAT immunohistofluorescence, nonspecific binding sites were blocked with 10% normal rabbit serum/0.2% Triton X-100 in PBS for 1 h. Sections were washed in PBS and incubated with goat anti-ChAT (1:500, Chemicon) in 2% normal rabbit serum/0.2% Triton X-100 overnight. After a wash with PBS, the sections were incubated with Cy3-conjugated rabbit anti-goat (1: 1000, Chemicon) in 1% normal rabbit serum/0.2% Triton X-100 in PBS for 1 h and rinsed with PBS.
No staining was observed when primary antibody was left out or when primary antibody was pre-absorbed with either SST or ChAT. After a wash with PBS, the sections were incubated in the TUNEL reaction cocktail at 37 °C for 1 h. The sections were washed in PBS, mounted on slides, and overlaid with a coverslip with Vectashield (Vector Laboratories). All incubations and washes were performed at room temperature unless otherwise stated. Images were viewed and digitized with a Nikon Eclipse E400 epifluorescence microscope attached to a Hamamatsu digital camera C4742-95 using rhodamine and FITC filters.
Cell counts and quantification
Histological quantification of METH-induced apoptosis in striatal neurons was as described by Zhu et al. (2005). Briefly, cells of interest were quantified from 20 μm thick coronal sections in an area of 0.26 mm2 for each aspect of the striatum (dorsal–medial [DM], dorsal–lateral [DL], ventral–medial [VM], and ventral–lateral [VL]) (see schematic diagram in Fig. 1 [adapted from Hof et al., 2000]). Average neuronal cell counts (neurons labeled with NeuN) were obtained from six control animals as a baseline for quantification of the percentage of TUNEL-positive neurons in experimental animals. TUNEL cell counts were averaged from five 20 μm serial sections per animal (see Fig. 2). The observer who did the cell counts and immunofluorescence quantitation was blind to treatment group.
Fig. 1.

Schematic of the striatum indicating the four regions selected for cell counts. DM, VM, DL, and VL. Black boxes indicate the 0.26 mm2 area where cells were manually counted after immunostaining. Reproduced from Hof et al. (2000).
Fig. 2.
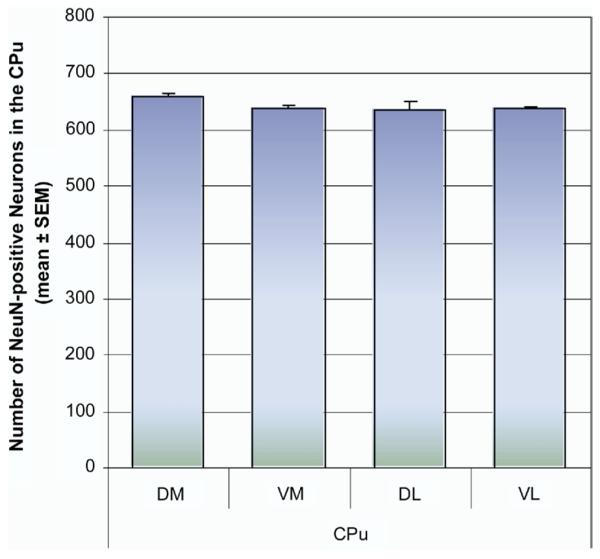
Estimation of the number of NeuN-positive neurons in the four quadrants of the striatum. Coronal sections through the striatum (bregma 0.38±0.1 mm) were processed for immunohistofluorescence with Cy3-labeled antibodies against the NeuN. NeuN-positive neurons were counted from 20 μm thick coronal sections in an area of 0.26 mm2 for each of the four quadrants of the caudate-putamen (CPu, DM, DL, VM, and VL). The mean number of NeuN-positive neurons was taken from an average of 30 sections from six control animals (five serials sections from each animal). No significant deviation in total cell counts was observed between animals or between regions of the striatum.
Nissl staining
Coronals sections of the striatum from bregma 0.38 mm±0.1 mm were fixed and defatted. Slides were then rinsed with water followed by 2 min incubation in 0.2% Cresyl Violet. Sections were then washed with water and dehydrated in a graded series of alcohol/water solutions. Tissue was placed in xylene for 5 min and a coverslip was gently overlaid.
Statistical analysis
Analysis is performed from mean±S.E.M. Differences between groups were analyzed by ANOVA followed by post hoc comparison using Fisher's protected least significance test. Significance criteria were set at P<0.05. Student's t-test was used to compare differences between two groups.
RESULTS
METH induces apoptosis in some neurons of the striatum
We subdivided the striatum into four compartments in order to assess potential differences between dorsal–ventral and lateral–medial aspects of this structure (Fig. 1). To determine the percentage of neurons undergoing METH-induced apoptosis, we first established a consistent and reliable baseline of the total number of neurons within each aspect of the striatum. This was accomplished by labeling the neurons with an antibody against the neuron-specific marker NeuN, followed by a secondary antibody conjugated to the chromophore Cy3. Five striatal sections (bregma 0.38 mm±0.1 mm) from each animal (total of six animals) were processed for immunohistofluorescence for NeuN and all fluorescent neurons were counted manually under the microscope. The average number of NeuN-positive neurons from the 30 sections was taken from each region of interest (Fig. 2) and later used to determine the percentage of TUNEL-positive neurons in the aftermath of METH. The total number of NeuN immunopositive neurons in each aspect of the striatum was very consistent between animals (Fig. 2).
We tested various doses of METH from 10 to 40 mg/kg of body weight on the induction of TUNEL-positive nuclei in the striatum. We chose TUNEL staining (a late marker of apoptosis) because it has been shown that some apoptosis can occur independently of activation of caspase-3, an early marker of apoptosis (Ferrer et al., 2003; Spalding et al., 2005). There was a sharp increase in the amount of TUNEL-positive nuclei going from 20 to 30 mg/kg, i.p. (Fig. 3A). Analysis of cell death by METH over time showed that TUNEL positive cells peaked 24 h post-treatment (Zhu et al., 2005). Continued increase of dosing at 40 mg/kg did not show any significant increase from 30 mg/kg. Thus, a 30 mg/kg dose of METH was chosen for subsequent studies. Although the DM aspect of the striatum consistently displayed lower levels of METH-induced apoptosis relative to other striatal quadrants, no significance was found between the four different regions of the striatum (Fig. 3B). The average amount of apoptosis induced by METH (30 mg/kg) was approximately 25%; however, there was a large inter-animal variability in the amount of METH-induced TUNEL-positive cells (Fig. 3C). We measured body core temperature and found that METH-induced hyperthermia does not display a large inter-animal variability (Zhu et al., 2006).
Fig. 3.


Induction of cell death by METH as a function of dose. Mice were injected once with METH (i.p.) at the doses indicated. The mice were killed 24 h after METH and sections of striatal tissue were processed for TUNEL immunohistofluorescence. (A) Scanning confocal micrographs of TUNEL staining in the striatum of mice treated with increasing doses of METH (a–e). Scale bar=50 μm. (B) Percentage of striatal neurons displaying TUNEL staining for DM, VM, DL, and VL regions of the striatum (mean S.E.M.). * P<0.005, ** P<0.01, *** P<0.05 compared with saline control. (C) Scatter graphs indicate percentage of TUNEL-positive neurons for each animal within each treatment group. Each dot represents one animal. Blue=saline, red=METH 10 mg/kg, green=METH 20 mg/kg, purple=METH 30 mg/kg, yellow=METH 40 mg/kg; n=10–11 per experimental group.
To determine whether METH induced apoptosis occurs selectively in neurons of the striatum instead of glial cells, we combined TUNEL with immunohistofluorescence for NeuN. This marker was chosen as opposed to other neuronal markers because the labeling of this marker occurs primarily in the nucleus and TUNEL-labeling is also a nuclear stain. Immunohistofluorescence showed that TUNEL-positive nuclei colocalized with NeuN (Fig. 4g–i). METH treatment caused NeuN staining to be less pronounced (Fig. 4d), probably an indication that these neurons have already incurred severe deficits in molecular markers such as receptors and peptides. We did not assess TUNEL with glial-specific stains because all the TUNEL staining colocalized with immunohistofluorescence for NeuN.
Fig. 4.

METH induces apoptosis in some striatal neurons of the striatum. Double-labeled epifluorescent micrographs of striatal tissue stained with Cy3-labeled antibodies against NeuN and TUNEL with FITC-conjugated dUTPs in control (a–c) and METH-treated (30 mg/kg, i.p., killed 24 h post-treatment) animals (d–i). Bottom panels (c, f, i) are overlays of both TUNEL and NeuN staining. Higher magnification of METH-treated animals indicates that NeuN-positive neurons overlap with TUNEL-positive cells (g–i). White arrows point to overlapping TUNEL and NeuN positive cells. Scale bar=100 μm (a–f), 20 μm (g–i).
Effects of METH on the different neuronal populations of the striatum
GABAergic projection neurons
We tried to combine TUNEL with immunohistofluorescence for various selective markers of the striatonigral and striatopallidal projection neurons; however, the results were unsatisfactory. For example, we attempted to colocalize TUNEL with immunohistofluorescence for dopamine D1 or D2 receptors and the neuropeptides substance P, dynorphin, and enkephalin (data not shown). Additionally, we injected a fluorescent retrograde tracer into the terminal field areas of striatopallidal (globus pallidum) and striatonigral (substantia nigra pars reticulata) projections but this approach was also unsuccessful (data not shown). We believe that all these attempts were unsuccessful because by the time some striatal neurons displayed METH-induced TUNEL staining (extensive nicking of the nuclear DNA), the levels of molecular markers such as receptors and neuropeptides reached levels below the limit of detection of immunohistochemistry with the commercial antibodies that we employed. In the light of these technical constraints, we decided to demonstrate the apoptosis of projection neurons by an indirect method using immunohistofluorescence for DARPP-32, a reliable marker for both striatopallidal and striatonigral projection neurons.
We did not observe colocalization of DARPP-32 with TUNEL-positive cells (Fig. 5A g–i). However, immunoreactivity of DARPP-32 showed significant decreases in the METH-treated group compared with control (Fig. 5A a–f). Closer observation showed that in a given area where there was TUNEL staining, it was very difficult to also find DARPP-32 staining (Fig. 5A d–f). This was dramatically different in controls where there was abundant DARPP-32 staining and no TUNEL (Fig. 5A a–c). Cell counts of DARPP-32-positive neurons showed that METH caused a 15.4%±2.5% decrease of DARPP-32-positive neurons in the DM region, 23.8%±3.8% in the VM region, 22.8%±3.8% in the VL region and 24.4%±3.9% in the DL region compared with controls (Fig. 5B). This decrease corresponded with the increases in percentage of TUNEL-positive neurons in Fig. 3B. The immunohistological approach used here demonstrated that the loss of DARPP-32 correlated with the increases in TUNEL-positive cells, consistent with the interpretation that METH induces the loss of some projection neurons of the striatum.
Fig. 5.

The number of DARPP-32-containing projection neurons is decreased by METH. Mice received one injection of METH (i.p.) at 30 mg/kg of body weight and were killed 24 h later. Sections of striatal tissue were processed for immunohistofluorescence. (A) Double-labeled epifluorescent micrographs of striatal tissue stained with Cy3-labeled antibodies against DARPP-32 and TUNEL with FITC-conjugated dUTPs in control (a–c) and METH-treated animals (d–i). Bottom panels (c, f, I, l) are overlays of both TUNEL and DARPP-32 staining. Higher magnification of METH-treated animals indicates that the two chromophores do not overlap (g–i). Scale bar=100 μm (a–f), 20 μm (g–i). (B) Counts of DARPP-32-positive neurons (mean±S.E.M.) demonstrate a significant decrease in all four quadrants of the striatum after exposure to METH. * P<0.05 compared with corresponding regions of saline control. CPu, caudate-putamen.
Parvalbumin-containing GABA interneurons
We did not observe colocalization of immunohistofluorescence of parvalbumin-containing GABAergic interneurons and TUNEL-positive neurons (Fig. 6A j–l). Despite this, immunoreactivity of parvalbumin-positive neurons was diminished (Fig. 6A d–f) and dendritic arbors appeared shorter (Fig. 6A j–l) in METH-treated animals compared with controls (Fig. 6A a–c, g–i). To detect the potential loss of some of these interneurons after exposure to METH, cell counts of parvalbumin-positive neurons were performed in the four quadrants of the striatum. Although all four quadrants showed a decrease, it was the dorsal region of the striatum where this GABAergic interneuron was concentrated and most affected by METH treatment; causing a 43.7%±5.5% decrease of the marker in the DM region and 49%±10.8% in the DL region (Fig. 6B).
Fig. 6.
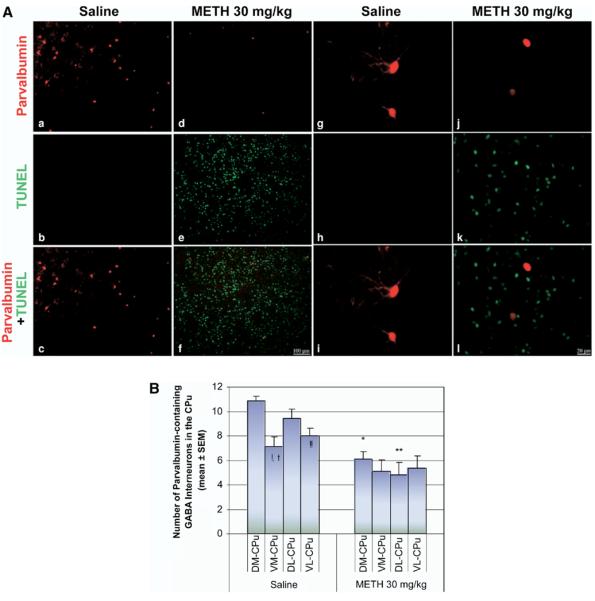
Immunohistofluorescence of parvalbumin interneurons is diminished by METH. Mice received a single injection of METH (i.p.) at a dose of 30 mg/kg and were killed 24 h after the treatment. Sections of striatal tissue were processed for immunohistofluorescence. (A) Double-labeled epifluorescent micrographs of striatal tissue stained with Cy3-labeled antibodies against parvalbumin and TUNEL with FITC-conjugated dUTPs in control (a–c) and METH-treated animals (d–f). Bottom panels (c, f, i, l) are overlays of both TUNEL and parvalbumin staining. Higher magnification of tissue from METH-treated animals demonstrated that the two chromophores do not overlap (g–i), although cell bodies showed significant loss of dendritic arborizations (j–l). Scale bar=100 μm (a–f), 20 μm (g–l). (B) Counts of parvalbumin-positive neurons (mean±S.E.M.) demonstrate a significant decrease of immunohistofluorescence in the dorsal regions of the striatum. * P<0.0001, ** P<0.005, *** P<0.05 compared with corresponding regions of saline control. ! P<0.0001, !! P<0.001 compared with the DM region of saline. † P<0.005 compared with the DL region of saline. CPu, caudate-putamen.
Cholinergic and SST interneurons
Colocalization of immunohistofluorescence between ChAT-containing cholinergic interneurons and TUNEL-positive neurons was not observed throughout the striatum. No obvious change was seen from the histology in the number and morphology of the cholinergic interneurons after METH treatment compared with control. Cell counts of the cholinergic interneurons confirmed these neurons to be unaffected in the ventral portions of the striatum. However, the dorsal regions of the striatum showed loss of this cell type. This is most significant for the DM portion of the striatum, which had a 28.9%±4.8% decrease from control (Fig. 7B). It should be noted that these deficits were observed in the animals that displayed high amounts of TUNEL.
Fig. 7.

Some cholinergic interneurons are vulnerable to METH. Animals were injected once with METH (i.p., 30 mg/kg) and killed 24 h after METH. Coronal sections through the striatum were processed for immunohistofluorescence. (A) Double-labeled epifluorescent micrographs of striatal tissue stained with Cy3-labeled antibodies against ChAT and TUNEL with FITC-conjugated dUTPs in control (a–c) and METH-treated mice (d–i). Bottom panels (c, f, i) are overlays of both TUNEL and ChAT staining. Higher magnification of METH-treated group demonstrates the lack of colocalization between the two chromophores (g–i). Scale bar=100 μm (a–f), 20 μm (g–i). (B) Counts of ChAT-positive neurons (mean±S.E.M.) demonstrate a significant decrease in the DM region of the striatum. * P<0.005 compared with the corresponding regions of saline control. ! P<0.0001, † P<0.005 compared with the DL region of saline. !! P<0.0005 compared with DM region of saline. # P<0.05 compared with the DM of METH. % P<0.05 compared with the DL region of METH. CPu, caudate-putamen.
No colocalization of immunohistofluorescence was found between SST and TUNEL-positive neurons (Fig. 8A). Like the cholinergic interneurons, there is no obvious morphological change observable throughout the striatum. Cell counts in the four quadrants of the striatum did not reveal the loss of immunohistofluorescence for SST (Fig. 8B), suggesting that this population of striatal interneuron is refractory to METH.
Fig. 8.

SST interneurons are refractory to METH-induced apoptosis. (A) Double-labeled epifluorescent micrographs of striatal tissue stained with antibody against SST (Cy3) and TUNEL with FITC-conjugated dUTPs in control (a–c) and METH-treated (30 mg/kg, i.p., killed 24 h post-treatment) mice (d–i). Bottom panels (c, f, i) are overlays of both TUNEL and SST staining. Higher magnification of METH-treated mice shows absence of overlap between the two chromophores (g–i). Scale bar=100 μm (a–f), 20 μm (g–i). (B) Counts of SST-positive neurons (mean±S.E.M.) demonstrate no statistical significance between the two treatment groups or between the regions within the treatment groups. CPu, caudate putamen.
Effect of METH on Nissl staining of striatal tissue
Because we did not observe colocalization between TUNEL and selective markers of striatal neurons, we stained striatal sections from control and METH-treated animals with Cresyl Violet. We found a significant decrease in the number of cells stained with Cresyl Violet in the METH-treated group (Fig. 9A and B). The METH-treated animals showed cells that appeared to have fragmented and clumped nuclei (Fig. 9A). The magnitude of cell loss observed with the Nissl stain correlated with the amount of TUNEL staining (compare Figs. 3B and 9B). Moreover, in all sections examined, a high density of TUNEL always appeared with a paucity of DARPP-32 staining. These observations demonstrate the loss of some striatal neurons in the aftermath of METH.
Fig. 9.

Effect of METH treatment on the cell density of the striatum. Mice were treated with a single METH 30 mg/kg (i.p.) injection and killed 24 h later. Coronal sections of the striatum were stained with Cresyl Violet. Light micrographs of Nissl staining in saline (A) and METH 30 mg/kg (B) treated animals. (C) Nissl-stained cells were counted in 20 μm thick coronal sections of the striatum in an area of 0.26 mm2 for each of the four quadrants of the caudate-putamen (CPu, DM, DL, VM, and VL). Results represent the mean±S.E.M. number of cells counted in 10 animals per experimental group. * P<0.05 compared with controls of each corresponding striatal region. No significance was found between regions.
DISCUSSION
None of the selected striatal markers for medium spiny neurons (DARPP-32) and interneurons (ChAT, SST and parvalbumin) exhibited colocalization with the TUNEL-positive cells. This would initially suggest that these neuronal cells were not undergoing apoptosis. But the reduction in population size for these cells is observed only in animals showing TUNEL-positive cells, suggesting that some striatal neurons die by apoptosis after METH. Furthermore, the magnitude of cell loss observed with Nissl-stained sections correlated with the amount of cell loss observed with TUNEL staining. Labeling by TUNEL marks the occurrence of DNA fragmentation, which occurs late in the apoptosis process. Thus, damage to neuronal markers may have occurred before DNA fragmentation. The agreement between TUNEL and the loss of Nissl staining in striatal tissue of animals treated with METH demonstrates that some neurons are lost in the aftermath of METH.
METH is a potent psychostimulant that induces excessively high levels of dopamine in the extracellular compartment. Excess release of endogenous striatal dopamine has been hypothesized to mediate neurotoxicity at dopaminergic terminals and cause neuronal damage to some striatal neurons (Davidson et al., 2001). Metabolites of dopamine generate toxic free radicals, which contribute to this damage (Cadet and Brannock, 1998). The connection between METH-induced dopamine terminal toxicity and elevated dopamine levels was established using pharmacological agents that blocked post-synaptic dopamine receptors or pre-synaptic dopamine transporters, such agents were found to protect from METH (Buening and Gibb, 1974; Gibb and Kogan, 1979; Hotchkiss and Gibb, 1980; Sonsalla et al., 1986; O'Dell et al., 1993; Xu et al., 2005). Microinjections of the dopamine agonist, SKF38393, administered centrally into the rat brain are toxic to striatal neurons (Kelley et al., 1990). Additionally, we have shown that blockade of dopamine receptors using the D1 receptor antagonist, SCH23390, or D2 receptor antagonist, raclopride, prior to METH administration can prevent apoptosis (Xu et al., 2005). Since excess dopamine is neurotoxic, the local anatomical organization of dopamine terminals and receptors plays an important role in where damage occurs following METH administration. Given that relatively high levels of dopamine receptors are located on the medium spiny neurons, we can thus speculate that these neurons would be more vulnerable to the deleterious effects of METH administration. Another type of neurotransmission that could account for the differential vulnerability of striatal neurons to METH involves the neurotransmitter glutamate.
Dopaminergic and glutamatergic systems display complex anatomical arrangements in the striatum. Microdialysis studies show that METH induces a delayed release of glutamate in the striatum (Nash and Yamamoto, 1992). Agents that block this delayed increase of extracellular glutamate can prevent METH-induced depletion of dopamine content (Stephans and Yamamoto, 1994). Glutamate analogues such as quinolinic acid and kainic acid have been shown to induce excitotoxic lesions of striatal neurons (Beal et al., 1991; Qin et al., 1996). Excitotoxicity is mediated by ionotrophic NMDA and AMPA/KA receptors (Beal, 1992; Nakanishi, 1992). Excessive activation of glutamate receptors is thought to contribute to neuronal death in neurological disorders such as trauma, epilepsy, Parkinson's disease, and Alzheimer's disease (Beal, 1992; Marino et al., 2003; Arundine and Tymianski, 2004; Hynd et al., 2004). In addition, activation of the NMDA receptor in the striatum leads to the activation of nitric oxide synthase (Szabo, 1996), which results in the synthesis of the diffusible second messenger nitric oxide, an agent that has been associated with damage of surrounding neurons in the brain (Dawson et al., 1991; Dawson and Snyder, 1994). Given the reciprocal interactions between dopamine and glutamate, the selective vulnerability of some striatal neuron subtypes may be due to the anatomical differences in the expression of glutamate receptor subtypes and subunits. Studies indicate that the majority of glutamate receptors are found on medium spiny neurons. These neurons contain the NMDA NR1 and NR2A/2b subunits in the striatum as well as AMPA GluR1, GluR2/3, and GluR4 subunits (Chen and Reiner, 1996; Chen et al., 1998; Hu et al., 2004; Wang et al., 2004). Cholinergic interneurons and half of the GABAergic parvalbumin-containing interneurons express NMDA NR2A/2B R while SST interneurons do not (Landwehrmeyer et al., 1995; Chen and Reiner, 1996; Chen et al., 1996). The different responses to glutamate receptor activation are reflected in the localization of these receptors within the striatum. Given that SST interneurons lack the receptor, it may account for the resistance to NMDA-mediated excitotoxicity on these neurons. Furthermore, damage to medium spiny neurons and parvalbumin interneurons may be due to the presence of these receptors.
The various populations of striatal neurons are intricately interconnected affecting each other within the striatum and at the targets of innervation of the medium spiny projection neurons. Thus, it is reasonable to assume that the loss of some projection and interneurons after METH may have an impact on the surviving neighboring neurons. The projection neurons exert inhibition on their targets in the substantia nigra and the globus pallidum via GABAA receptors (Precht and Yoshida, 1971). In addition, the GABAergic medium spiny neurons can inhibit each other via their recurrent collaterals within the striatum (Wilson and Groves, 1980; Yung et al., 1996). In the light of this, the loss of some projection neurons after METH could be interpreted to result in attenuation of inhibition within the striatum and in the targets of the projection neurons. However, the state of the projection neurons of the striatum is affected by the activity of the interneurons. Our results demonstrate that some cholinergic and GABA-parvalbumin interneurons may also be killed by acute METH.
Our data suggest that approximately half of the GABA-parvalbumin interneurons of the dorsal striatum are killed by METH. These interneurons have a high content of GABA (Bolam et al., 1983) and receive glutamatergic excitation from the cortex (Koos and Tepper, 1999). Thus, in the aftermath of cortical transmission these interneurons exert inhibition on striatal neurons via GABA, and it is noteworthy that METH ablates nearly half of this population of interneuron in the dorsal striatum. We demonstrate here that another population of striatal interneuron impacted by METH is the cholinergic interneuron. These interneurons affect the output of the projection neurons. For example, cholinergic transmission augments striatal dopamine release (Zhou et al., 2001) and increases the responses of the medium spiny projection neurons to glutamate (Calabresi et al., 1998). The present study demonstrates that in the post-METH striatum the interneuron balance is skewed in favor of transmission by the SST/nitric oxide synthase interneurons because the latter population is refractory to METH. The nitric oxide made by the SST/nitric oxide synthase interneurons is known to increase the release of glutamate and dopamine in the striatum (Kawaguchi et al., 1995). The striatal SST interneurons also express the neuropeptide NPY (Kawaguchi et al., 1995). These neuropeptides may play a neuroprotective role in the striatum. For example, SST significantly attenuated ischemic neuronal damage induced by middle cerebral artery occlusion in the rat brain (Rauca et al., 1999). Similarly, SST attenuated NMDA-induced cell death in cultured fetal rat cortical neurons (Forloni et al., 1997). In addition, a recent study showed that the intraventricular administration of the neuropeptide NPY protected the mouse striatum from METH-induced apoptosis (Thiriet et al., 2005). These studies suggest that SST and NPY belong to a class of neuroprotective peptides in the brain. It needs to be investigated how these neuropeptides confer protection on the neuron expressing them, since our results demonstrate that this type of interneuron is resistant to METH-induced damage.
CONCLUSION
In summary, our results demonstrate the loss of some striatal neurons in the aftermath of METH. This conclusion is supported by the appearance of TUNEL staining in the striatum, the loss of immunostaining for selective markers of striatal neurons, and decreased density of Nissl staining after METH. The affected phenotypes are: projection neurons, cholinergic and GABA-parvalbumin interneurons. Of note is the observation that the SST/NPY interneurons are refractory to METH. More work is needed to investigate the functional state, namely the output, of the striatal neurons that survive the ravages of METH.
Acknowledgments
We thank Dr. Jenny Zhu and Gertrude Rivera for editorial assistance in preparation of this manuscript. This work was supported by ‘Specialized Neuroscience Research Program’ grant NS41073 from the National Institute for Neurological Disorders and Stroke JAA. Support has also come from the ‘Research Centers in Minority Institutions’ award to Hunter College for infrastructure.
Abbreviations
- ChAT
choline acetyltransferase
- DARPP-32
dopamine- and cAMP-regulated phosphoprotein, of apparent Mr 32,000
- DL
dorsal–lateral
- DM
dorsal–medial
- METH
methamphetamine
- NADPH
nicotinamide adenine dinucleotide phosphate
- NeuN
neuron-specific nuclear protein
- PBS
phosphate-buffered saline
- SST
somatostatin
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
- VL
ventral–lateral
- VM
ventral–medial
REFERENCES
- Albin RL, Reiner A, Anderson KD, Penney JB, Young AB. Striatal and nigral neuron subpopulations in rigid Huntington's disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann Neurol. 1990;27:357–365. doi: 10.1002/ana.410270403. [DOI] [PubMed] [Google Scholar]
- Albin RL, Reiner A, Anderson KD, Dure LS, Handelin B, Balfour R, Whetsell WO, Jr, Penney JB, Young AB. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol. 1992;31:425–430. doi: 10.1002/ana.410310412. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Mechanisms of excitotoxicity in neurologic diseases. FASEB J. 1992;6:3338–3344. [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington's disease. J Neurosci. 1991;11:1649–1659. doi: 10.1523/JNEUROSCI.11-06-01649.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Clarke DJ, Smith AD, Somogyi P. A type of aspiny neuron in the rat neostriatum accumulates [3H]gamma-aminobutyric acid: combination of Golgi-staining, autoradiography, and electron microscopy. J Comp Neurol. 1983;213:121–134. doi: 10.1002/cne.902130202. [DOI] [PubMed] [Google Scholar]
- Buening MK, Gibb JW. Influence of methamphetamine and neuroleptic drugs on tyrosine hydroxylase activity. Eur J Pharmacol. 1974;26:30–34. doi: 10.1016/0014-2999(74)90070-3. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Brannock C. Free radicals and the pathobiology of brain dopamine systems. Neurochem Int. 1998;32:117–131. doi: 10.1016/s0197-0186(97)00031-4. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Jayanthi S, Deng X. Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB J. 2003;17:1775–1788. doi: 10.1096/fj.03-0073rev. [DOI] [PubMed] [Google Scholar]
- Calabresi P, De Murtas M, Bernardi G. The neostriatum beyond the motor function: experimental and clinical evidence. Neuroscience. 1997;78:39–60. doi: 10.1016/s0306-4522(96)00556-8. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Endogenous ACh enhances striatal NMDA-responses via M1-like muscarinic receptors and PKC activation. Eur J Neurosci. 1998;10:2887–2895. doi: 10.1111/j.1460-9568.1998.00294.x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Reiner A. Cellular distribution of the NMDA receptor NR2A/2B subunits in the rat striatum. Brain Res. 1996;743:346–352. doi: 10.1016/s0006-8993(96)01098-0. [DOI] [PubMed] [Google Scholar]
- Chen Q, Veenman CL, Reiner A. Cellular expression of ionotropic glutamate receptor subunits on specific striatal neuron types and its implication for striatal vulnerability in glutamate receptor-mediated excitotoxicity. Neuroscience. 1996;73:715–731. doi: 10.1016/0306-4522(96)00011-5. [DOI] [PubMed] [Google Scholar]
- Chen Q, Veenman L, Knopp K, Yan Z, Medina L, Song WJ, Surmeier DJ, Reiner A. Evidence for the preferential localization of glutamate receptor-1 subunits of AMPA receptors to the dendritic spines of medium spiny neurons in rat striatum. Neuroscience. 1998;83:749–761. doi: 10.1016/s0306-4522(97)00452-1. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Gould PV, Parent A. Sparing of striatal neurons coexpressing calretinin and substance P (NK1) receptor in Huntington's disease. Brain Res. 1996;730:232–237. doi: 10.1016/0006-8993(96)00307-1. [DOI] [PubMed] [Google Scholar]
- Davidson C, Gow AJ, Lee TH, Ellinwood EH. Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev. 2001;36:1–22. doi: 10.1016/s0165-0173(01)00054-6. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Snyder SH. Gases as biological messengers: nitric oxide and carbon monoxide in the brain. J Neurosci. 1994;14:5147–5159. doi: 10.1523/JNEUROSCI.14-09-05147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Cadet JL. Methamphetamine-induced apoptosis is attenuated in the striata of copper-zinc superoxide dismutase transgenic mice. Brain Res Mol Brain Res. 2000;83:121–124. doi: 10.1016/s0169-328x(00)00169-8. [DOI] [PubMed] [Google Scholar]
- Deng X, Wang Y, Chou J, Cadet JL. Methamphetamine causes widespread apoptosis in the mouse brain: evidence from using an improved TUNEL histochemical method. Brain Res Mol Brain Res. 2001;93:64–69. doi: 10.1016/s0169-328x(01)00184-x. [DOI] [PubMed] [Google Scholar]
- Deng YP, Albin RL, Penney JB, Young AB, Anderson KD, Reiner A. Differential loss of striatal projection systems in Huntington's disease: a quantitative immunohistochemical study. J Chem Neuroanat. 2004;27:143–164. doi: 10.1016/j.jchemneu.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Marshall JF. Methamphetamine neurotoxicity: dissociation of striatal dopamine terminal damage from parietal cortical cell body injury. Synapse. 1998;30:433–445. doi: 10.1002/(SICI)1098-2396(199812)30:4<433::AID-SYN10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington's disease. Science. 1985;230:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Friguls B, Dalfo E, Justicia C, Planas AM. Caspase-dependent and caspase-independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathol Appl Neurobiol. 2003;29:472–481. doi: 10.1046/j.1365-2990.2003.00485.x. [DOI] [PubMed] [Google Scholar]
- Forloni G, Lucca E, Angeretti N, Chiesa R, Vezzani A. Neuroprotective effect of somatostatin on nonapoptotic NMDA-induced neuronal death: Role of cyclic GMP. J Neurochem. 1997;68:319–327. doi: 10.1046/j.1471-4159.1997.68010319.x. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- Gibb JW, Kogan FJ. Influence of dopamine synthesis on methamphetamine-induced changes in striatal and adrenal tyrosine hydroxylase activity. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:185–187. doi: 10.1007/BF00500283. [DOI] [PubMed] [Google Scholar]
- Guyot MC, Hantraye P, Dolan R, Palfi S, Maziere M, Brouillet E. Quantifiable bradykinesia, gait abnormalities and Huntington's disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience. 1997;79:45–56. doi: 10.1016/s0306-4522(96)00602-1. [DOI] [PubMed] [Google Scholar]
- Hanson GR, Rau KS, Fleckenstein AE. The methamphetamine experience: a NIDA partnership. Neuropharmacology. 2004;47(Suppl 1):92–100. doi: 10.1016/j.neuropharm.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Harvey DC, Lacan G, Melegan WP. Regional heterogeneity of dopaminergic deficits in vervet monkey striatum and substantia nigra after methamphetamine exposure. Exp Brain Res. 2000;133:349–358. doi: 10.1007/s002210000386. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Chesselet MF. Apoptosis in Huntington's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:255–265. doi: 10.1016/S0278-5846(03)00021-6. [DOI] [PubMed] [Google Scholar]
- Hof P, Young W, Bloom F, Belichenko P, Celio MR. Comparative cytoarchitectonic atlas of the C57BL/6 and 129/Sv mouse brains. Elsevier; New York: 2000. [Google Scholar]
- Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- Hu HJ, Chen LW, Yung KK, Chan YS. Differential expression of AMPA receptor subunits in substance P receptor-containing neurons of the caudate-putamen of rats. Neurosci Res. 2004;49:281–288. doi: 10.1016/j.neures.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int. 2004;45:583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Imam SZ, Crow JP, Newport GD, Islam F, Slikker W, Jr, Ali SF. Methamphetamine generates peroxynitrite and produces dopaminergic neurotoxicity in mice: protective effects of peroxynitrite decomposition catalyst. Brain Res. 1999;837:15–21. doi: 10.1016/s0006-8993(99)01663-7. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kelley AE, Delfs JM, Chu B. Neurotoxicity induced by the D-1 agonist SKF 38393 following microinjection into rat brain. Brain Res. 1990;532:342–346. doi: 10.1016/0006-8993(90)91781-b. [DOI] [PubMed] [Google Scholar]
- Koos T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Landwehrmeyer GB, Standaert DG, Testa CM, Penney JB, Jr, Young AB. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci. 1995;15:5297–5307. doi: 10.1523/JNEUROSCI.15-07-05297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang NY, Rutledge CO. Comparison of the release of [3H]dopamine from isolated corpus striatum by amphetamine, fenfluramine and unlabelled dopamine. Biochem Pharmacol. 1982a;31:983–992. doi: 10.1016/0006-2952(82)90332-x. [DOI] [PubMed] [Google Scholar]
- Liang NY, Rutledge CO. Evidence for carrier-mediated efflux of dopamine from corpus striatum. Biochem Pharmacol. 1982b;31:2479–2484. doi: 10.1016/0006-2952(82)90057-0. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Loonam TM, Noailles PA, Yu J, Zhu JP, Angulo JA. Substance P and cholecystokinin regulate neurochemical responses to cocaine and methamphetamine in the striatum. Life Sci. 2003;73:727–739. doi: 10.1016/s0024-3205(03)00393-x. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Valenti O, Conn PJ. Glutamate receptors and Parkinson's disease: opportunities for intervention. Drugs Aging. 2003;20:377–397. doi: 10.2165/00002512-200320050-00006. [DOI] [PubMed] [Google Scholar]
- McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci. 1998;18:8417–8422. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann UD, Ricaurte GA. Amphetamine neurotoxicity: accomplishments and remaining challenges. Neurosci Biobehav Rev. 2004;27:821–826. doi: 10.1016/j.neubiorev.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Mitchell IJ, Cooper AJ, Griffiths MR. The selective vulnerability of striatopallidal neurons. Prog Neurobiol. 1999;59:691–719. doi: 10.1016/s0301-0082(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Nash JF, Yamamoto BK. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992;581:237–243. doi: 10.1016/0006-8993(92)90713-j. [DOI] [PubMed] [Google Scholar]
- O'Dell SJ, Weihmuller FB, Marshall JF. Methamphetamine-induced dopamine overflow and injury to striatal dopamine terminals: attenuation by dopamine D1 or D2 antagonists. J Neurochem. 1993;60:1792–1799. doi: 10.1111/j.1471-4159.1993.tb13405.x. [DOI] [PubMed] [Google Scholar]
- Precht W, Yoshida M. Blockage of caudate-evoked inhibition of neurons in the substantia nigra by picrotoxin. Brain Res. 1971;32:229–233. doi: 10.1016/0006-8993(71)90171-5. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Chase TN. Stimulation of N-methyl-D-aspartate receptors induces apoptosis in rat brain. Brain Res. 1996;725:166–176. doi: 10.1016/0006-8993(96)00200-4. [DOI] [PubMed] [Google Scholar]
- Raiteri M, Cerrito F, Cervoni AM, Levi G. Dopamine can be released by two mechanisms differentially affected by the dopamine transport inhibitor nomifensine. J Pharmacol Exp Ther. 1979;208:195–202. [PubMed] [Google Scholar]
- Rauca C, Schafer K, Hollt V. Effects of somatostatin, octreotide and cortistatin on ischaemic neuronal damage following permanent middle cerebral artery occlusion in the rat. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:633–638. doi: 10.1007/s002109900136. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Maguire-Zeiss KA, Vonkeman HE, Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington's disease patients. Ann Neurol. 1995;38:852–861. doi: 10.1002/ana.410380605. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Vonsattel JP, MacDonald ME, Sun Z, Deng YP, Reiner A. Selective loss of striatal preprotachykinin neurons in a phenocopy of Huntington's disease. Mov Disord. 2002;17:327–332. doi: 10.1002/mds.10032. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- Seiden LS, Fischman MW, Schuster CR. Long-term methamphetamine induced changes in brain catecholamines in tolerant rhesus monkeys. Drug Alcohol Depend. 1976;1:215–219. doi: 10.1016/0376-8716(76)90030-2. [DOI] [PubMed] [Google Scholar]
- Singer HS, Minzer K. Neurobiology of Tourette's syndrome: concepts of neuroanatomic localization and neurochemical abnormalities. Brain Dev. 2003;25(Suppl 1):S70–S84. doi: 10.1016/s0387-7604(03)90012-x. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Gibb JW, Hanson GR. Roles of D1 and D2 dopamine receptor subtypes in mediating the methamphetamine-induced changes in monoamine systems. J Pharmacol Exp Ther. 1986;238:932–937. [PubMed] [Google Scholar]
- Spalding KL, Dharmarajan AM, Harvey AR. Caspase-independent retinal ganglion cell death after target ablation in the neonatal rat. Eur J Neurosci. 2005;21:33–45. doi: 10.1111/j.1460-9568.2004.03826.x. [DOI] [PubMed] [Google Scholar]
- Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- Stumm G, Schlegel J, Schafer T, Wurz C, Mennel HD, Krieg JC, Vedder H. Amphetamines induce apoptosis and regulation of bcl-x splice variants in neocortical neurons. FASEB J. 1999;13:1065–1072. doi: 10.1096/fasebj.13.9.1065. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Physiological and pathophysiological roles of nitric oxide in the central nervous system. Brain Res Bull. 1996;41:131–141. doi: 10.1016/0361-9230(96)00159-1. [DOI] [PubMed] [Google Scholar]
- Thiriet N, Deng X, Solinas M, Ladenheim B, Curtis W, Goldberg SR, Palmiter RD, Cadet JL. Neuropeptide Y protects against methamphetamine-induced neuronal apoptosis in the mouse striatum. J Neurosci. 2005;25:5273–5279. doi: 10.1523/JNEUROSCI.4893-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, Simon SL, Geaga JA, Hong MS, Sui Y, Lee JY, Toga AW, Ling W, London ED. Structural abnormalities in the brains of human subjects who use methamphetamine. J Neurosci. 2004;24:6028–6036. doi: 10.1523/JNEUROSCI.0713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura Y, Kowall NW, Beal MF. Selective sparing of NADPH-diaphorase-somatostatin-neuropeptide Y neurons in ischemic gerbil striatum. Ann Neurol. 1990;27:620–625. doi: 10.1002/ana.410270606. [DOI] [PubMed] [Google Scholar]
- Villemagne V, Yuan J, Wong DF, Dannals RF, Hatzidimitriou G, Mathews WB, Ravert HT, Musachio J, McCann UD, Ricaurte GA. Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable to those recreationally abused by humans: evidence from [11C]WIN-35,428 positron emission tomography studies and direct in vitro determinations. J Neurosci. 1998;18:419–427. doi: 10.1523/JNEUROSCI.18-01-00419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- Wang WW, Cao R, Rao ZR, Chen LW. Differential expression of NMDA and AMPA receptor subunits in DARPP-32-containing neurons of the cerebral cortex, hippocampus and neostriatum of rats. Brain Res. 2004;998:174–183. doi: 10.1016/j.brainres.2003.11.034. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Groves PM. Fine structure and synaptic connections of the common spiny neuron of the rat neostriatum: a study employing intracellular inject of horseradish peroxidase. J Comp Neurol. 1980;194:599–615. doi: 10.1002/cne.901940308. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- Xu W, Zhu JPQ, Angulo JA. Induction of striatal pre- and postsynaptic damage by methamphetamine requires the dopamine receptors. Synapse. 2005;58:110–121. doi: 10.1002/syn.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Cadet JL, Angulo JA. Neurokinin-1 (NK-1) receptor antagonists abrogate methamphetamine-induced striatal dopaminergic neurotoxicity in the murine brain. J Neurochem. 2002;83:613–622. doi: 10.1046/j.1471-4159.2002.01155.x. [DOI] [PubMed] [Google Scholar]
- Yu J, Wang J, Cadet JL, Angulo JA. Histological evidence supporting a role for the striatal neurokinin-1 receptor in methamphetamine-induced neurotoxicity in the mouse brain. Brain Res. 2004;1007:124–131. doi: 10.1016/j.brainres.2004.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung KK, Smith AD, Levey AI, Bolam JP. Synaptic connections between spiny neurons of the direct and indirect pathways in the neostriatum of the rat: evidence from dopamine receptor and neuropeptide immunostaining. Eur J Neurosci. 1996;8:861–869. doi: 10.1111/j.1460-9568.1996.tb01573.x. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- Zhu JPQ, Cadet JL, Angulo JA. Blockade of neurokinin-1 receptors attenuates methamphetamine-induced TUNEL staining. Soc Neurosci Abstr. 2002;28:701–709. [Google Scholar]
- Zhu JPQ, Xu W, Angulo JA. Disparity in the temporal appearance of methamphetamine-induced apoptosis and depletion of dopamine terminal markers in the striatum of mice. Brain Res. 2005;1049:171–181. doi: 10.1016/j.brainres.2005.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JPQ, Xu W, Angulo N, Angulo JA. Methamphetamine-induced striatal apoptosis in the mouse brain: Comparison of a binge to an acute bolus drug administration. Neurotoxicology. 2006;27:131–136. doi: 10.1016/j.neuro.2005.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]