

Abstract
Background:
A pivotal developmental question is whether tubers in tuberous sclerosis complex (TSC) form by germline and somatic TSC1 or TSC2 gene mutations. Loss of TSC1 or TSC2 in vitro and in vivo leads to mTORC1 cascade activation and ribosomal protein S6 phosphorylation (P-S6). Giant cells (GCs) in tubers exhibit S6 phosphorylation, suggesting cell-specific loss of TSC gene function.
Methods:
TSC1 and TSC2 gene mutations were investigated in DNA extracted from tuber sections (n = 6) and microdissected P-S6–labeled GCs by sequencing and loss of heterozygosity (LOH) analysis to define germline and somatic mutations.
Results:
A germline TSC1 mutation was defined in 1 case and TSC2 mutations were defined in 5 cases. LOH was not detected in whole tuber sections or microdissected P-S6–labeled GCs. TSC1 and TSC2 were sequenced in microdissected P-S6–immunolabeled GCs. In 5 specimens, a somatic mutation was identified in single GCs that was not detected in whole tuber sections or leukocyte DNA. Four somatic mutations were novel variants (1 nonsense and 3 missense mutations) and 1 additional nonsense somatic mutation was previously reported as a germline mutation. In 1 case, no somatic mutation was identified. There was reduced expression of TSC1 or TSC2 transcripts in the TSC1 or TSC2 associated specimens. In the cases containing a nonsense mutation, no transcript mRNA was detected, suggesting nonsense-mediated degradation.
Conclusions:
We provide evidence to support the hypothesis that tubers form by biallelic TSC1 or TSC2 gene inactivation reflecting a “2-hit” mechanism of germline and somatic mutational events.
GLOSSARY
- AML
= angiomyolipoma;
- DN
= dysplastic neuron;
- FFPE
= formalin fixed, paraffin embedded;
- GC
= giant cell;
- H-E
= hematoxylin and eosin;
- LOH
= loss of heterozygosity;
- TSC
= tuberous sclerosis complex.
Tuberous sclerosis complex (TSC) results from mutations in the TSC1 or TSC2 (TSC2) genes.1,2 Tubers are developmental malformations of the cerebral cortex in TSC that are characterized by disorganized lamination and the presence of a unique cell type, giant cells (GCs), which exhibit cytomegaly.3 Adjacent to GCs are dysplastic neurons (DNs) characterized by a dysmorphic cell body and aberrant dendritic arborizations.
An intensive debate surrounds the molecular mechanisms governing tuber formation during brain development. Loss of heterozygosity (LOH) at either TSC gene locus demonstrated in hamartomas in other organ systems from patients with TSC reflects a biallelic or “2-hit” mutational mechanism in which lesion formation depends on the combined effects of germline and somatic mutations.4–6 Biallelic TSC gene inactivation leads to activation of the mammalian target of rapamycin complex 1 (mTORC1) cascade as evidenced by phosphorylation of p70 S6 kinase1 and ribosomal S6 proteins.7–10 LOH has been demonstrated in TSC subependymal giant cell astrocytomas6,11 but not confirmed in tubers.4,11–13 Variably reduced TSC1 or TSC2 protein expression14,15 and enhanced protein S6 phosphorylation16,17 in tubers suggests that loss of TSC1 or TSC2 function and enhanced mTORC1 signaling likely underlies tuber formation. Thus, while a 2-hit mechanism may be responsible for formation of other TSC lesions, it is not known whether tubers form as a result of biallelic gene inactivation. We hypothesized that tubers form by biallelic gene inactivation and we analyzed the TSC1 and TSC2 genes by LOH assay and sequencing in 6 tubers as a strategy to define germline and somatic mutations.
METHODS
Tubers were obtained from 6 patients (n = 4 male, 2 female; average age at surgery or death 16.7 years) with a clinical diagnosis of TSC.18 In 5 patients, the tubers were resected as part of epilepsy surgery for refractory seizures and the sixth patient died of cardiopulmonary arrest. In all of the patients, germline TSC1 or TSC2 mutations were defined in leukocyte DNA as part of clinical evaluation. In 1 additional postmortem specimen (male, age 35 years), only frozen tuber and angiomyolipoma (AML) tissue were available.
Human tissue specimens.
Formalin fixed, paraffin embedded (FFPE) TSC brain specimens were sectioned at 7 μm and mounted on poly-l-lysine–coated coverslips. Histologic examination of the tubers by hematoxylin and eosin (H-E) staining revealed abnormal cortical lamination, numerous GCs, and DNs. Control frontal lobe neocortex was obtained postmortem from 2 patients who died of nonneurologic causes without a history of TSC or epilepsy (1 male, age 22; 1 female, age 26; average postmortem interval to autopsy, 14 hours).
Immunohistochemistry.
All FFPE tissue sections were hydrated through graded ethanols, probed with phospho-ribosomal S6 antibodies (Ser235/236; 1:500 dilution; Cell Signaling, New England Biolabs, Beverly, MA) and visualized using avidin-biotin conjugation (Vectastain ABC Elite; Vector Labs, Burlingame, CA) with 3,3′-diaminobenzidine. Sections used for histologic analysis were dehydrated through graded ethanols and xylenes and coverslip mounted (Permount). Those used for DNA or mRNA extraction were maintained in diethylpyrocarbonate (DEPC)-treated water at 4°C.
Laser microdissection and DNA extraction.
Single P-S6–immunoreactive GCs (n = 100 per case) as well as pyramidal neurons devoid of P-S6 labeling in control frontal cortex (n = 100 per case) were visualized under light microscopy and microdissected with a CellPixIITM laser capture system (Arcturus) or Veritas™ (Westburg) laser capture microdissection and laser cutting systems (see e-Methods on the Neurology® Web site at www.neurology.org).
LOH analysis, PCR, TSC gene sequencing, and TSC mRNA expression.
DNA extracted from cortical tuber sections and single P-S6–labeled GCs was analyzed for LOH and TSC gene sequence (see e-Methods). We carried out fluorescent DNA sequencing using the Big-DyeTM Terminator system (Applied Biosystems) and ABI Prism sequencing. Individual gene sequence for TSC1 and TSC2 was compared with either the Leiden Open Variation TSC database (http://chromium.liacs.nl/LOVD2/TSC/home.php) or NCBI SNP database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp). TSC1 and TSC2 mRNA levels were determined as described previously (see e-Methods).16
Standard protocol approvals, registrations, and patient consents.
All human tissue was obtained in accordance with protocols approved by Academic Medical Center, University of Amsterdam, and the University of Pennsylvania Institutional Review Board and Committee on Human Research. Tissue procurement was deemed exempt from patient consent requirement because all patient data were de-identified prior to receipt of tissue in the laboratory.
RESULTS
Detection of germline mutations in tuber sections and LOH analysis.
We first demonstrated intact TSC1 and TSC2 sequence in genomic DNA extracted from whole sections of control cortex and then from microdissected pyramidal cells (n = 100) to confirm the validity and integrity of our PCR primers and amplification conditions from FFPE brain sections. Analysis of TSC1 and TSC2 in normal cortex revealed intact gene sequence compared with NCBI GenBank (http://www.ncbi.nlm.nih.gov/Genbank/index.html) and the Leiden Open Variation TSC databases. Additionally, the sequencing results from FFPE tissue were reliable for several reasons. First, in both control and TSC associated cases, we identified TSC1 and TSC2 SNPs that were previously entered into the NCBI SNP database demonstrating that well-characterized SNPs could be detected in our samples. Second, TSC1 or TSC2 mutations were not detected in control samples. Third, germline mutations identified in tubers were also found in leukocyte DNA from the same patients, thus confirming the identity of the germline mutation.
TSC1 and TSC2 gene sequences were analyzed in DNA extracted from whole tuber sections (n = 6 TSC cases). Germline mutations were identified in all 6 specimens (TSC1, 1 case, TSC2, 5 cases; table) and among these, 5 mutations had been previously entered into the Leiden Open Variation TSC database as known disease-causing mutations.19–22 The remaining case mutation likely represented a novel mutation variant since this sequence variation was not identified as a SNP in either NCBI or Leiden databases, and it was not identified in any of our control specimens. LOH analysis performed in genomic DNA extracted from the 6 tuber specimens did not reveal loss of heterozygosity at either gene locus. Indeed, as evidence that our LOH assay was valid, in 1 additional case we identified LOH in TSC2 in a renal AML but not a tuber from the same patient (figure 1). These findings supported previous results13 that unlike AMLs, large somatic TSC1 or TSC2 gene deletions do not commonly occur in tubers.
Table Summary of germline (G) and somatic (S) mutations detected by sequencing of DNA extracted from a pooled sample of microdissected P-S6–immunoreactive giant cells in tubers
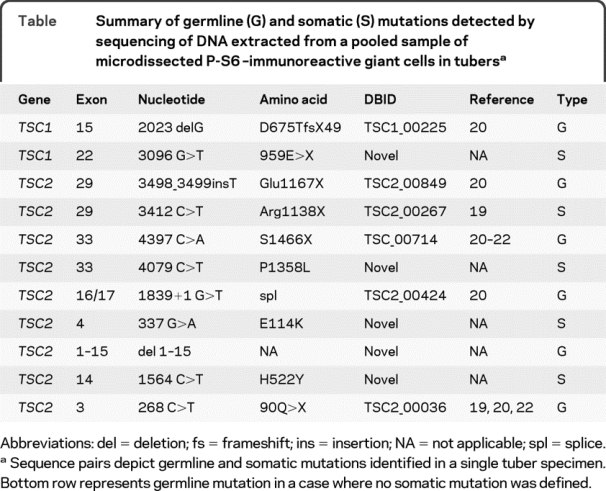

Figure 1 Loss of heterozygosity is not a feature of cortical tubers
(A) Note mutation detected in leukocyte DNA from patient with tuberous sclerosis complex (TSC) (arrow, TSC2 exon 28 delC) compared with control leukocyte DNA sample. Both wild-type and mutated alleles are detected in cortical tuber homogenates but there is loss of the wild-type allele in angiomyolipoma DNA. (B) Hematoxylin and eosin labeled (H&E) and P-S6–labeled giant cells within a tuber and cells in the angiomyolipoma.
S6 protein phosphorylation in tubers.
GCs in tubers express P-S6 protein.16,17 In keeping with these findings, there was robust expression of P-S6 in GCs and large DNs in all tuber specimens (figure 2). Morphologically normal neurons near the edge of the resection margin did not exhibit P-S6 labeling. These cells did express NeuN. There was minimal P-S6 immunoreactivity in control cortex specimens.16,17 In the postmortem TSC case, we analyzed an area without obvious cortical lamination defects (normal perituberal cortex), and there were no P-S6–labeled cells observed.

Figure 2 Cell-selective activation of the mTORC1 pathway in tubers and biallelic TSC1 mutations
(A) P-S6–labeled giant cells (GCs) (arrows) in human tuber specimen. (B) Schematic depicting single P-S6–labeled GCs (arrows, holes) removed by laser capture microdissection. Scale bar 250 μm. (C) Germline and somatic TSC1 mutations in microdissected P-S6–labeled GCs. (a) Germline mutation, exon 15 delG2023 (arrow) in P-S6–labeled GCs. The same mutation was identified in leukocyte DNA from this patient. (b) Exon 22 nonsense mutation (3096G>T, 959E>X) in single P-S6–labeled GCs was not detected in leukocyte DNA from this patient.
Detection of somatic mutations in P-S6–labeled giant cells.
To further investigate whether large deletions did not affect individual cell populations, single P-S6–labeled GCs (n = 100 cells per case) were microdissected from all tuber sections and genomic DNA was extracted. LOH was not detected at either locus in single microdissected cells (data not shown). These findings supported, at the single cell level, the hypothesis that large somatic TSC1 or TSC2 gene deletions do not occur in P-S6–immunoreactive GCs.
We focused on the TSC specimens with confirmed disease-causing germline mutations (n = 5 cases; 4 surgical, 1 postmortem). All exons of TSC1 and TSC2 were sequenced in DNA extracted from the pooled sample of 100 microdissected GCs in each specimen from each case. In one specimen (figure 2), the germline exon 15 TSC1 mutation (exon 15 delG2023) that led to a frameshift and premature stop codon, detected in blood and whole tuber DNA from this patient, was found in all P-S6–labeled GCs (figure 2). This mutation had been previously reported20 in the Leiden Open Variation TSC database (database ID: TSC1_00225; http://chromium.liacs.nl/LOVD2/TSC/home.php). A previously unreported somatic nonsense TSC1 mutation was detected in exon 22 (3096G>T, 959E>X) in DNA extracted from microdissected P-S6–labeled GCs (figure 2) that was not detected in leukocyte DNA nor in DNA extracted from the whole tuber in this patient. Sequencing of TSC2 in P-S6–labeled GCs from this specimen did not reveal mutations.
In another surgical specimen, a previously reported germline TSC2 mutation (exon 29, 3498_ 3499insT, Glu1167X, Leiden database ID: TSC2_ 00849)20 was identified in DNA extracted from a whole tuber section. A nonsense TSC2 mutation (exon 29, 3412C>T, Arg1138X, Leiden database ID: TSC2_00267)18 was detected in single microdissected GCs (figure e-1). This mutation was not detected in leukocyte DNA or total DNA extracted from the whole tubers (figure e-1) and thus likely reflected a somatic mutation. Sequencing of TSC1 in P-S6–labeled GCs from this case did not reveal mutations.
In a third surgical specimen, a previously reported germline TSC2 mutation (exon 33, 4397C>A, 1466Ser>X; Leiden database ID: TSC2_00714)20–22 was identified in DNA extracted from a whole tuber section and in all P-S6–labeled GCs. A new missense TSC2 mutation (exon 33, C>T, 1358P>L), not found as a SNP in either the Leiden or NCBI databases, was detected in single microdissected GCs. This missense mutation was not detected in leukocyte DNA or total DNA extracted from the whole tubers and thus likely reflected a novel somatic mutation. Sequencing of TSC1 in P-S6–labeled GCs from this case did not reveal mutations.
In the postmortem TSC specimen, a previously reported germline TSC2 mutation was identified in the splice site between exons 16 and 17 (c.1839 + 1G>T; Leiden database ID: TSC2_00424)20 in whole tuber and perituberal tissue homogenates (table). The germline splice site mutation was found in GCs microdissected from the tuber in this specimen. A novel somatic TSC2 missense mutation (exon 4, 337 G>A, E114K) was detected in single microdissected GCs. Sequencing of TSC1 in P-S6–labeled GCs from this case did not reveal mutations.
In the fourth surgical specimen, DNA sequencing from the whole lesion revealed a previously reported nonsense mutation in TSC2 exon 3 (268C>T; 90Q>X; Leiden database ID: TSC2_00036).18,20,22 There was no somatic second hit mutation identified and only 1 known synonymous SNP (exon 22, 2686C>T, 860F>F; rs13337626) was found.
In 1 additional surgical specimen, a previously unreported germline deletion of TSC2 exons 1–15 was detected. A previously unreported missense mutation in TSC2 exon 14 (1564C>T, 522H>Y) was detected in single microdissected GCs. Analysis of existing NCBI and Leiden databases did not identify this sequence change as a known SNP (of note, 3 known SNPs were detected in this case). Sequencing of TSC1 in P-S6–labeled GCs from this case did not reveal mutations.
Reduced expression of TSC1 and TSC2 mRNA in P-S6–labeled GCs.
mRNA was amplified from single microdissected P-S6–labeled GCs as well as pyramidal cells from control cortex and was used to probe arrays containing TSC1 and TSC2 cDNAs. TSC1 mRNA levels were reduced in GCs dissected from the TSC1-associated cases and reduced TSC2 mRNA was detected in GCs from TSC2-associated cases when compared with control pyramidal neurons (figure 3). These results are consistent with prior reports, for example, demonstrating reduced TSC1 gene transcript in individuals with germline TSC1 mutations23 and in mouse Tsc1 knockout strains.24 In one TSC2-associated specimen, TSC2 protein levels were reduced in GCs by immunohistochemistry (figure 3).

Figure 3 Altered TSC1 and TSC2 mRNA expression in giant cells (GCs)
(A) Reduced levels of TSC1 and TSC2 transcripts in GCs microdissected from tubers with either TSC1 (a) or TSC2 (b) germline mutations compared with control pyramidal neurons (Con; *p < 0.05). (B) TSC2 protein expression in control pyramidal neurons (a) and Purkinje cells in the cerebellum (b). In (c), there is selective loss of TSC2 protein in GCs (large arrows) compared with surrounding dysmorphic neurons (thin arrow). (d) Failure to detect the exon 15 (lane 2) and exon 22 (lane 3) TSC1 transcripts containing a nonsense mutation but detection of exon 22 in control pyramidal cells (lane 4).
We further asked whether transcripts containing nonsense mutations could be detected since previous studies have demonstrated that nonsense mutation–containing transcripts are subject to nonsense-mediated mRNA degradation in TSC.23 Thus, we postulated that, for example, the exon 15 and exon 22 transcripts in the first surgical case might not be detected in single GCs since they could be removed by nonsense-mediated mRNA degradation. Indeed, neither mRNA species (figure 3) was detected by RT-PCR in single microdissected, phospho-S6 immunoreactive GCs (n = 20), whereas the germline-encoded transcript was easily detected by RT-PCR in control neurons. These results suggest a putative mechanism to account for reduced TSC transcript levels in GCs.
DISCUSSION
We demonstrate evidence for somatic second hit TSC1 or TSC2 gene mutations in P-S6–immunoreactive GCs in cortical tubers and suggest that biallelic gene inactivation is a plausible genetic mechanism underlying the pathogenesis of tuber formation in TSC. Since TSC gene inactivation leads to mTORC1 activation, selective expression of P-S6 in GCs in a human tuber specimen provides a compelling anatomic image of a select cell type in tubers that lacks normal TSC1–TSC2 function. Sequencing of genomic DNA from single GCs exhibiting mTORC1 activation identified somatic TSC mutations not detected in whole tuber or leukocyte DNA suggesting that somatic mutations occur within a select cell population within tubers. We focused on cases in which the germline mutations were known disease-causing mutations, and show that in 5 of 6 specimens, a somatic mutation can be defined. While a 2-hit or somatic mutational mechanism has been proposed for a variety of hamartomas in TSC, this is the first report to use single cell gene sequencing to investigate this genetic mechanism in a developmental cortical malformation.
There are several caveats to our results. First, our results leave open the intriguing possibility that haploinsufficiency alone,13 e.g., the effects of germline mutation by itself, may be adequate for tuber formation, since in one specimen, a somatic mutation could not be identified. Our findings suggest that the developmental pathogenesis of tubers may be more genomically complex than previously believed. These results support the hypothesis that tubers are formed as a consequence of the combinatorial effects of germline and somatic mutational events detected in a single cell type4 but that the effects of haploinsufficiency alone in a particular cell type, i.e., a cell autonomous effect, may be adequate to induce altered cortical cytoarchitecture. Our results are both similar to and distinct from previous studies that have investigated somatic mutations in tubers.4–6,13 For example, in 2 prior reports, somatic mutations could not be detected in tubers; however, these investigators did not use single cell microdissection or gene sequencing.5,6,13 In contrast, in 1 previous report, LOH was detected in a single tuber specimen,4 whereas in our study, LOH was not identified in any of our specimens, either in homogenates or single microdissected cells. We trust the integrity of our technical approaches since germline mutations detected in tubers were confirmed in blood from the same patients, and in no case were somatic mutations found in GCs identified in blood. LOH was not identified in any of the tubers but was easily identified in a single AML specimen. Further evidence to support the possibility of biallelic gene inactivation is that there was reduced transcript expression of either TSC1 or TSC2 in single GCs. Furthermore, the failure to detect a transcript containing a nonsense mutation also suggests that there are reduced TSC1 or TSC2 mRNA levels in cells that likely lead to reduced or even absent protein levels. A similar finding has been demonstrated in existing conditional Tsc1 mouse knockout strains.24 A final limitation to our results is that we could not screen all possible cell types in tubers, e.g., DNs, astrocytes, oligodendrocytes, for somatic mutations and cannot conclusively determine how many different cell populations may contain somatic mutations. These findings have important implications for understanding autism and epilepsy in TSC and future studies to examine somatic mutations at the single cell level in distinct cell populations in tubers are clearly warranted.
There are both affected and unaffected brain regions in TSC. Thus, our results allow us to postulate a theoretical view of how tubers may form during brain development in which a somatic TSC gene mutation occurs in a single neuroglial progenitor cell in the embryonic brain, and in combination with the effects of the germline mutation, results in functional TSC protein inactivation (figure 4). Subsequent activation of mTORC1 signaling leads to cytomegaly (giant cells) in the progeny of this progenitor cell as has been demonstrated in several experimental systems.25,26 While the cell of origin for GCs remains unknown, GCs likely derive from progenitors in the telencephalic ventricular zone27 and thus could migrate, albeit abnormally, to cortex. Cells lacking functional TSC1 or TSC2 do not migrate appropriately into the cortical plate leading to a focal malformation or tuber.

Figure 4 Developmental model for tuber formation
Far left, a somatic TSC gene mutation occurs in a neuroglial progenitor cell (red circle) superimposed on an existing germline mutation present in all progenitor cells in each patient with tuberous sclerosis complex (blue). Next, cell proliferation results in generation of more cells containing 2 mutational hits. Both haploinsufficient cells (blue) and cells with biallelic mutations (red) migrate into the embryonic cortical plate. Inactivation of the TSC gene leads to mTORC1 pathway activation and progressive cytomegaly. A tuber forms when both haploinsufficient cells and cells sustaining 2 mutations arrive in the cortical plate resulting in a focal, genetic mosaic lesion. There is persistent activation of the mTORC1 cascade as evidenced by enhanced phosphorylation of ribosomal S6 protein and cytomegaly. In contrast, haploinsufficient cells representing structurally normal neurons in perituberal cortex adjacent to tubers (blue triangles) form intact laminae and exhibit preserved cytoarchitecture.
When examined in toto, tubers likely reflect a mosaic of cell-autonomous and cell-non-autonomous events, that is, direct cellular consequences of altered TSC1/TSC2 protein function and indirect effects of these cells on neighboring cells. We postulate that a small (or large) expansion of a single cell lineage by cell division could give rise to a subset of cells with altered migratory or laminar destinations. Since correct cortical lamination is a sequential process in which each wave of migration may be dependent on the success of the previous wave, altered migration or differentiation in a small population of cells could disrupt a focal group of cells. If neighboring cells, for example, depend on structural support or migratory cues from these abnormal cells, it is reasonable to believe that lamination in this region could be altered for both mutation-containing and non-mutation-containing cells. Future studies will be needed to determine how cells with reduced TSC1 or TSC2 levels disrupt the migratory pathways of adjacent cells lacking a somatic mutation.
In morphologically normal cerebral cortex surrounding tubers, the mTOR pathway is not activated in cells that contain only germline TSC gene mutations, e.g., haploinsufficient cells, and these progeny migrate appropriately into the cortical plate (figure 4). Thus, single somatic inactivating mutations may provide one mechanism by which tubers can form directly adjacent to morphologically normal cortex. Future studies will be needed to determine if a distinct somatic mutation occurs in each tuber in any single patient or if multiple second hit mutations can be detected in 1 tuber specimen.
ACKNOWLEDGMENT
The authors thank Jia Yu, MD, and Marianna Baybis for technical assistance, and Karin Boer (Department of Neuropathology, Academic Medical Center, University of Amsterdam) and Mark Nellist (Department of Clinical Genetics, Erasmus Medical Center, the Netherlands) for assistance with TSC gene sequencing.
DISCLOSURE
Dr. Crino has received travel expenses and/or honoraria for lectures or educational activities not funded by industry; is a coinventor of US Patent 6,326,146, issued 1999: Method of determining multiple mRNAs in dying cells; and receives research support from the NIH (5R01NS048557-05 [PI] and 5R01NS045021-07 [PI]), the Tuberous Sclerosis Alliance, and from the Department of Defense TSC CDMRP. Dr. Aronica receives research support from the National Epilepsy Fund, Hersenstichting Nederland, and Stichting Michelle (M06.011and M07 01 [PI]). Dr. Baltuch reports no disclosures. Dr. Nathanson receives research support from the NIH (5R01CA114478-02 [PI] and 5R01CA118871-02 [PI]).
Supplementary Material
Address correspondence and reprint requests to Dr. Peter B. Crino, Department of Neurology, University of Pennsylvania, 3 West Gates Bldg., 3400 Spruce St., Philadelphia, PA 19104 peter.crino@uphs.upenn.edu
Supplemental data at www.neurology.org
Disclosure: Author disclosures are provided at the end of the article.
Received October 30, 2009. Accepted in final form February 17, 2010.
REFERENCES
- 1.European TS Consortium (ECTS). Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993;75:1305–1315. [DOI] [PubMed] [Google Scholar]
- 2.van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997;277:805–808. [DOI] [PubMed] [Google Scholar]
- 3.Richardson EP. Pathology of tuberous sclerosis. Ann NY Acad Sci 1991;615:128–139. [DOI] [PubMed] [Google Scholar]
- 4.Green AJ, Smith M, Yates JR. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet 1994;6:193–196. [DOI] [PubMed] [Google Scholar]
- 5.Henske E, Scheithauerβ, Short M, et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet 1996;59:400–406. [PMC free article] [PubMed] [Google Scholar]
- 6.Henske E, Wessner L, Golden J, et al. Loss of TSC2 in both subependymal giant cell astrocytomas and angiomyolipomas supports a two-hit model for the pathogenesis of tuberous sclerosis tumors. Am J Pathol 1997;151:1639–1647. [PMC free article] [PubMed] [Google Scholar]
- 7.Tee AR, Fingar DC, Manning BD, et al. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA 2002;99:13571–13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoki K, Li Y, Zhu T, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signaling. Nat Cell Biol 2002;4:648–657. [DOI] [PubMed] [Google Scholar]
- 9.Arrazola P, Hino O, Kobayashi T, et al. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 2002;4:699–704. [DOI] [PubMed] [Google Scholar]
- 10.Kenerson HL, Aicher LD, True LD, Yeung RS. Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res 2002;62:5645–5650. [PubMed] [Google Scholar]
- 11.Chan JA, Zhang H, Roberts PS, et al. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol 2004;63:1236–1242. [DOI] [PubMed] [Google Scholar]
- 12.Wolf HK, Normann S, Green AJ, et al. Tuberous sclerosis-like lesions in epileptogenic human neocortex lack allelic loss at the TSC1 and TSC2 regions. Acta Neuropathol 1997;93:93–96. [DOI] [PubMed] [Google Scholar]
- 13.Niida Y, Stemmer-Rachamimov AO, Logrip M, et al. Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am J Hum Genet 2001;69:493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerfoot C, Wienecke R, Menchine M, et al. Localization of tuberous sclerosis 2 mRNA and its protein product TSC2 in normal human brain and in cerebral lesion of patients with tuberous sclerosis. Brain Pathol 1996;6:367–377. [DOI] [PubMed] [Google Scholar]
- 15.Johnson MW, Emelin JK, Park SH, Vinters H. Co-localization of TSC1 and TSC2 gene products in tubers of patients with tuberous sclerosis. Brain Pathol 1999;9:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baybis M, Yu J, Lee A, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol 2004;56:478–487. [DOI] [PubMed] [Google Scholar]
- 17.Miyata H, Chiang AC, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol 2004;56:510–519. [DOI] [PubMed] [Google Scholar]
- 18.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;335:1345–1356. [DOI] [PubMed] [Google Scholar]
- 19.Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 2001;68:64–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Au KS, Williams AT, Roach ES, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 2007;9:88–100. [DOI] [PubMed] [Google Scholar]
- 21.Choi JE, Chae JH, Hwang YS, Kim KJ. Mutational analysis of TSC1 and TSC2 in Korean patients with tuberous sclerosis complex. Brain Dev 2006;28:440–446. [DOI] [PubMed] [Google Scholar]
- 22.Hung CC, Su YN, Chien SC, et al. Molecular and clinical analyses of 84 patients with tuberous sclerosis complex. BMC Med Genet 2006;7:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeganathan D, Fox MF, Young JM, Yates JR, Osborne JP, Povey S. Nonsense-mediated RNA decay in the TSC1 gene suggests a useful tool pre- and post-positional cloning. Hum Genet 2002;111:555–565. [DOI] [PubMed] [Google Scholar]
- 24.Meikle L, Talos DM, Onda H, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 2007;27:5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 2005;8:1727–1734. [DOI] [PubMed] [Google Scholar]
- 26.Uhlmann EJ, Li W, Scheidenhelm DK, Gau CL, Tamanoi F, Gutmann DH. Loss of tuberous sclerosis complex 1 (Tsc1) expression results in increased Rheb/S6K pathway signaling important for astrocyte cell size regulation. Glia 2004;47:180–188. [DOI] [PubMed] [Google Scholar]
- 27.Lamparello P, Baybis M, Pollard J, et al. Developmental lineage of cell types in cortical dysplasia with balloon cells. Brain 2007;130:2267–2276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.