

Abstract
Objective:
Diffusion tensor imaging (DTI) quantifies Brownian motion of water within tissue. The goal of this study was to test whether, following a remote episode of optic neuritis (ON), breakdown of myelin and axons within the optic nerve could be detected by alterations in DTI parameters, and whether these alterations would correlate with visual loss.
Methods:
Seventy subjects with a history of ON ≥6 months prior underwent DTI of the optic nerves, assessment of visual acuities (VA) and contrast sensitivities (CS), and laboratory measures of visual evoked potentials (VEP) and optical coherence tomography (OCT).
Results:
Radial diffusivity (RD) correlated with visual acuity (r = −0.61), Pelli-Robson CS (r = −0.60), 5%CS (r = 0.61), OCT (r = −0.78), VEP latency (r = 0.61), and VEP amplitude (r = −0.46). RD differentiated the unaffected fellow nerves from affected nerves in all visual outcome categories. RD also discriminated nerves with recovery to normal from mild visual impairment, and those with mild impairment from profound visual loss. RD differentiated healthy controls from both clinically affected nerves and unaffected fellow nerves after ON. RD differentiated all categories of 5%CS outcomes, and all categories of Pelli-Robson CS with the exception of normal recovery from mildly affected.
Conclusions:
Increased optic nerve radial diffusivity (RD) detected by diffusion tensor imaging (DTI) was associated with a proportional decline in vision after optic neuritis. RD can differentiate healthy control nerves from both affected and unaffected fellow nerves. RD can discriminate among categories of visual recovery within affected eyes. Optic nerve injury as assessed by DTI was corroborated by both optical coherence tomography and visual evoked potentials.
GLOSSARY
- 5%CS
= 5% contrast sensitivity;
- CI
= confidence interval;
- CIS
= clinically isolated syndrome;
- CS
= contrast sensitivity;
- DTI
= diffusion tensor imaging;
- FA
= fractional anisotropy;
- MR
= magnetic resonance;
- MS
= multiple sclerosis;
- NMO
= neuromyelitis optica;
- OCT
= optical coherence tomography;
- ON
= optic neuritis;
- PR
= Pelli-Robson contrast sensitivity;
- RD
= radial diffusivity;
- RNFL
= retinal nerve fiber layer;
- ROI
= region of interest;
- VA
= visual acuity;
- VEP
= visual evoked potential.
In multiple sclerosis (MS), cumulative CNS injury increases the risk for future disability and disease progression.1 While conventional MRI sequences in clinical practice provide valuable information about the location and volume of white matter lesions, standard imaging does not adequately quantify the underlying tissue destruction.2 This inability to differentiate the severity of tissue injury within lesions may contribute to the clinico-radiologic paradox of MS.3
Diffusion tensor imaging (DTI) can quantitatively assess CNS lesions to reveal alterations in tissue structure. As a proof-of-concept regarding the clinical relevance of DTI, this study evaluated whether quantitative DTI of the optic nerve could differentiate gradations of visual recovery for those with a previous history of optic neuritis (ON). Our hypothesis was that breakdown of tissue integrity would alter DTI parameters, particularly radial diffusivity (RD), in proportion to visual loss. For DTI to serve as a surrogate of myelin and axon integrity, DTI should correlate with VEP latency, reflecting demyelination, and retinal nerve fiber layer (RNFL) thickness and visual evoked potential (VEP) amplitude, reflecting axonal loss.
This report expands upon our previous work by enrolling a larger number of subjects with ON due to several etiologies in addition to MS, by assessing the relationship of DTI to different categories of vision loss, and by providing a detailed analysis of the relationship between RNFL and VEP to visual function.4
METHODS
Standard protocol approvals and patient consent.
This cohort study was approved by the local Human Research Protection Office/Institutional Review Board, and all subjects provided written informed consent.
Subject protocol.
Inclusion criteria were 1) ≥1 episode of ON, at least 6 months prior, 2) age 18–65, and 3) no other condition affecting vision. Subjects with severe onset (visual acuity [VA] ≤20/200 or 0.1) and either good (VA ≥20/40 or 0.5) or poor recovery (≤20/70 or 0.29) were recruited to include the full spectrum of clinical outcomes.
Magnetic resonance protocol.
Magnetic resonance (MR) data were acquired using a custom fabricated transmitter coil and a 4-element phased-array flexible receiver coil on a 3T MR scanner (Allegra, Siemens AG, Germany). A vacuum-molded pillow minimized head movement. Subjects were instructed to minimize any movement and to close their eyes during the scan.
A single shot spin-echo echoplanar imaging diffusion sequence was employed with fat-suppression, and reduced field of view technique with twice refocused diffusion weighting.5,6 Diffusion-weighted images were acquired transaxially (field of view 168 mm × 84 mm, matrix 128 × 64, partial Fourier 6/8, echo time 65 msec) with 2 collated groups of 1.3-mm-thick slices. Each slice group of 5 interleaved slices (10 slices total) was cardiac gated (150 msec delay by pulse oximetry), yielding a repetition time of 4–6 seconds. Eight to 12 image sets, each with 1 b = 0 (b0) and 12 diffusion-weighted images on 12 diffusion encoding directions with b = 600 s/mm2, were acquired for each slice group.7 Total scan time was 40 minutes.
DTI calculation.
Each DTI data set was motion corrected.8 Images with excessive movement (≥3 mm translation) were excluded. All transforms were rigid body affine and computed by vector gradient measure maximization.9 The b0 volumes of each DTI data set were aligned using intensity correlation maximization. The final motion-corrected result was obtained by algebraically composing all transforms, and then averaging all data sets after application of the composed transforms using cubic spline interpolation. The final resampling step output 13 volumes with interpolated resolution of 0.65 × 0.65 × 0.65 mm3. The diffusion tensor matrix at each voxel was estimated by linear least-squares fitting of the motion-corrected and resampled DTI dataset.10
Region of interest analysis.
The region of interest (ROI) was selected manually on the b0 image to include 15–20 consecutive voxels (9.75–13.0 mm in length) within the nerve center, starting 12–15 voxels (about 8.0 mm) posterior to the retina (figure e-1 on the Neurology® Web site at www.neurology.org). Outlying voxels having FA greater than 2 SD from the mean were discarded to avoid magnetic field inhomogeneity induced artifacts and incorrect diffusion calculations. Excluded outlying voxels constituted <1% of total ROI voxels, all within 6 individual nerves with a median 2 voxels involved (range 1–6). Voxels with signal-to-noise ratio (optic nerve signal vs the background noise) lower than 32 were not included.
Clinical testing.
Vision tests were performed with a Snellen 20-foot wall chart, a 5% contrast sensitivity (5%CS) chart in an illuminated cabinet at 3 meters (Precision Vision, IL), and Pelli-Robson contrast sensitivity (PR) chart at 1 meter (Metropia Ltd., Cambridge, UK). Contrast tests were scored per manufacturer's instructions, with PR by number of letters correct, and 5%CS as the line with the majority of correctly identified letters. 5%CS vision subgroups were unaffected, normal/mildly affected (<0.4 logMar), moderately affected (0.5–0.7 logMar), and severely affected (0.8–1.0 logMar). PR subgroups were unaffected, normal (>1.60 logMar), mildly affected (1.55–1.35 logMar based upon 2–4 SD below normal), moderately affected (1.30–1.05 logMar based upon 4–6 SD below normal), and severely affected (≤1.00 logMar based upon >6 SD below normal). Best-corrected vision was achieved with glasses or pinhole occluder. Visual evoked potentials (VEP) P100 latency (normal mean 98.9 msec, upper limit 112.9 msec) and N75:P100 amplitude (normal mean 8.34 mV ± 3.32) were read in blinded fashion. If the waveform was unobtainable due to poor fixation from inadequate vision, the maximal latency of 170 msec and the minimal amplitude of 1.5 mV were used, representing the most prolonged waveform and the lowest discernible amplitude for this machine, respectively. Optical coherence tomography (OCT) fast RNFL thickness was obtained on a Zeiss Stratus OCT III with v4.0 software by a trained and certified technician. For eyes with poor visual function, OCT was obtained with external fixation of the good eye as the technician assessed the quality of the scan. OCT scans with signal strength <6 were excluded. The nadir VA during the acute phase of the ON was determined from chart review when available. If unavailable, the onset nadir VA was classified as severe if the subject “could not recognize a spouse or loved one at conversation distance.”
Statistical analyses.
Linear mixed modeling accounted for 2 eyes within a single individual (SAS Institute Inc.). OCT was evaluated as the average overall RNFL for each individual eye. Visual acuity after ON was categorized based upon the Ranges of Vision Loss by the International Council of Ophthalmology.11 Due to low numbers in the moderate (n = 6) and severe (n = 8) categories, these nerves were combined and categorized as severe. Rank correlation coefficients were obtained by randomly selecting a single nerve from each subject, with 1,000 repetitions.
RESULTS
Baseline demographics.
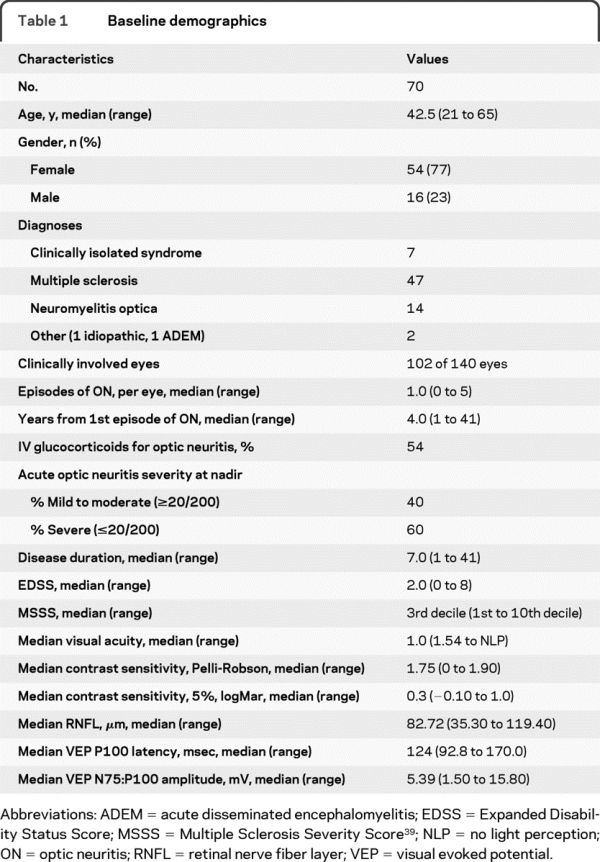
Demographics for the 70 subjects who contributed 102 clinically affected eyes are shown in table 1. There were 47 subjects with MS and 22 subjects with other diagnoses (7 clinically isolated syndrome [CIS], 14 neuromyelitis optica [NMO], 1 acute disseminated encephalomyelitis, and 1 idiopathic ON). Severe onset (VA ≤0.1) of ON was noted in 62 (60%) eyes with ON, and 57 (54%) eyes with ON received IV glucocorticoids (1,000 mg/day of methylprednisolone or 200 mg/day dexamethasone for at least 3 days). The population spanned the spectrum of visual outcomes after ON, with recovery VA ranging from 20/13 to no light perception, and 24% having recovered to worse than 20/50. Time from ON to the clinical study was a median 4.0 years (range 1–41).
Table 1 Baseline demographics
All diffusion parameters correlated strongly with visual outcomes.
Of the DTI parameters, RD had particularly high correlations with all visual outcomes, including VA (r = −0.61, p < 0.001), PR (r = −0.60, p < 0.001), and 5%CS (r = 0.61, p < 0.001) (table 2). Mean diffusivity correlations with visual outcomes were similar, as radial and mean diffusivities were highly correlated (r = 0.98, p < 0.001). Axial diffusivity and fractional anisotropy (FA) displayed strong correlations with visual outcomes, but lower than those found for radial and mean diffusivities (table 2).
Table 2 Summary of clinical and laboratory correlations for diffusion tensor imaging parameters
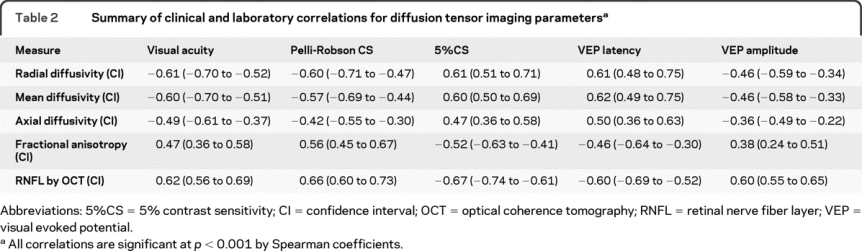
The 4 DTI parameters revealed a similar hierarchy of discrimination among VEP measures and OCT, with radial and mean diffusivities again having the highest correlations. In particular, RD strongly correlated with RNFL thickness (r = −0.78, p < 0.001), VEP latency (r = 0.61, p < 0.001), and VEP amplitude (r = −0.46, p < 0.001) (table 2, figure 1).
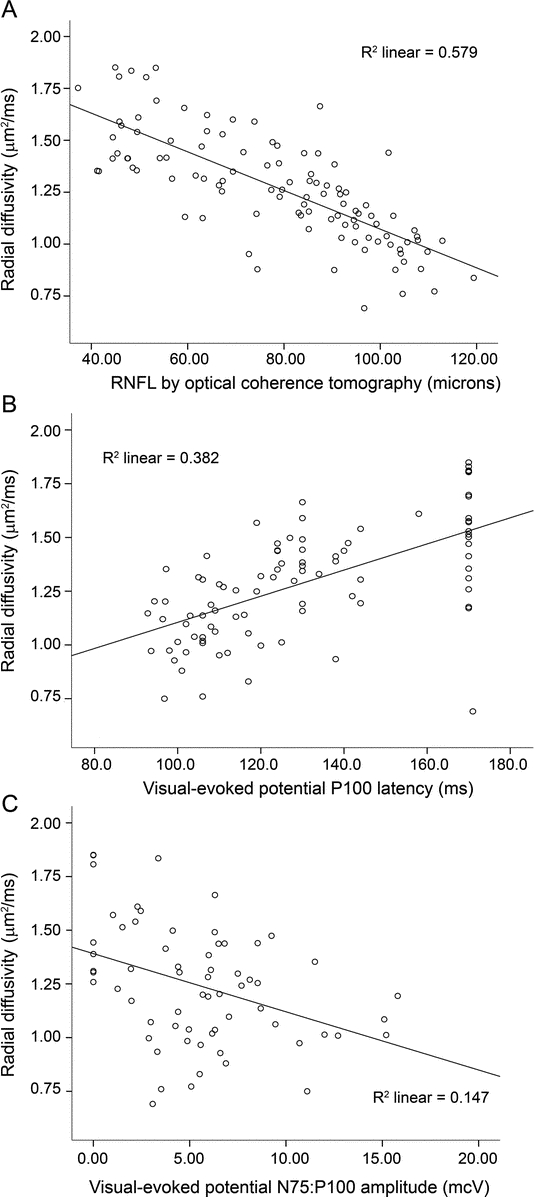
Figure 1 Scatterplots of radial diffusivity to demonstrate correlations between optical coherence tomography and visual evoked potentials (VEP)
Radial diffusivity demonstrates high correlations with retinal nerve fiber layer (RNFL) thickness (A) and VEP measures (B, C). For VEP latency, an upper limit of 170 msec was used.
All diffusion parameters discriminated visual recovery.
RD displayed a strong relationship with visual outcome, after categorizing subjects into VA severity subgroups, as defined by the International Council of Ophthalmology of normal (≥20/25), mild (20/30–20/60), combined moderate/severe (20/70–20/400), and profound (≤20/800) (figure 1, mixed modeling p < 0.0001). RD discriminated control nerves (adjusted mean [confidence interval (CI)] by mixed modeling: 0.72 [0.63–0.80]) from unaffected fellow nerves (1.08 [1.02–1.14]), unaffected from the affected nerves with normal recovery (1.23 [1.18–1.27]), normal from mild impairment (1.42 [1.32–1.52]), and mild from profound visual loss (1.65 [1.57–1.75]). RD did not differentiate the mild vs moderate/severe categories (1.61 [1.50– 1.71]), and the moderate/severe vs profound categories. The abilities of the remaining 3 DTI parameters to differentiate VA categories are shown in figure 2.

Figure 2 Boxplots of diffusion tensor imaging parameters by visual acuity recovery after optic neuritis
(A) Radial diffusivity, (B) axial diffusivity, (C) fractional anisotropy, and (D) mean diffusivity. Visual acuity categories: normal = 20/13–20/25, mild = 20/30–20/50, moderate/severe = 20/70–20/400, profound = 20/800–no light perception (NLP).
The ability of DTI to differentiate visual function was further evaluated using 5%CS and PR, subgrouped into clinical categories. When visual recovery was determined based on 5%CS, increasing RD again displayed a strong increasing relationship with worsening categories of vision (figure 3, mixed modeling p < 0.0001). RD could distinguish control (0.74 [0.66–0.83]) from unaffected fellow nerves (1.08 [1.02–1.13]), unaffected from affected nerves with normal/mild recovery (1.21 [1.16–1.25]), normal/mild from moderate (1.37 [1.28–1.46]), and moderate from severe (1.61 [1.54–1.68]). The abilities of the other 3 DTI parameters in differentiating recovery categories based on 5%CS are shown in figure 3.

Figure 3 Boxplots of diffusion tensor imaging parameters by 5% contrast sensitivity recovery after optic neuritis
(A) Radial diffusivity, (B) axial diffusivity, (C) fractional anisotropy, and (D) mean diffusivity. 5% Sloan contrast sensitivity categories (logMar): mild = <0.5, moderate = 0.5–0.7, severe = 0.8–1.0.
When recovery of visual function was determined by PR, increasing RD was again strongly correlated with worsening categories of vision (figure e-2, mixed modeling p < 0.0001). RD distinguished control (0.72 [0.63–0.80]) from unaffected fellow nerves (1.07 [1.01–1.13]), unaffected from affected nerves with normal recovery (1.22 [1.17–1.27]), mild recovery (1.27 [1.20–1.35]) from moderate impairment (1.46 [1.37–1.56]), and moderate from severe (1.65 [1.57–1.74]). RD did not discriminate normal recovery from mild visual loss defined by PR. The results for the remaining 3 DTI parameters in differentiating PR-defined categories of vision are provided in figure e-2.
RD increased with acute ON nadir severity and recurrence.
Increasing severity of the ON visual nadir during the acute setting was associated with an increasing RD after at least 6 months follow-up, after categorizing the nadir into mild (1.17 [1.07–1.26]), moderate (1.28 [1.18–1.38]), and severe (1.42 [1.36–1.48]) visual loss (repeated-measures analysis of variance, p < 0.001). Even when analyzing eyes with full visual recovery (VA ≥0.8, n = 68), increasing RD continued to increase based upon increasing severity of vision loss at the acute nadir (repeated- measures analysis of variance, p = 0.01). For eyes with recurrent clinical episodes of ON, each additional historical episode was associated with increasing RD (repeated-measures analysis of variance, p < 0.001).
Correlations among the 4 diffusion parameters and visual recovery were observed regardless of the etiology of the ON (MS vs CIS vs NMO). There were no discernible differences in the correlations of DTI parameters and visual recovery in patients with MS based upon differing immunomodulatory therapies.
DISCUSSION
This study establishes that all DTI parameters in the anterior visual pathway correlate with clinically meaningful visual outcomes following ON due to several etiologies. Specifically, RD differentiated optic nerves of healthy subjects from the clinically unaffected fellow eye in subjects with remote ON. RD was highly correlated with all clinical outcomes and had high discriminatory ability among categories of visual recovery. We have also verified that RD demonstrates strong correlations with accepted electrophysiologic parameters (VEP latency and amplitude) and with an accepted structural surrogate of optic nerve injury (RNFL thickness by OCT).
The general lack of correlation between MRI measures and clinical outcomes in MS has limited the clinical and research utility of standard MRI sequences. The clinico-radiologic paradox may be due to variability in lesion severity and in lesion location, involvement of poorly imaged cortex and deep gray matter structures, inadequate assessment of spinal cord lesions, underestimation of abnormalities within seemingly normal-appearing white matter, and individual variability in repair potential and neural plasticity. Also contributing to this “paradox” is that the clinical outcome measures for this complex neurologic disease are suboptimal. DTI can improve upon standard MRI by its ability to quantify lesion severity, as demonstrated in this study.
Studying the optic nerves removes some of the confounding variables which contribute to the MRI paradox.12–20 Prior studies have reported that T1- and T2-weighted imaging of the optic nerves after ON have a low correlation with clinical outcomes, although imaging measurements of optic nerve area and magnetization transfer ratio have demonstrated good correlations.21–23 As a relatively straight tract of single-ordered axons, whose clinical function is readily measured, the anterior visual pathway has special advantages for analyzing the relationship between DTI and outcomes compared to other CNS regions. This translational study of DTI in human optic nerve follows upon DTI studies of optic nerve injury in animals that had direct histopathologic correlation.24–26 White matter tracts consisting of parallel axons tightly packed with myelin are anisotropic, or directional, to the diffusion of water. Chronic injury due to loss of myelin and axons leads to reduced anisotropy. This results in increased diffusion perpendicular to the white matter tract (analogous to RD), increased overall diffusivity (mean diffusivity), and decreased tissue directionality (FA).27,28
Axial diffusivity, as a surrogate of axonal integrity, did not correlate as well with visual and laboratory outcomes in comparison to radial and mean diffusivities. When assessed in acute injury, axial diffusivity in white matter tracts has been shown in both animal models and humans to decrease in proportion to worsening recovery.4,24,29,30 However, our prior study revealed that pathologic changes within human CNS tissue from the acute to the chronic stage resulted in axial diffusivity becoming less informative over time.4 As myelin debris is cleared and inflammation and edema resolve, we speculate that the demyelinated axons are no longer tightly packed, and the widening interstitial space dilutes the ability of DTI to detect and measure anisotropic diffusion within axons. Hence, the present study of remote ON found a modest correlation between axial diffusivity and VEP amplitude (r = −0.35), an electrophysiologic surrogate for axon loss. In MS, the dynamic evolution of inflammation, axonal injury, and myelin loss creates a challenging situation for imaging with pathology specificity, especially when the timing of inflammation relative to tissue injury is not always known. In addition to the temporal aspect, the pathology in MS is also complex and variable, with axon and myelin injury strongly linked.31,32
VEP was utilized in this study to help distinguish the contributions of axonal and myelin injury within optic nerves. RD was strongly correlated with VEP latency (r = 0.61), a parameter associated with myelin integrity, and moderately correlated to amplitude (r = −0.46), a measure associated with axonal integrity. A high correlation was also noted between RD and RNFL thickness (r = −0.78). RNFL thickness also demonstrated strong correlations for both VEP measures (r = −0.60 for latency, r = 0.60 for amplitude). Although VEP latency is highly sensitive to confirm previous ON, VEP is not an ideal measure of clinical outcome (figure e-3). Both DTI and OCT, although less sensitive for ON detection, have advantages over VEP as a surrogate of clinical outcome and in longitudinal assessments.
Currently, OCT is more practical in the clinical setting and within clinical trials compared to optic nerve DTI, based on the greater time, cost, and technical skill required for the latter. The present study confirms that RNFL thickness is an excellent surrogate marker for optic nerve integrity. However, the 2 techniques can provide different and complementary information. Of prime distinction, DTI is not limited to the optic nerve, and can have application within white matter of the brain and spinal cord. In addition, the role of OCT during acute clinical episodes has not been defined, as recurrent retrobulbar ON may obscure the true RNFL due to retinal edema.33 RNFL measures the retina and not the optic nerve directly and thus can be affected by other retinal diseases. The present study indicated that OCT displays a floor effect (figure e-2), whereby progressive decreases in vision among moderate through profound categories may not be associated with further thinning of the RNFL.34 However, this floor effect with OCT may be less of an issue in a general MS population, where most individuals do not have a devastating outcome after ON.
Although very strong correlations were noted between DTI and clinical outcomes, unexplained variability remained. As an example, several optic nerves displayed both elevated RD and reduced RNFL by OCT, yet tested in the normal range on all clinical vision testing. Also, this study primarily measured central vision in its visual outcomes, not peripheral vision. Visual fields were not performed because ON preferentially affects central vision.35 In this study, lesions within the postchiasmal visual system that may affect vision were not assessed by DTI.36
The present study supports the ability for DTI to assess tissue injury by demonstrating a proportional relationship to functional outcomes in remote ON. DTI has demonstrated good correlation to histopathology in both animal models and humans.37 DTI of acute enhancing lesions within the brain may also predict severe tissue injury determined by a persistent T1 hypointensity.38 Taken together, these studies suggest that regions of elevated diffusivity within the CNS, whether symptomatic or clinically asymptomatic, correlate with functional outcomes and CNS tissue breakdown.
DTI merits further study as a clinical trial endpoint and predictor of clinical outcome in demyelinating disease of the CNS. One goal of the present study was to provide a foundation for future studies of human systems having less straightforward clinical endpoints, such as in motor control, cognition, and sensation. Although the present proof-of-concept study in optic nerve used a long scanning time and a custom-made transmitting research coil, DTI of the brain has several attributes, including practical scanning time, resolution, and complete brain coverage, and it can be implemented on current clinical scanners. DTI may have a future role in the evaluation of new therapeutics, determining prognosis by quantifying the amount of injury within an MS plaque, and monitoring the response to treatment.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Kathryn Trinkaus.
DISCLOSURE
Dr. Naismith has received travel expenses and/or honoraria for lectures and educational activities from Bayer Schering Pharma, Biogen Idec, Teva Pharmaceutical Industries Ltd., and Elan Corporation; serves on speakers' bureaus for Bayer Schering Pharma, Biogen Idec, Elan Corporation, and Teva Pharmaceutical Industries Ltd.; and receives research support from Acorda Therapeutics Inc., the NIH (K23NS052430-01A1 [PI] and K12RR02324902 [PI]), and the National MS Society. Dr. Xu, N.T. Tutlam, and Dr. Trinkaus report no disclosures. Dr. Cross serves on scientific advisory boards for Eli Lilly and Company, Genentech, Inc., and Biogen Idec; serves on the editorial boards of Brain Pathology and the Journal of Neuroimmunology and as an editor and contributor to CONTINUUM; receives royalties from the publication of Handbook of Multiple Sclerosis, 4th Ed. (Taylor & Francis Group, 2006); serves on speakers' bureaus for Bayer Schering Pharma and Biogen Idec; has received speaker honoraria from Amgen and Pfizer Inc.; and receives research support from Sanofi-Aventis, Acorda Therapeutics Inc., Genentech Inc., Biogen Idec, the NIH (NINDS PO1 NS059560-01 [PI], NINDS UO1 NS45719-01A1 [Co-I], RO1 NS047592 [Co-I], NINDS RO1 NS 051591 [PI]), ICTS Washington University, the National MS Society USA, Consortium of Multiple Sclerosis Centers, and the Barnes-Jewish Hospital Foundation. Dr. Song reports no disclosures.
Supplementary Material
Address correspondence and reprint requests to Dr. Robert T. Naismith, Neurology, Washington University, Box 8111, 660 S. Euclid Ave., St. Louis, MO 63110 naismithr@neuro.wustl.edu
See also page 1694
Supplemental data at www.neurology.org
*The two first authors contributed equally on this project and manuscript.
‡The two senior authors contributed equally on this project and manuscript.
Study funding: Supported by National Institutes of Health (K23NS052430-01A1 to R.T.N., K12RR02324902 to R.T.N., K24 RR017100 to A.H.C.); the National MS Society (FG1782A1 to J.X., CA1012 to A.H.C. and S.K.S., RG 3670 to S.K.S.); and the Manny and Rosalyn Rosenthal-Dr. John L. Trotter Chair in Neuroimmunology from Barnes-Jewish Hospital Foundation (A.H.C.). This publication was made possible by grant number UL1 RR024992 from the National Center for Research Resources (NCRR), a component of the NIH and NIH Roadmap for Medical Research. This research was also supported in part by NIH grants CO6 RR020092 and RR024992 (Washington University Institute of Clinical and Translational Sciences–Brain, Behavioral and Performance Unit). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
Disclosure: Author disclosures are provided at the end of the article.
Received November 2, 2009. Accepted in final form February 17, 2010.
REFERENCES
- 1.Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol 1999;12:295–302. [DOI] [PubMed] [Google Scholar]
- 2.Barkhof F, Bruck W, De Groot CJ, et al. Remyelinated lesions in multiple sclerosis: magnetic resonance image appearance. Arch Neurol 2003;60:1073–1081. [DOI] [PubMed] [Google Scholar]
- 3.Barkhof F. The clinico-radiological paradox in multiple sclerosis revisited. Curr Opin Neurol 2002;15:239–245. [DOI] [PubMed] [Google Scholar]
- 4.Naismith RT, Xu J, Tutlam NT, et al. Disability in optic neuritis correlates with diffusion tensor-derived directional diffusivities. Neurology 2009;72:589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeong EK, Kim SE, Guo J, Kholmovski EG, Parker DL. High-resolution DTI with 2D interleaved multislice reduced FOV single-shot diffusion-weighted EPI (2D ss-rFOV-DWEPI). Magn Reson Med 2005;54:1575–1579. [DOI] [PubMed] [Google Scholar]
- 6.Xu J, Sun SW, Naismith RT, Snyder AZ, Cross AH, Song SK. Assessing optic nerve pathology with diffusion MRI: from mouse to human. NMR Biomed 2008;21:928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasan KM, Parker DL, Alexander AL. Comparison of gradient encoding schemes for diffusion-tensor MRI. J Magn Reson Imaging 2001;13:769–780. [DOI] [PubMed] [Google Scholar]
- 8.Shimony JS, Burton H, Epstein AA, McLaren DG, Sun SW, Snyder AZ. Diffusion tensor imaging reveals white matter reorganization in early blind humans. Cereb Cortex 2006;16:1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rowland DJ, Garbow JR, Laforest R, Snyder AZ. Registration of [18F]FDG microPET and small-animal MRI. Nucl Med Biol 2005;32:567–572. [DOI] [PubMed] [Google Scholar]
- 10.Shimony JS, McKinstry RC, Akbudak E, et al. Quantitative diffusion-tensor anisotropy brain MR imaging: normative human data and anatomic analysis. Radiology 1999;212:770–784. [DOI] [PubMed] [Google Scholar]
- 11.ICO. ICO World Recommendations for Defining Visual Loss. Available at: www.icoph.org/pdf/visualstanres.pdf. Accessed November 2, 2009.
- 12.Wheeler-Kingshott CA, Trip SA, Symms MR, Parker GJ, Barker GJ, Miller DH. In vivo diffusion tensor imaging of the human optic nerve: pilot study in normal controls. Magn Reson Med 2006;56:446–451. [DOI] [PubMed] [Google Scholar]
- 13.Trip SA, Wheeler-Kingshott C, Jones SJ, et al. Optic nerve diffusion tensor imaging in optic neuritis. Neuroimage 2006;30:498–505. [DOI] [PubMed] [Google Scholar]
- 14.Hickman SJ, Wheeler-Kingshott CA, Jones SJ, et al. Optic nerve diffusion measurement from diffusion-weighted imaging in optic neuritis. AJNR Am J Neuroradiol 2005;26:951–956. [PMC free article] [PubMed] [Google Scholar]
- 15.Wheeler-Kingshott CA, Parker GJ, Symms MR, et al. ADC mapping of the human optic nerve: increased resolution, coverage, and reliability with CSF-suppressed ZOOM-EPI. Magn Reson Med 2002;47:24–31. [DOI] [PubMed] [Google Scholar]
- 16.Kolbe S, Chapman C, Nguyen T, et al. Optic nerve diffusion changes and atrophy jointly predict visual dysfunction after optic neuritis. Neuroimage 2009;45:679–686. [DOI] [PubMed] [Google Scholar]
- 17.Hickman SJ, Toosy AT, Jones SJ, et al. A serial MRI study following optic nerve mean area in acute optic neuritis. Brain 2004;127:2498–2505. [DOI] [PubMed] [Google Scholar]
- 18.Hickman SJ, Toosy AT, Jones SJ, et al. Serial magnetization transfer imaging in acute optic neuritis. Brain 2004;127:692–700. [DOI] [PubMed] [Google Scholar]
- 19.Trip SA, Schlottmann PG, Jones SJ, et al. Optic nerve atrophy and retinal nerve fibre layer thinning following optic neuritis: evidence that axonal loss is a substrate of MRI-detected atrophy. Neuroimage 2006;31:286–293. [DOI] [PubMed] [Google Scholar]
- 20.Trip SA, Schlottmann PG, Jones SJ, et al. Optic nerve magnetization transfer imaging and measures of axonal loss and demyelination in optic neuritis. Mult Scler 2007;13:875–879. [DOI] [PubMed] [Google Scholar]
- 21.Kupersmith MJ, Alban T, Zeiffer B, Lefton D. Contrast-enhanced MRI in acute optic neuritis: relationship to visual performance. Brain 2002;125:812–822. [DOI] [PubMed] [Google Scholar]
- 22.Thorpe JW, Barker GJ, Jones SJ, et al. Magnetisation transfer ratios and transverse magnetisation decay curves in optic neuritis: correlation with clinical findings and electrophysiology. J Neurol Neurosurg Psychiatry 1995;59:487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hickman SJ, Toosy AT, Miszkiel KA, et al. Visual recovery following acute optic neuritis–a clinical, electrophysiological and magnetic resonance imaging study. J Neurol 2004;251:996–1005. [DOI] [PubMed] [Google Scholar]
- 24.Song SK, Sun SW, Ju WK, Lin SJ, Cross AH, Neufeld AH. Diffusion tensor imaging detects and differentiates axon and myelin degeneration in mouse optic nerve after retinal ischemia. Neuroimage 2003;20:1714–1722. [DOI] [PubMed] [Google Scholar]
- 25.Sun SW, Liang HF, Cross AH, Song SK. Evolving Wallerian degeneration after transient retinal ischemia in mice characterized by diffusion tensor imaging. Neuroimage 2008;40:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun SW, Liang HF, Le TQ, Armstrong RC, Cross AH, Song SK. Differential sensitivity of in vivo and ex vivo diffusion tensor imaging to evolving optic nerve injury in mice with retinal ischemia. Neuroimage 2006;32:1195–1204. [DOI] [PubMed] [Google Scholar]
- 27.Song SK, Sun SW, Ramsbottom MJ, Chang C, Russell J, Cross AH. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage 2002;17:1429–1436. [DOI] [PubMed] [Google Scholar]
- 28.Song SK, Yoshino J, Le TQ, et al. Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage 2005;26:132–140. [DOI] [PubMed] [Google Scholar]
- 29.Sun SW, Liang HF, Trinkaus K, Cross AH, Armstrong RC, Song SK. Noninvasive detection of cuprizone induced axonal damage and demyelination in the mouse corpus callosum. Magn Reson Med 2006;55:302–308. [DOI] [PubMed] [Google Scholar]
- 30.Budde MD, Xie M, Cross AH, Song SK. Axial diffusivity is the primary correlate of axonal injury in the experimental autoimmune encephalomyelitis spinal cord: a quantitative pixelwise analysis. J Neurosci 2009;29:2805–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278–285. [DOI] [PubMed] [Google Scholar]
- 32.Kornek B, Storch MK, Weissert R, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol 2000;157:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costello F, Hodge W, Pan YI, Eggenberger E, Coupland S, Kardon RH. Tracking retinal nerve fiber layer loss after optic neuritis: a prospective study using optical coherence tomography. Mult Scler 2008;14:893–905. [DOI] [PubMed] [Google Scholar]
- 34.Naismith RT, Tutlam NT, Xu J, et al. Optical coherence tomography is less sensitive than visual evoked potentials in optic neuritis. Neurology 2009;73:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keltner JL, Johnson CA, Spurr JO, Beck RW. Comparison of central and peripheral visual field properties in the optic neuritis treatment trial. Am J Ophthalmol 1999;128:543–553. [DOI] [PubMed] [Google Scholar]
- 36.Wu GF, Schwartz ED, Lei T, et al. Relation of vision to global and regional brain MRI in multiple sclerosis. Neurology 2007;69:2128–2135. [DOI] [PubMed] [Google Scholar]
- 37.Schmierer K, Wheeler-Kingshott CA, Tozer DJ, et al. Quantitative magnetic resonance of postmortem multiple sclerosis brain before and after fixation. Magn Reson Med 2008;59:268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naismith RT, Xu J, Tutlam NT, et al. Increased diffusivity in acute multiple sclerosis lesions predicts risk of black hole. Neurology 2010;74:1694–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roxburgh RH, Seaman SR, Masterman T, et al. Multiple Sclerosis Severity Score: using disability and disease duration to rate disease severity. Neurology 2005;64:1144–1151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.