

Abstract
Glutamate racemases (EC 5.1.1.3) catalyze the cofactor-independent stereoinversion of D- and L-glutamate and are important for viability in several Gram-negative and -positive bacteria. As the only enzyme involved in the stereoinversion of L- to D-glutamate for peptidoglycan biosynthesis, glutamate racemase is an attractive target for the design of antibacterial agents. However, the development of competitive tight-binding inhibitors has been problematic and highly species specific. Despite a number of recent crystal structures of cofactor-independent epimerases and racemases, cocrystallized with substrates or substrate analogues, the source of these enzymes’ catalytic power and their ability to acidify the Cα of amino acids remains unknown. The present integrated computational and experimental study focuses on the glutamate racemase from Bacillus subtilis (RacE). A particular focus is placed on the interaction of the glutamate carbanion intermediate with RacE. Results suggest that the reactive form of the RacE–glutamate carbanion complex, vis-à-vis proton abstraction from Cα, is significantly different than the RacE–D-glutamate complex on the basis of the crystal structure and possesses dramatically stronger enzyme–ligand interaction energy. In silico and experimental site-directed mutagenesis indicates that the strength of the RacE–glutamate carbanion interaction energy is highly distributed among numerous electrostatic interactions in the active site, rather than being dominated by strong hydrogen bonds. Results from this study are important for laying the groundwork for discovery and design of high-affinity ligands to this class of cofactor-independent racemases.
Introduction
All Gram-positive bacteria incorporate D-glutamate into their thick peptidoglycan-based cell wall, which provides the structural stability required to prevent osmotic lysis.1,2 In addition, several pathogenic bacteria, including Bacillus anthracis, also synthesize a γ-linked D-glutamic acid homopolymeric capsule, 3–6 which has recently been shown to be a major determinant of pathogenicity for inhalation anthrax.3,5,7 Bacteria directly generate D-glutamate from L-glutamate via a class of enzymes called glutamate racemases.
Glutamate racemases (EC 5.1.1.3) catalyze the stereoinversion of D- and L-glutamate. Glutamate racemase is important for viability in several Gram-negative and -positive bacteria, including Bacillus subtilis, and is thought to be the principal enzyme involved in stereoinversion of L- to D-glutamate for peptidoglycan biosynthesis in these organisms.8–11 This family of enzymes has emerged as an attractive target for the design of antibacterial agents.12–16 However, the development of tight-binding reversible inhibitors has been largely unsuccessful, with notable species-specific exceptions.13
There have been detailed structural, mechanistic, and computational investigations that successfully describe how pyridoxal 5′-phosphate-dependent racemases,17–26 Mg2+ ion-assisted racemases, and enolases27–34 acidify the Cα proton of their substrates. However, cofactor-independent racemases and epimerases have been much more of an enigma. Seminal studies by Knowles and co-workers on both proline35–37 and glutamate racemases38–40 established that the enzymes employ a “two-base” racemization mechanism, in which two Cys residues flank the substrate’s Cα (Scheme 1). In the D→L direction, one Cys thiolate functions as the catalytic base, abstracting the Cα proton to generate a carbanion intermediate, which is then protonated by the second Cys residue’s thiol to yield the antipode. In the reverse direction, L→D, the conjugate base of the second Cys residue abstracts the Cα proton from the L-stereoisomer, while the conjugate acid of the first Cys residue donates a proton to the Cα to yield D-isomer product. This general mechanism is also operative for diaminopimelate epimerase.41
Scheme 1.

Stepwise Double Proton Transfer Recemization
Kinetic isotope effect (KIE) studies by Knowles and coworkers on the glutamate racemase from Lactobacillus fermenti showed that the enzyme exhibited a substantial primary KIE on Vmax/KM and Vmax,40 indicating that C–H bond cleavage is at least partially rate-determining and that substrate binding is not partially rate-determining. Furthermore, an “oversaturated” regime was not detected for glutamate racemase (as opposed to proline racemase35), which is indicative of a rapid interconversion of the two free enzymic forms (i.e., the form that binds L-glutamate and the form that binds D-glutamate).39
The publication of a number of studies describing the structures of cofactor-independent racemases, including the glutamate racemase from B. subtilis (RacE-D-glu),42 B. anthracis (RacE1- and RacE2-D-glu),43 Helicobacter pylori and Escherichia coli,44 and proline racemase with the 2-pyrrolecarboxylic acid45 and diaminopimelate epimerase with azidiaminopimelate (DAP) stereoisomers,46 has resulted in significant advances toward understanding how this class of enzymes works. The only other previously liganded crystal structure was the glutamate racemase from Aquifex pyrophilus, MurI, cocrystallized with the weak inhibitor D-glutamine.47 However, the structures of B. subtilis RacE liganded with D-glutamate and MurI liganded with D-glutamaine were radically different, leading to the hypothesis that the MurI structure may be a noncatalytic form of the enzyme (i.e., representative of the enzyme in the absence of any glutamate).42 There have been a number of computational studies based on the MurI structure,48–50 in which the position of the D-glutamate ligand was docked into the active site as an initial starting point for the calculations. Only studies by Puig et al.49 have focused on proton-transfer transition states in the MurI enzyme, which required a protonated form of the substrate α-carboxylate in order for racemization to occur. The nature of the substrate–enzyme interactions observed by Puig et al. is significantly different from those observed in the current study.
The physicochemical rationale underlying Cα proton acidification and the catalytic acceleration of proton abstraction remain poorly understood. Carbanion stabilization may occur via delocalization of negative charge through many strong hydrogen bond donors to the α-carboxylate. Alternatively, the α-carboxylate may be directly protonated. Additional stabilization may be provided by an ylide-type interaction (between the carbanionic intermediate’s ammonium and the negatively charged Cα), which is significantly strengthened by desolvation.49,51,52 The B. subtilis RacE-D-glutamate structure strongly disfavors the possibility for a general acid that can protonate the ligand’s α-carboxylate, due to the lack of any general acid candidate in the active site.42 Another important question is how the catalytic bases specifically deprotonate the Cα (pKa ~29 for the zwitterion)52 in preference to the ammonium (pKa ~9.6) of the zwitterionic glutamate, especially given the very close distances between the sulfur of Cys74 (catalytic base in the D→L direction) and the N of the D-glutamate in the crystal structure of RacE (3.28 Å between N and S, vs 3.46 Å between Cα and S).42
These questions are ideally addressed by considering the dynamic properties of the enzyme–substrate complex, particularly directed to studies employing high-energy intermediates that are stabilized by the enzyme. The current study focuses on the B. subtilis RacE enzyme and employs both computational methods (MD-QM/MM and docking simulations using the RacE structure) and experimental approaches (mutagenesis of key hydrogen-bonding and polar contacts around substrate α-carboxylate) to probe the nature of the transition-state structure of the enzyme–substrate complex.
This work provides a starting point for utilizing the transition-state binding energy of glutamate racemase (which in principle should yield a rate acceleration of ~1013)52 in ligand discovery. The approach taken in this study was to assess the dynamic properties of the intermediate glutamate racemase–glutamate carbanion complexes, with the objective of identifying active-site residues predicted to stabilize these intermediates. Site-directed mutagenesis and kinetic analysis were used in conjunction with the computational studies to provide a framework for rationalizing the catalytic power and strength of ligand binding in glutamate racemase. These results provide an important starting point for exploiting the transition-state binding energy of glutamate racemase in ligand discovery.
This approach may be used in conjunction with the very powerful methods of designing transition-state analogues based on transition-state structures validated by comparison of calculated and experimental KIE values, as carried out by Schramm and co-workers, which have led to unprecedented advances in the development of reversible inhibitors of high affinity and specificity.53–58
Materials, Methods, and Computational Procedures
Computational Details
The computational details are given in the Supporting Information. Insightful quantum mechanical-molecular mechanical (QM/MM) approaches have been used to successfully investigate the energetics and dynamical aspects of binding of substrates and transition-state analogues in enzymes59–61 and the role of dynamics in the control of transition-state barriers in enzymatic reactions.62 However, such approaches are computationally expensive and are not necessarily amenable for exploratory computational investigations. The computational procedures employed in the current study utilize a layered approach, beginning with a force field parametrization of the carbanion and subsequent use of classical molecular dynamics (MD) trajectories to identify four possible reactive geometries, which are then ported for use in semiempirical PM3 geometry optimizations. Two structures, identified by semiempirical calculations as likely leading to Cys/Cα proton transfer, were investigated using ab initio QM/MM methods. The MD simulations in this study primarily focus on the interaction of the enzyme with the high-energy carbanionic intermediate, generating trajectories of reactive poses for proton transfer between Cα and both catalytic Cys residues. Semiempirical methods are used to examine the probability of proton transfer of selected enzyme–carbanion MD snapshots, which may then be investigated by ab initio QM/MM methods, in order to determine more accurate ground- and transition-state structures for proton transfers. This method is somewhat similar to the work of Alhambra and co-workers63 on the mechanism of yeast enolase, in which classical MD trajectories are used to obtain reasonable sampling of enzyme–ligand conformations, which are then employed in more detailed QM/MM methods to investigate the chemistry of proton transfer. However, rather than defining a reaction coordinate, the approach of the current study is to examine the MD trajectory of the carbanionic intermediate itself, from which an ensemble of structures may be examined by QM/MM methods.
Experimental Details
Materials
DNA PCR purification kit, DNA gel extraction kit, and plasmid miniprep kit were acquired from Qiagen (Valencia, CA). DNA oligonucleotides were synthesized at Integrated DNA Technologies Inc. (Coralville, IA). Picomaxx DNA polymerase for PCR-amplification was acquired from Stratagene (La Jolla, CA). Restriction endonucleases were obtained from New England Biolabs (Ipswich, MA). Ni-NTA His • Bind metal affinity resin and pET-15b were purchased from Novagen (Madison, WI). DNA sequencing was performed at the Roy J. Carver Biotechnology Center (Urbana, IL). The plasmid miniprep kit and gel filtration standards were obtained from Bio-Rad (Hercules, CA). Isopropyl β-D-thiogalactoside (IPTG) was obtained from Fisher Scientific (Fair Lawn, NJ). L-Glutamate, ampicillin, dithiothreitol (DTT), and 2,6-pyridinedicarboxylic acid (or dipicolinic acid, DPA) were obtained from Sigma Aldrich (St. Louis, MO). Compound 118 was purchased from Sigma Aldrich’s Rare Chemical Library. Amicon centrifugal filter devices with a MWCO of 10 000 Da were obtained from Millipore (Billerica, MA). Iodonitrotetrazolium chloride, nicotinamide adenine dinucleotide hydrate (NADH), diaphorase from Clostridium kluyveri, and L-glutamate dehydrogenase from bovine liver (obtained as an ammonium sulfate suspension) were purchased from Sigma Aldrich. Adenosine-5′-diphophate disodium salt dihydrate was obtained from USB Corp. (Cleveland, OH).
Cloning of Glutamate Racemase (RacE) from Bacillus subtilis
Bacillus subtilis DB10464 was obtained from Dr. Robert L. Switzer (Urbana, IL). Genomic DNA was obtained from midlog cultures of B. subtilis DB104 utilizing a DNeasy Tissue Kit from Qiagen (Valencia, CA). B. subtilis racE (GenBank BSU2835) was PCR-amplified using primers (see Supporting Information) corresponding to the 5′ and 3′ ends of the gene. These primers were engineered such that 5′ XhoI and 3′ BamHI restriction sites were incorporated. The PCR product was then purified using a PCR purification kit from Qiagen (Valencia, CA). The purified amplicons were incubated with XhoI and BamHI to generate directional annealing sites and then ligated with pET-15b, to replace the XhoI-BamHI fragment within the polylinker region. The ligation mixtures were introduced into E. coli XL1-Blue from Stratagene (La Jolla, CA) by electroporation. The integrity of each gene from individual clones was confirmed by DNA sequencing. pET-15b-racE was isolated using a plasmid miniprep kit and introduced by electroporation into E. coli T7 lysogen [BL21(DE3)] from Novagen (Madison, WI).
Expression and Purification of Hexahistidine-Tagged Mutant and Wild-Type RacE
pET-15b-racE plasmid was electroporated into chemically competent E. coli BL21(DE3) cells and grown overnight at 37 °C in 5 mL of LB medium supplemented with 100 μg/mL ampicillin. A 5 mL starter culture was then back-diluted into 500 mL of LB containing 100 μg/mL ampicillin and grown at 37 °C on an orbital shaker set to 220 rpm. Cells were grown until the optical density at 600 nm reached ~0.5. IPTG was then added to a final concentration of 0.1 mM to induce expression, and cells were grown for an additional 16 h at 15 °C. After induction, cells were harvested by centrifugation at 5000g for 15 min. The cell pellets were resuspended in 7.5 mL of bind buffer (50 mM phosphate, 500 mM NaCl, 10 mM imidazole, 5 mM β-mercaptoethanol, pH 8.0) and lysed by sonication (three 20 s cycles at 23 kHz and 20 W) using a model 100 Sonic Dimembrator from Fisher Scientific. Cell lysate was clarified by centrifugation at 30000g for 30 min. Clarified lysate was filtered using a 0.22 μm Millex GP syringe tip filter from Millipore before employing a GE Healthcare AKTAprime chromatographic system to load the clarified lysate onto a 5 mL HiTrap Chelating Cartridge charged with 0.1 M NiSO4 at a flow rate of 2.0 mL/min. Bound protein was washed with ~50 mL of wash buffer (50 mM phosphate, 250 mM NaCl, 50 mM imidazole, 5 mM β-mercaptoethanol, pH 8.0). Bound protein was eluted with elution buffer (50 mM phosphate, 1000 mM NaCl, 500 mM imidazole, 5 mM β-mercaptoethanol, pH 8.0), and the collected eluant was exchanged into protein storage buffer (50 mM Tris, 100 mM NaCl, 0.2 mM DTT, pH 8.0) utilizing a 10 000 MWCO Amicon centrifugal filter device from Millipore.
After four buffer exchanges, absorbance spectroscopy was used to assay protein concentration based on previously employed methods.65 An extinction coefficient of 24 410 M−1 cm−1 at 280 nm was calculated from the primary amino acid sequence and used to quantitate the concentration of mutant and wild-type protein on the basis of absorbance readings acquired using a Cary 300 Bio UV–visible spectrophotometer from Varian Inc. (Palo Alto, CA). Protein stocks were stored in 20% glycerol at −20 °C at a concentration of 10 mg/mL.
Site-Directed Mutagenesis
Mutagenesis of the pET-15b-racE plasmid was performed using the QuikChange mutagenesis kit from Stratagene. Forward and reverse mutagenic primers (see Supporting Information for sequences) were designed with the desired mutation in the center of the primer and containing 10–20 bases of complementary plasmid sequence on either side. Each thermocycling reaction carried out to effect mutant strand synthesis contained 1x PfuUltra reaction buffer, 1.25 U PfuUltra High-Fidelity DNA polymerase, 0.2 mM dNTP mix, 125 ng forward and reverse primers containing desired mutation, 0.2 μg/mL pET-15b-racE template, and ultrapure water to a final volume of 25 μL. Reactions were cycled 18 times in a thermocycler and subsequently run on a 1% agarose gel for visualization. One microliter of DPN I restriction enzyme was added to successful reactions and incubated at 37 °C overnight to degrade parental DNA. The resulting mixture was electroporated into chemically competent E. coli DH5-α cells and grown in LB medium containing 100 μg/mL ampicillin. Plasmid DNA containing the putative mutation was subsequently isolated from transformed DH5-α cells using a Qiagen miniprep kit and sequenced to ensure the desired mutation had been introduced.
Circular Dichroism Spectroscopy of Purified Mutant and Wild-Type Protein
Circular dichroism (CD) spectra were obtained for mutant and wild-type proteins in the far-UV region utilizing a J-720 CD spectrometer from JASCO (Easton, MD). A quartz cylindrical cuvette with a total volume of 350 μL and a path length of 0.1 cm was employed for each assay. CD spectra were collected from 260 to 190 nm for wild type (2.88 μM) and T76A (3.28 μM) in optically clear borate buffer (50 mM potassium borate, pH 8.0) at a scan rate of 50 nm/s with a 1 nm wavelength step and with seven accumulations.
JASCO Spectra Manager v1.54a software was used for data acquisition, and raw data files were uploaded onto the DICHROWEB online server (http://dichroweb.cryst.bbk.ac.uk). The CDSSTR algorithm with reference set 4, optimized for analyzing spectra collected in the range from 240 to 190 nm, was used to quantitate secondary structural elements from collected CD spectra.66
Determination of Steady-State Kinetic Parameters Using a Coupled Enzyme Assay
A previously described coupled enzyme assay utilizing L-glutamate dehydrogenase and diaphorase67 was developed to monitor the rate of racemization of D-glutamate by glutamate racemase. Briefly, the L-glutamate produced from the stereoisomerization of D-glutamate by glutamate racemase is oxidatively deaminated by L-glutamate dehydrogenase to produce 2-oxoglutarate and NADH. The NADH formed is used by the second coupling enzyme diaphorase to reduce iodonitrotetrazolium chloride (INT), which has an absorbance maximum at 500 nm. Each reaction (1.30 mL) was performed at 25 °C in disposable PLAS-TIBRAND cuvettes containing 50 mM Tris-HCl (pH 8.0), 5 mM NAD+, 0.5 mM INT, 2.5 mM ADP, 20 units of L-glutamate dehydrogenase, 2 units of diaphorase, and D-glutamate at varying concentrations. Reactions were initiated by the addition of mutant or wild-type RacE, and the formation of reduced INT was monitored at 500 nm using a Cary 300 Bio UV–visible spectrophotometer from Varian Inc. (Palo Alto, CA), equipped with an 8 × 6 water thermostatted multicell holder. Data acquisition was coordinated using the CaryWinUV Kinetics Application v3.00, and a nonlinear curve fit was applied using GraphPad Prism v4.03 from GraphPad Software (San Diego, CA).
The relative molar absorptivity of reduced INT was calculated as previously described.67
Kinetic Assays Using Circular Dichroism Spectroscopy
Enzyme-catalyzed stereoinversion of L-glutamate was assayed using a J-720 circular dichroism spectrometer from JASCO. A thermostatted, cylindrical cuvette with a capacity of 700 μL and a path length of 1 cm was used for reactions below 5 mM, and a 350 μL cuvette with a 0.1 cm path length was used for reactions with higher substrate concentrations. L-Glutamate in an optically clear potassium borate buffer (50 mM boric acid, 100 mM KCl, 0.2 mM DTT, pH 8.0) was incubated at 25 °C in the presence of glutamate racemase (0.22 μM) at the indicated concentrations. L-Glutamate (concentrations ranged from 5 to 200 mM) was monitored by recording the CD signal 225 nm. Data acquisition was performed using the JASCO Spectra Manager v1.54A software, and a nonlinear curve-fit was applied using Kaleidagraph v3.6 (Synergy Software Inc.). For assays involving DPA or Compound 118, conditions are as above except that a 350 μL cylindrical cell with a 1 mm path length was employed in order to reduce the high absorbance due to the compounds. Initial rate data were globally fitted with GraphPad Prism v4.03 from GraphPad Software (San Diego, CA).
Results
Generating Structures That Are “Reactive” for Glutamate Racemization
These studies correspond to I from the flowchart in Figure 1.
Figure 1.
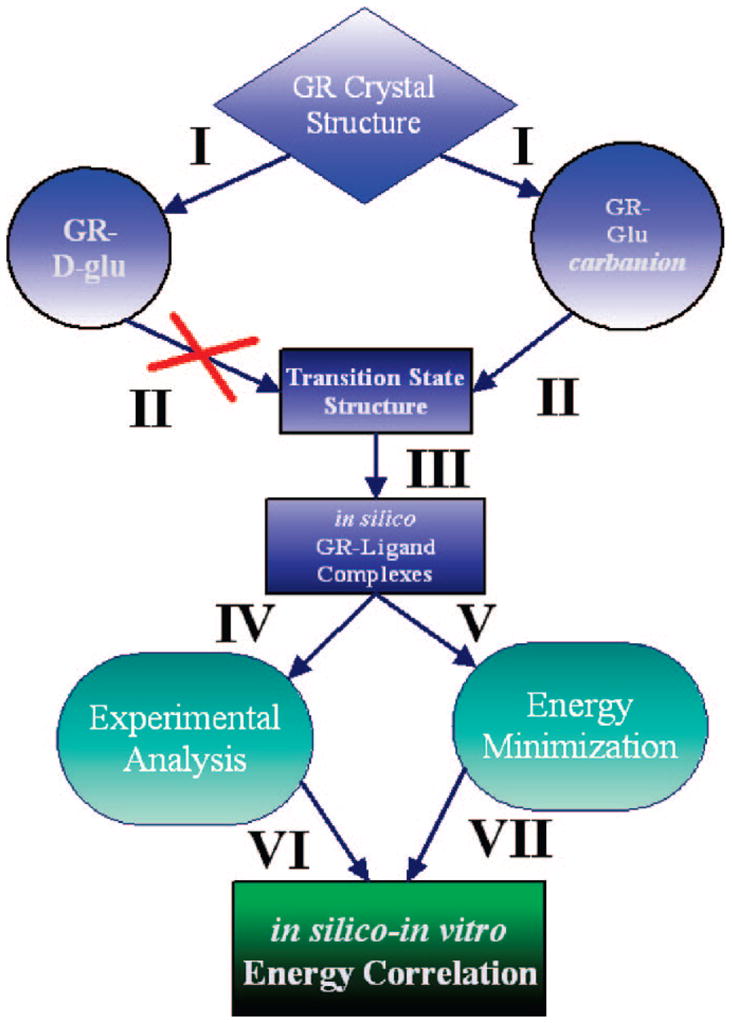
Overall method employed for prediction of ligand–enzyme binding energies for glutamate racemase. We start with the coordinates from the crystal structure of B. subtilis glutamate racemase (RacE) complexed with D-glutamate (PDB 1ZUW). Molecular dynamics simulations85 (I) are performed on this species or on RacE complexed with a Cα-deprotonated glutamate carbanion. For details of the parametrization of the glutamate carbanion and the conditions of the MD simulations, see Computational Procedures in the Supporting Information. Snapshots from MD simulations are used in geometry optimizations (II) to attempt to locate proton-transfer transition states between catalytic acid/bases (Cys74 or Cys185) and Cα of glutamate. (Both semiempirical PM386 and QM/MM 6-31G**/OPLS87 are employed; see Computational Procedures for details.) Snapshots from the simulation using the parametrized glutamate carbanion, but not the D-glutamte from the crystal structure, yielded a number of snapshots that were productive for locating transition states. One of these snapshots is then used as a target for high-throughput flexible ligand docking (III) against a database of over 1 million compounds, which included the Chemical Computing Group’s Lead-Like library of compounds in addition to a number of promising glutamate analogues. A docking rank was assigned to each ligand (based on the LigandFit88,89 module of the Cerius2 program, Accelrys Inc.), and the top 200 complexes are then energy-minimized (V). Experimental data on complexes of interest are either directly determined or extracted from the literature (IV). The experimental data and the in silico interaction energies are then directly compared to test the robustness of the computational procedure.
Extended simulations (>1 ns) of RacE starting from the D-glutamate complex (i.e., Cα is tetrahedral), with Cys74 as a thiolate, where the ligand is in the original conformation (an extended or “noncyclic” form as in the crystal structure), yielded geometries that were not consistent with Cα proton abstraction. Specifically, both the starting crystal structure itself and the equilibrium geometries yielded conformations in which the ammonium N of the D-glutamate was ~3.5 Å from Cys74’s thiolate sulfur atom, such that proton abstraction at the ammonium would be the strongly favored reaction, not abstraction of the Cα proton. Rather than perform a simulation of this system for an unspecified time, in hopes of capturing a potentially reactive geometry for Cα proton abstraction, we opted for an alternative strategy (vide infra).
An alternative approach to obtaining reactive geometries (i.e., those that are proximal to Cα proton-transfer transition states) for the glutamate racemase reaction from the ground-state Michaelis complex is to perform classical MD simulations in the presence of a probable reactive intermediate. The most likely high-energy intermediate, which may approximate the transition state, is the Cα-deprotonated glutamate, the glutamate carbanion. The change in ligand positioning and electrostatic environment may more accurately reflect the reactive form of the enzyme and provide an ensemble of reactive structures that may be investigated for proton transfer by various QM geometry optimization methods. However, the limitation with this approach is the need to perform a parametrization of the glutamate carbanion.
Parameterization of a Glutamate Carbanion
The details of the parametrization are given in the Supporting Information section. Figure SI-1 and Table SI-1 show that the molecular mechanics calculations using the parametrized (for MMFF94 force field) molecule reproduce the semiempirical geometry and the carbanion/water interaction energies within 1 kcal/mol of the semiempirical results (i.e., within 5.5% of the interaction energy calculated with semiempirical methods). In contrast, nonparameterized charges (assigned automatically) underestimate the strength of the water/carbanion interaction by about 27.5% of the semiempirical results and fail to replicate the water/carbanion energy-minimized geometry. AM1-level energy minimization of the glutamate carbanion form of the ligand led to a low-energy minimum that exhibits planar Cα, Cβ, N, and Cγ(COOH), and a hydrogen-bonding distance between the –NH3 and Cδ oxygen, forming a six-member-like “cyclic” species. Figure SI-2 juxtaposes the conformations of the planar glutamate carbanion and the extended form of D-glutamate found in the RacE crystal structure.
Classical MD Simulations Using the “Cyclic” and “Noncyclic” forms of Glutamate
Two dihedral angles capture the essential features of the internal geometry differences between the conformation of glutamate in the crystal structure and the energy-minimized glutamate carbanion. The N/Cα/Cβ/Cγ dihedral angle is 72.1° in the crystal structure (extended conformation of glutamate) and −47.5° in the energy-minimized “cyclic” structure. The Cα/Cβ/Cγ/Cδ dihedral angle is 59.3° in the crystal structure and 83.6° in the energy-minimized “cyclic” carbanionic structure. Furthermore, a salient difference between the RacE-D-Glu complex from the crystal structure and the cyclic carbanion obtained from MD snapshots (described below) is the hydrogen bond network with the enzyme. A ligand map of the RacE-D-Glu (Figure 2A) shows a number of backbone amide H-bonds, which are largely replaced by functional side chains in the reactive RacE-Glu-carbanion (Figure 2B). The active site of a representative reactive snapshot of RacE-Glu-carbanion, which highlights the ligand’s cyclic nature and the orientations of the proximal residues, is shown in Figure 3.
Figure 2.

Ligand map for RacE-D-Glu from crystal structure vs ligand map for RacE-Glu-carbanion “reactive” form with glutamate carbanion. The key to the ligand maps is indicated at the bottom of the figure. (A) Ligand map for the crystal structure (PDB 1ZUW) and (B) map derived from one of the MD snapshots with the glutamate carbanion that yielded proton-transfer transition states upon geometry optimization. In the reactive form (B), the glutamate carbanion receives its hydrogen bonds from water and the functional groups of the active site amino acids, taking on a cyclic shape to form an intramolecular hydrogen bond. The glutamate carbanion also accepts a weaker hydrogen bond from the backbone amide of Asn75 (not shown for clarity). In the crystal structure (A), the contacts are from a mixture of backbone amides, functional groups, and waters.
Figure 3.

Selective active-site contacts between glutamate carbanion and glutamate racemase. Glutamate racemase carbons are shown in gray, while those of glutamate carbanion are shown in light green hues. The van der Waals surface of the glutamate carbanion is indicated. Hydrogen bonds are depicted in yellow. (A) View looking down the NH3–Cα bond of the planar glutamate carbanion. The two catalytic acid/bases (Cys185 and Cys74) are shown above and below the plane of the carbanion, respectively. (B) Cyclic nature of the glutamate carbanion, which forms an intramolecular hydrogen bond between γ-carboxylate oxygen and NH3+.
In short, there is indirect evidence to support an alternative conformation of the substrate, which may be the reactive form (vis-à-vis proton abstraction from Cα). This is the impetus for a side-by-side investigation of the cyclic and noncyclic forms of glutamate carbanion. Figure SI-3 (Supporting Information) shows a superposition of the RacE-D-glutamate complex with both the RacE–noncyclic carbanion and RacE–cyclic carbanion complexes. The subtle realignment of the glutamate carbanion in the cyclic form produces a complex that is more amenable to in silico proton transfer from Cys185 and/or Cys74 to Cα, due to distances and angles between proton donor and acceptor. Figure SI-4A shows a plot of the distances between Cα and Cys(185 or 74) sulfur as a function of time, for the classical MD simulations of both cyclic and noncyclic carbanion conformations, respectively. Cα–Cys74 sulfur distances are about 0.7 Å closer for the cyclic than for the noncyclic conformation, but Cα–Cys185 sulfur distances are similar for the two conformations. However, the most useful metric was to analyze the MD snapshots for both the transfer angle and the distance between proton donor and acceptor. Figure 4 shows a plot of both the proton-transfer distance between donor (Cys185 or 74) and acceptor (Cα Glu-carbanion) and the proton-transfer angle. The snapshots selected for further analysis (i.e., transition-state geometry optimizations) were those with the most optimal distances and angles for proton transfer, and they are indicated by circled points in Figure 4. The distributions of coordinates from the simulation with the noncyclic carbanion only yielded reasonable proton transfer geometries between Cys185 and Cα, while distances between Cys74 and Cα did not exhibit geometries consistent with proton transfer (Figure SI-4B).
Figure 4.

Identifying optimal angles and distances for proton transfer (cyclic glutamate carbanion) from MD simulations. Plots of Cα–H–S angles and Cα–S distances from structures obtained from MD trajectories of the cyclic glutamate racemase–glutamate carbanion complex. The upper panel corresponds to coordinates of Cys74, and the lower panel corresponds to coordinates of Cys185. Structures that yielded angles and distances most appropriate for proton transfer were selected for semiempirical and QM/MM transition-state searches. The specific structures that were selected are circled in red.
Location of Transition States for Proton Transfer in RacE–Glutamate Carbanion Complexes Obtained from Classical MD Simulations
These studies correspond to II from the flowchart in Figure 1.
Semiempirical Transition-State Structures from MD Snapshots
In order to make an initial assessment of the potential for a given MD snapshot to yield a transition-state geometry for Cα proton transfer, we used small active-site models (<70 heavy atoms) of these MD snapshots, at the semiempirical PM3 level. Active-site models were constructed by using the coordinates from snapshots from the MD simulations indicated in Figure 4 (see Computational Procedures in the Supporting Information for details). Four structures were tested using the cyclic carbanion simulation (two with Cys185, Figure 4, lower panel, and two with Cys74, Figure 4, upper panel, proximal to Cα, respectively). Three of the four cyclic models converged on Cα proton-transfer transition states in the PM3 geometry optimization, two with proximal Cys185 and one with proximal Cys74. Figure SI-5A,B shows two of those semiempirical transition-state structures, for the proximal Cys185 (corresponding to what would be the transition state for Cα deprotonation in the L→D direction in the enzyme) and Cys74 (corresponding to what would be the transition state for the Cα deprotonation in the D→L direction in the enzyme).
The models constructed from two MD snapshots using the noncyclic glutamate carbanion (Figure SI-4B) did not yield any semiempirical transition states for proton transfer, using the same methodology as used for the cyclic snapshots. This may be due to a nonoptimal positioning of the S–H bond of Cys185 relative to the planar glutamate carbanion; in the case of the cyclic carbanion simulations, the S–H bonds of the Cys(185 or 74) were approximately perpendicular to the plane of the glutamate carbanion, while the noncyclic snapshots produced structures that were nearly parallel to the plane of the glutamate carbanion.
Ab Initio QM/MM Transition States from MD Snapshots
Two of the MD snapshots selected from the cyclic glutamate carbanion MD simulation (Figure 4), which were confirmed to yield PM3 transition states for proton transfer between Cys(185 or 74) and Cα, were further examined in QM/MM HF/6-31G**/OPLS geometry optimizations. In this case the entire system of enzyme, solvent, and ligand was included in the calculations. Transition states for proton transfer were located for both the proximal Cys74 (D→L direction) and Cys185 (L→D direction), as depicted in Figure SI-7. The salient features of the transition states relative to the crystal structure are summarized in the Supporting Information (Table SI-2). These data show (1) large movement of His187 imidazolium toward the oxygens of the γ-carboxylate, (2) large movement of the Asp10 carboxylate away from the ammonium nitrogen of the glutamate ligand, (3) closer distance between γ-carboxylate oxygen and nitrogen ammonium to form a more planar cyclic glutamate, and (4) repositioning of the distances of a number of the key hydrogen bond donors to the α- and γ-carboxylates.
There are a number of common features between the D→L and L→D transition-state structures. The QM/MM transition states are both late (relative to abstraction of Cα proton from tetrahedral glutamate, such that the transition state looks more like product than reactant), with S(Cys)–H–Cα angles of 179.2° and 171.2° for the D→L and L→D directions, respectively. For the D→L reaction the proton would be equidistant between donor and acceptor at 1.6 Å, which means that it is 0.15 Å late, relative to the equidistance point between heavy atoms. The equidistance point is a reasonable reference, due to the close proximity of stretching force constants for S–H and C–H bonds.68 Similarly, the L→D transition state is 0.25 Å late, relative to the equidistance point. This intuitively makes sense, as one would expect from the Hammond postulate for the high-energy carbanion intermediate to more resemble the transition state than the enzyme–D/L-glutamate ground state. Nevertheless, the glutamate substrate has some tetrahedral (i.e., chiral) character in both transition states. The 3.48 and 2.72 Å movement of the δ-N+ of His187 toward the O of the γ-carboxylate ligand in the D→L and L→D directions, respectively, relative to its location in the crystal structure is a striking feature of both transition states. The other common feature is the large displacement of the Asp10 carboxylate away from the ammonium of the ligand. The Asp10 carboxylate forms a short H-bond with a local crystal water in both transition states (2.39 and 2.44 Å between heteroatoms in the D→L and L→D directions, respectively).
Flexible Ligand Docking against a Reactive Target
These studies correspond to III and V from the flowchart in Figure 1.
High-throughput docking calculations using binding-site geometries from the reactive MD snapshots (i.e., using protein target structures that led to proton-transfer transition states) were performed against a lead-like library of over 1 million compounds, as indicated in the Supporting Information. A docking rank was assigned to each ligand (using the LigScore scoring function of the Cerius2 program, Accelrys Inc.). Importantly, the glutamate carbanion ranked ninth, overall. The top 200 scored compounds were globally energy-minimized with the protein target, explicit solvent, and ligand, and the ligand–protein interaction energy was determined. Those compounds that had strong ligand–protein interaction energies and which were commercially available were slated for further investigation. A number of compounds from this list were found to be inhibitors of RacE. Two of these are described in this study: dipicolinate dianion (or DPA) and Compound 118. Ligand interaction maps of these compounds in the active sites of RacE are illustrated in Figure SI-12A,B in the Supporting Information.
Determination of Ligand Binding Affinities of DPA and Compound 118
These studies correspond to IV from the flowchart in Figure 1.
DPA and Compound 118, which were identified in the docking studies above, were investigated for their inhibition properties on RacE. A CD-based assay of L-glutamate to D-glutamate stereoinversion was employed. DPA was found to be a competitive inhibitor, as determined by global kinetic analysis (Figure SI-10A) and Lineweaver–Burk plots (Figure SI-10B). The Ki for DPA was determined by globally fitting the initial rate kinetic data to the Michaelis–Menten equation for competitive inhibition and found to have a value of 1.97 ± 1.16 mM. For Compound 118, a structural analogue of DPA, the IC50 value was determined to be 2.9 ± 1.9 mM (Figure SI-11).
Determination of Cataltyic Parameters for Wild-Type and Mutants of RacE
These studies correspond to IV from the flowchart in Figure 1.
Determination of Steady-State Kinetic Parameters for Wild-Type RacE
The coupled enzyme assay (described in the Experimental Details section) was used to determine the wild-type RacE steady-state kinetic parameters in the D→L direction. Figure 5A shows the fit of the initial rate data from the wild-type enzyme to the Michaelis–Menten equation, which yields kcat = 1.3 ± 0.06 s−1 and KM = 0.25 ± 0.04 mM. The circular dichroism assay was used to determine the kinetic parameters in the L→D direction, as there is no coupled enzyme assay available in this direction. The fit of the initial rate data to the Michaelis–Menten equation in the L→D direction is shown in Figure SI-9, which yields kcat = 87 ± 9 s−1 and KM = 14 ± 6 mM. Importantly, the Haldane relationship, which asserts that the overall equilibrium for the reaction should be equal to kcat-AKM-B/kcat-BKM-A (A and B represent the forward and reverse reactions), is demonstrated in these studies. For a racemase the overall equilibrium is unity, so kcat/KM D→L should be within error of kcat/KM L→D, which is borne out in the data.
Figure 5.

Steady-state kinetic analysis of stereoinversion in the D→L glutamate direction for wild type and mutants of B. subtilis RacE. Where error bars are indicated, the measurement was averaged from three independent readings. The data were fitted to the Micahelis–Menten equation, and the fitted values are reported in Table SI-4. Data and curve fit (A) for the wild-type RacE and (B) for the T76A mutant. Analysis of the protein secondary structure of both wild-type and mutant enzymes was performed by circular dichroism spectroscopy. The secondary structural distributions of the wild type and T76A were found to be within error of one another (data are reported in Table SI-3).
Determination of Steady-State Kinetic Parameters for T76A RacE
The coupled enzyme assay (described in the Experimental Details section) was used to determine the T76A RacE steady-state kinetic parameters in the D→L direction. Figure 5B shows the fit of the initial rate data to the Michaelis–Menten equation, which yields kcat = 0.43 ± 0.05 s−1 and KM = 8.6 ± 1.4 mM.
The Energy Correlation: Experimental and Computational Energies
In order to assess the accuracy of the computational RacE–ligand interaction energy to report on binding free energies, a plot of the former as a function of the latter was approximated, using a number of assumptions, which are outlined here. All experimental binding energies were calculated using the standard ΔG = −RT ln(K) relationship. As one cannot directly measure the binding of a transition state/high-energy intermediate by binding studies, one must employ the thermodynamic relationship between the rate of the noncatalyzed reaction vs the rate of the enzyme-catalyzed reaction,69 K‡ = knon/(kcat/KM), where K‡ is the dissociation constant for the enzyme–transition state complex, knon is the rate constant for the uncatalyzed reaction (~10−11 s−1 for α amino acids52), kcat is the experimentally determined kcat, and KM is the experimentally determined KM for RacE (both kcat and KM are taken from the current study). A 1 M standard state is used for both the transition state and inhibitor dissociation constants. A graph of the linear fit between these trends is shown in Figure 6. The fitted slope was 7.9 ± 0.5; R = 0.986.
Figure 6.

Experimental binding free energy vs computational interaction energies of glutamate racemase–ligand complexes. Experimental binding energies are all ΔG values, while computational values are ligand–enzyme interaction energies calculated with MMFF94 from energy-minimized structures. The black line is a fit (slope = 7.6 ± 0.5; R = 0.989) to all data. All experimental binding energies were calculated using the standard ΔG = −RT ln(K) relationship. As one cannot directly measure the binding of a transition state/high-energy intermediate by direct binding, one must employ the thermodynamic relationship between the rate of the noncatalyzed reaction vs the rate of the enzyme-catalyzed reaction,69 K‡ = knon/(kcat/KM), where K‡ is the dissociation constant for the enzyme–transition state complex, knon is the rate constant for the uncatalyzed reaction (~10−11 s−1 for α amino acids52), kcat is the experimentally determined kcat, and KM is the experimentally determined KM (both kcat and KM are taken from the current study). This same procedure is used for both wild-type and T76A RacE. The Ki and IC50 for DPA (2.7 mM) and Compound 118 (2.9 mM), respectively, were taken from the current study. N-Hydroxyglutamate forms an elimination product (structure shown above) upon abstraction of its Cα proton by the Lactobacillus fermenti MurI, with Ki = 54 μM.14 D-Gln is a weak competitive inhibitor of Aquifex pyrophilus, with Ki = 50 mM.47 The KM values of D- and L-Glu, from the current study, were used to estimate the equilibrium dissociation constants for D- and L-Glu, respectively. A 1 M standard state is used for both the transition-state and inhibitor dissociation constants.
An interesting corollary to the strong relationship between RacE–ligand interaction energy and binding free energy is that the parametrized glutamate carbanion has very strong interaction energy (from the electrostatic interaction) with RacE. Furthermore, this strong RacE–glutamate carbanion interaction energy is notably dissipated with the T76A mutant, in such a way that it mirrors the calculated loss in transition-state binding energy based on the experimentally determined catalytic proficiency of T76A.
In order to parse the source for the strong electrostatic interaction between the glutamate carbanion and RacE, the contribution of each active-site chemical moiety to the interaction energy was individually examined. Figure 7 shows a bar graph of the contribution that each moiety makes to the glutamate carbanion–RacE interaction energy. It can be seen that the primary contributors to the interaction energy are the functional groups of Thr76, Asn75, and His187 and the three waters bound in the active site.
Figure 7.

Electrostatic contribution of individual moieties in the active site of RacE to the glutamate carbanion interaction energy. The calculations were performed using the MMFF94 force field with the parametrized glutamate carbanion. The employed RacE–glutamate carbanion complex was obtained from a snapshot from MD simulations, which was also used in QM/MM geometry optimizations for location of proton-transfer transition states. The negative energies in the bar graph all correspond to productive binding of the glutamate carbanion. Water 1 (Wat_1) is hydrogen-bonding to the α-carboxylate of the glutamate carbanion, water 2 is hydrogen-bonding to the ammonium group of glutamate carbanion, and water 3 is hydrogen-bonding to the γ-carboxylate of the glutamate carbanion.
Discussion
Obtaining and Evaluating Reactive RacE Structures
Previous computational studies on glutamate racemase have used the coordinates from the Aquifex pyrophilus MurI structure as a model. In some of those studies, it was necessary to protonate the Cα-carboxylate in order to obtain valid transition states for Cα proton abstraction, by assigning a general acid in the active site.49,50 Other studies focused on the distribution of reactive ground-state conformations using MD simulations rather than examining transition states for proton transfer.48 The goal of the present work was to employ computational methods to capture the dynamics of reactive enzyme–substrate interactions as well as the details of transition-state structures, focusing on how B. subtilis RacE glutamate racemase may catalyze proton abstraction at the Cα of glutamate. More specifically, the means by which the enzyme stabilizes the transition state is investigated by examining how the enzyme accommodates a high-energy carbanionic glutamate intermediate. There is strong experimental evidence that glutamate racemase proceeds by a stepwise mechanism, in which the intermediate would have carbanionic character.40,70 It is likely that this carbanionic intermediate is a good approximation to the transition state for proton abstraction from Cα, and it is expected to be much higher in energy than a glutamate–enzyme ground-state complex. In this study, snapshots of the RacE–glutamate carbanion complex were investigated to determine its ability to generate proton-transfer transition states in geometry optimizations.
The overall computational method used in the current study is designed to reduce computational costs by avoiding long exploratory MD simulations that may or may not lead to a reactive form of the substrate–enzyme complex, while at the same time not introducing the bias of a defined reaction coordinate for Cα proton transfer. These two goals are achieved by (1) starting the search for reactive intermediates using MD simulations with a probable high-energy intermediate–enzyme complex and (2) introducing filters (in the selection of reactive conformations) at the MM and semiempirical levels before attempting the more intensive ab initio searches.
A potential strength of this method is that there is no sampling bias along a predefined reaction coordinate. However, if one cannot obtain Boltzmann sampling to populate the reactive conformations, then the current method would not be beneficial. In such cases, enhanced sampling methods (e.g., umbrella sampling, free energy perturbation, etc.) are very important approaches. A drawback of the approach presented here is that it necessitates the parametrization of the reactive intermediate, the glutamate carbanion in this case, which may be time-consuming.
The cocrystal structure of B. subtilis RacE with D-glu is not in a catalytically competent conformation for removal of the glutamate Cα proton by the Cys74 thiol/thiolate (vide supra). Furthermore, low nanosecond MD simulations of the D-Glu–RacE complex, from the crystal structure, did not generate any catalytically competent geometries, as assessed by semiempirical and ab initio geometry optimizations. However, MD simulations of RacE with the glutamate carbanion yielded an ensemble of snapshots that were proximal to actual transition states, located by geometry optimization. Interestingly, MD snapshots of RacE–glutamate carbanion that did yield Cα proton-transfer transition states had an rmsd (all atom) vs the RacE–D-glu crystal structure of approximately 2.0 Å. The reactive snapshots from MD simulations produced Cα proton donor/acceptor angles and distances (illustrated in Figure 4) significantly more conducive to proton transfer than the crystal structure–glutamate carbanion complex, and also yielded more negative enzyme–glutamate carbanion interaction energies. These features of the “reactive” form of RacE—being proximal to geometries of Cα proton-transfer transition states and possessing strong enzyme–ligand interaction energy–could not be obtained by simply energy-minimizing the RacE–glutamate carbanion complex. Rather, MD simulation was necessary to traverse the local minima to reorganize the active site and achieve a proximal transition-state structure.
Significance of the “Carboxylate Hole”
A striking feature of the substrate liganded form of crystal structures for the three cofactor-independent racemases (and epimeraes) of glutamate racemase,42 DAP epimerase,46 and proline racemase45 is that there are numerous hydrogen bond donors surrounding the ligand carboxylate moieties. Before the publication of these structures, it was hypothesized that the α-carboxylate group of these amino acids would most likely need to be protonated before Cα abstraction.51 However, glutamate racemase has its carboxylates saturated with hydrogen bond donors, such that negative charge may be delocalized, in a manner similar to the “oxyanion hole” of serine proteases. This pocket is populated with a number of key threonine residues, which lack any general acids that would be capable of protonating the carboxylate.
In Vitro and In Silico Mutageneses Reveal the Strength and Distribution of the Electrostatic Interactions around the Carbanionic Intermediate
The strong interaction energy of the glutamate carbanion with the active site of glutamate racemase may be parsed by examining the individual electrostatic contributions from each active-site moiety (Figure 7). The bar graph predicts the electrostatic contribution (in kcal/mol) for each of the salient active-site moieties in this “reactive intermediate” pharmacophore. Figure 7 provides a strong foundation from which to design site-directed mutants for RacE and to interpret the results of any observed reduction in the steady-state parameters of said mutants relative to the wild-type enzyme. Figure 7 predicts that the source of the electrostatic component of the strong interaction energy between RacE and the glutamate carbanion is distributed, in a relatively even fashion, between a number of active-site moieties. It is interesting to note that most of the salient contacts from this reactive snapshot complex (except for Asn75 backbone amide) are through the residues’ functional groups, which means that we are able to test these hypotheses by simple mutagenesis. This hydrogen-bonding/electrostatic pattern is quite different than that observed for the ground-state pharmacophore (from the cocrystal structure with D-glu), which has numerous contacts with both backbone amides and functional groups (i.e., to Thr and Asn), which can be seen by comparing the ligand maps of Figure 2A,B. Only one of these mutants, T76A, was tested in this study.
There is a growing body of evidence that optimal hydrogen bonding in enzyme active sites may offer a significant contribution to catalytic accelerations.71 The importance of hydrogen bonds in stabilizing the enolate anion formed from deprotonated carbon acids was originally recognized by Gerlt and Gassman72 and Cleland and Kreevoy.73 Strong support for the enhancement of hydrogen bond strength in an enzyme’s active site was determined by the model studies of Herschlag,74,75 in which this reduction in free energy was quantified as being worth 0.93 kcal/mol/ΔpKa74 for a single hydrogen bond. This reduction in free energy would be substantial when considering the large increase in the pKa of the hydrogen bond acceptor (carboxylate carbonyl oxygen) as the Cα proton is removed, and the glutamate develops carbanionic/enolate anion character. Experimental validation for this effect is found in the mutagenesis study of Tonge and co-workers on enoyl-CoA hydratase,76 in which a glycine (whose amide nitrogen) is a hydrogen bond donor in the oxyanion hole is mutagenized to a proline, resulting in a 106-fold decrease in kcat (8.4 kcal/mol). Surprisingly, no such effect is observed in the current study.
The T76A mutant accounted for an increase in ΔΔG‡ of only 2.8 kcal/mol, which is within the range of a normal or “weak” hydrogen bond (in this case the β-hydroxyl of T76 to Cα carboxylate oxyanion of the carbanionic transition state). It has been suggested that the hydrogen bond donors in the Cα carboxylate binding pocket of cofactor-independent racemases constitute a “threonine pocket” reminiscent of the “oxyanion holes” of hydratases.43 However, it appears that the strength of the enzyme–transition state complex is much more distributed and weaker than the strong and focused contacts in proteases. It would not be unprecedented for hydrogen-bonding to the transition state to be the dominant driving force for catalysis. The classic studies on tyrosyl-tRNA synthetases by Fersht77–80 established that this enzyme could achieve the bulk of its catalytic power (15–20 kcal/mol of transition-state stabilization) by saturating the tyrosyl-AMP substrate with normal (i.e., not strong or low barrier) hydrogen bonds from enzymatic side chains in the active site.
Using the Reactive RacE Structure as an In Silico Screening Target
There exists a dearth of information about reversible binding of competitive inhibitors to glutamate racemase in the literature. There have been a number of recent studies on uncompetitive inhibitors of glutamate racemases that act at an allosteric position in selected species.44,81–83 The most detailed work on competitive inhibitors is that of de Dios et al., who utilized a family of 4-S modified D-glutamic acid analogues.13 These inhibitors were highly species-specific and relied on the presence of a nonconserved residue that lies at the interface of the active-site pocket and an adjacent hydrophobic pocket. As RacE possesses a key Val149 residue, which blocks access to this pocket, these inhibitors are very likely ineffective. The determinants of specificity for these 4-S modified D-glutamic acid inhibitors have been investigated.42,43,84 Given the strong interaction energy between the glutamate carbanion and RacE, and the particular complementary structure of this complex, it would be useful to identify inhibitors that possess features that support these characteristics. Docking of lead compounds into reactive active sites is one approach to this problem. Docking studies of a large lead-like library, of over 1 million compounds, did not yield any potential tight-binding inhibitors, as determined by RacE–ligand interaction energies from global energy minimization of the top 200 scoring compounds. Nevertheless, competitive inhibitors with a common motif (containing multiple carboxylate or sulfonic acid moieties) did emerge from the docking study, as indicated in Figure 6, albeit having Ki and IC50 values in the low millimolar range. A more direct approach would be to pursue de novo ligand discovery from the reactive RacE–Glu carbanion complex by docking chemical fragments into the target and identifying potential adducts that would most closely emulate the reactive complex.
Supplementary Material
Acknowledgments
This work was supported by NIH AI076830 (M.A.S.) and NIH AI057156 (S.R.B.).
Footnotes
Supporting Information Available: Computational procedures, Tables SI-1–SI-5, and Figures SI-1–SI-12. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bugg TD, Walsh CT. Nat Prod Rep. 1992;9:199–215. doi: 10.1039/np9920900199. [DOI] [PubMed] [Google Scholar]
- 2.Walsh CT. J Biol Chem. 1989;264:2393–2396. [PubMed] [Google Scholar]
- 3.Drysdale M, Heninger S, Hutt J, Chen Y, Lyons CR, Koehler TM. Embo J. 2005;24:221–227. doi: 10.1038/sj.emboj.7600495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keppie J, Smith H, Harris-Smith PW. Br J Exp Pathol. 1953;34:486–496. [PMC free article] [PubMed] [Google Scholar]
- 5.Makino S, Uchida I, Terakado N, Sasakawa C, Yoshikawa M. J Bacteriol. 1989;171:722–730. doi: 10.1128/jb.171.2.722-730.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Candela T, Fouet A. Mol Microbiol. 2006;60:1091–1098. doi: 10.1111/j.1365-2958.2006.05179.x. [DOI] [PubMed] [Google Scholar]
- 7.Uchida I, Makino S, Sasakawa C, Yoshikawa M, Sugimoto C, Terakado N. Mol Microbiol. 1993;9:487–496. doi: 10.1111/j.1365-2958.1993.tb01710.x. [DOI] [PubMed] [Google Scholar]
- 8.Baltz RH, Hoskins JA, Solenberg PJ, Treadway PJ. Method for knockout mutagenesis in Streptococcus pneumoniae. 5,981,281. US Patent. 1999
- 9.Doublet P, van Heijenoort J, Bohin JP, Mengin-Lecreulx D. J Bacteriol. 1993;175:2970–2979. doi: 10.1128/jb.175.10.2970-2979.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi K, Ehrich SD, Albertini A, et al. Proc Natl Acad Sci USA. 2003:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song JH, Ko KS, Lee JY, Baek JY, Oh WS, Yoon HS, Jeong JY, Chun J. Mol Cells. 2005;19:365–374. [PubMed] [Google Scholar]
- 12.Ashiuchi M, Yoshimura T, Esaki N, Ueno H, Soda K. Biosci Biotech Biochem. 1993;57:1978–1979. [Google Scholar]
- 13.de Dios A, Prieto L, Martin JA, Rubio A, Ezquerra J, Tebbe M, Lopez de Uralde B, Martin J, Sanchez A, LeTourneau DL, McGee JE, Boylan C, Parr TR, Jr, Smith MC. J Med Chem. 2002;45:4559–4570. doi: 10.1021/jm020901d. [DOI] [PubMed] [Google Scholar]
- 14.Glavas S, Tanner ME. Bioorg Med Chem Lett. 1997:2265–2270. [Google Scholar]
- 15.Kimura K, Tran LS, Itoh Y. Microbiology. 2004;150(Pt 9):2911–2920. doi: 10.1099/mic.0.27045-0. [DOI] [PubMed] [Google Scholar]
- 16.Tanner ME, Miao S. Tetrahedron Lett. 1994:4073–4076. [Google Scholar]
- 17.Faraci WS, Walsh CT. Biochemistry. 1988;27:3267–3276. doi: 10.1021/bi00409a022. [DOI] [PubMed] [Google Scholar]
- 18.Shaw JP, Petsko GA, Ringe D. Biochemistry. 1997;36:1329–1342. doi: 10.1021/bi961856c. [DOI] [PubMed] [Google Scholar]
- 19.Spies MA, Toney MD. Biochemistry. 2003;42:5099–5107. doi: 10.1021/bi0274064. [DOI] [PubMed] [Google Scholar]
- 20.Spies MA, Woodward JJ, Watnik MR, Toney MD. J Am Chem Soc. 2004;126:7464–7475. doi: 10.1021/ja049579h. [DOI] [PubMed] [Google Scholar]
- 21.Stamper CGF, Morollo AA, Ringe D. Biochemistry. 1998;37:10438–10445. doi: 10.1021/bi980692s. [DOI] [PubMed] [Google Scholar]
- 22.Stamper GF, Morollo AA, Ringe D. Biochemistry. 1999;38:6714. doi: 10.1021/bi995075y. [DOI] [PubMed] [Google Scholar]
- 23.Sun S, Toney MD. Biochemistry. 1999;38:4058–4065. doi: 10.1021/bi982924t. [DOI] [PubMed] [Google Scholar]
- 24.Watababe A, Kurokawa Y, Yoshimura T, Kurihara T, Soda K, Esaki N. J Biol Chem. 1999;274:4189–4194. doi: 10.1074/jbc.274.7.4189. [DOI] [PubMed] [Google Scholar]
- 25.Major DT, Gao J. J Am Chem Soc. 2006;128:16345–16357. doi: 10.1021/ja066334r. [DOI] [PubMed] [Google Scholar]
- 26.Major DT, Nam K, Gao J. J Am Chem Soc. 2006;128:8114–8115. doi: 10.1021/ja062272t. [DOI] [PubMed] [Google Scholar]
- 27.Babbitt PC, Hasson MS, Wedekind JE, Palmer DR, Barrett WC, Reed GH, Rayment I, Ringe D, Kenyon GL, Gerlt JA. Biochemistry. 1996;35:16489–16501. doi: 10.1021/bi9616413. [DOI] [PubMed] [Google Scholar]
- 28.Gerlt JA, Babbitt PC. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 29.Gerlt JA, Raushel FM. Curr Opin Chem Biol. 2003;7:252–264. doi: 10.1016/s1367-5931(03)00019-x. [DOI] [PubMed] [Google Scholar]
- 30.Kenyon GL, Gerlt JA, Petsko GA, Kozarich JW. Acc Chem Res. 1995;28:178–186. [Google Scholar]
- 31.Landro JA, Kallarakal AT, Ransom SC, Gerlt JA, Kozarich JW, Neidhart DJ, Kenyon GL. Biochemistry. 1991;30:9274–9281. doi: 10.1021/bi00102a020. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Zhang Y, Yang W. J Am Chem Soc. 2000;122:6560–6570. [Google Scholar]
- 33.Sims PA, Larsen TM, Poyner RR, Cleland WW, Reed GH. Biochemistry. 2003;42:8298–8306. doi: 10.1021/bi0346345. [DOI] [PubMed] [Google Scholar]
- 34.St Maurice M, Bearne SL. Biochemistry. 2002;41:4048–4058. doi: 10.1021/bi016044h. [DOI] [PubMed] [Google Scholar]
- 35.Fisher LM, Albery WJ, Knowles JR. Biochemistry. 1986;25:2529–2537. doi: 10.1021/bi00357a037. [DOI] [PubMed] [Google Scholar]
- 36.Fisher LM, Albery WJ, Knowles JR. Biochemistry. 1986;25:2538–2542. doi: 10.1021/bi00357a038. [DOI] [PubMed] [Google Scholar]
- 37.Fisher LM, Belasco JG, Bruice TW, Albery WJ, Knowles JR. Biochemistry. 1986;25:2543–2551. doi: 10.1021/bi00357a039. [DOI] [PubMed] [Google Scholar]
- 38.Gallo KA, Knowles JR. Biochemistry. 1993;32:3981–3990. doi: 10.1021/bi00066a019. [DOI] [PubMed] [Google Scholar]
- 39.Gallo KA, Tanner ME, Knowles JR. Biochemistry. 1993;32:3991–3997. doi: 10.1021/bi00066a020. [DOI] [PubMed] [Google Scholar]
- 40.Tanner ME, Gallo KA, Knowles JR. Biochemistry. 1993;32:3998–4006. doi: 10.1021/bi00066a021. [DOI] [PubMed] [Google Scholar]
- 41.Koo CW, Blanchard JS. Biochemistry. 1999;38:4416–4422. doi: 10.1021/bi982911f. [DOI] [PubMed] [Google Scholar]
- 42.Ruzheinikov SN, Taal MA, Sedelnikova SE, Baker PJ, Rice DW. Structure (Cambridge) 2005;13:1707–1713. doi: 10.1016/j.str.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 43.May M, Mehboob S, Mulhearn DC, Wang Z, Yu H, Thatcher GR, Santarsiero BD, Johnson ME, Mesecar AD. J Mol Biol. 2007;371:1219–1237. doi: 10.1016/j.jmb.2007.05.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lundqvist T, Fisher SL, Kern G, Folmer RH, Xue Y, Newton DT, Keating TA, Alm RA, de Jonge BL. Nature. 2007;447:817–822. doi: 10.1038/nature05689. [DOI] [PubMed] [Google Scholar]
- 45.Buschiazzo A, Goytia M, Schaeffer F, Degrave W, Shepard W, Gregoire C, Chamond N, Cosson A, Berneman A, Coatnoan N, Alzari PM, Minoprio P. Proc Natl Acad Sci USA. 2006;103:1705–1710. doi: 10.1073/pnas.0509010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pillai B, Cherney MM, Diaper CM, Sutherland A, Blanchard JS, Vederas JC, James MN. Proc Natl Acad Sci USA. 2006;103:8668–8673. doi: 10.1073/pnas.0602537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang KY, Cho CS, Kim SS, Sung HC, Yu YG, Cho Y. Nat Struct Biol. 1999;6:422–426. doi: 10.1038/8223. [DOI] [PubMed] [Google Scholar]
- 48.Mobitz H, Bruice TC. Biochemistry. 2004;43:9685–9694. doi: 10.1021/bi049580t. [DOI] [PubMed] [Google Scholar]
- 49.Puig E, Garcia-Viloca M, Gonzalez-Lafont A, Lluch JM. J Phys Chem A. 2006;110:717–725. doi: 10.1021/jp054555y. [DOI] [PubMed] [Google Scholar]
- 50.Puig E, Garcia-Viloca M, Gonzalez-Lafont A, Lluch JM, Field MJ. J Phys Chem B. 2007;111:2385–2397. doi: 10.1021/jp066350a. [DOI] [PubMed] [Google Scholar]
- 51.Rios A, Crugeiras J, Amyes TL, Richard JP. J Am Chem Soc. 2001;123:7949–7950. doi: 10.1021/ja016250c. [DOI] [PubMed] [Google Scholar]
- 52.Williams G, Maziarz EP, Amyes TL, Wood TD, Richard JP. Biochemistry. 2003;42:8354–8361. doi: 10.1021/bi0345992. [DOI] [PubMed] [Google Scholar]
- 53.Kline PC, Schramm VL. Biochemistry. 1993;32:13212–13219. doi: 10.1021/bi00211a033. [DOI] [PubMed] [Google Scholar]
- 54.Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. Biochemistry. 1998;37:8615–8621. doi: 10.1021/bi980658d. [DOI] [PubMed] [Google Scholar]
- 55.Schramm VL. Acc Chem Res. 2003;36:588–596. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
- 56.Schramm VL. Arch Biochem Biophys. 2005;433:13–26. doi: 10.1016/j.abb.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 57.Li CM, Tyler PC, Furneaux RH, Kicska G, Xu Y, Grubmeyer C, Girvin ME, Schramm VL. Nat Struct Biol. 1999;6:582–587. doi: 10.1038/9367. [DOI] [PubMed] [Google Scholar]
- 58.Scheuring J, Berti PJ, Schramm VL. Biochemistry. 1998;37:2748–2758. doi: 10.1021/bi972594x. [DOI] [PubMed] [Google Scholar]
- 59.Guo H, Rao N, Xu Q, Guo H. J Am Chem Soc. 2005;127:3191–3197. doi: 10.1021/ja0439625. [DOI] [PubMed] [Google Scholar]
- 60.Xu Q, Guo H. J Phys Chem B. 2004;108:2477–2483. [Google Scholar]
- 61.Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. Proc Natl Acad Sci USA. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agarwal PK. J Am Chem Soc. 2005;127:15248–15256. doi: 10.1021/ja055251s. [DOI] [PubMed] [Google Scholar]
- 63.Alhambra C, Gao J, Corchado JC, Villà J, Truhlar DG. J Am Chem Soc. 1999;121:2253–2258. [Google Scholar]
- 64.Meng Q, Switzer RL. J Bacteriol. 2001;183:5513–5522. doi: 10.1128/JB.183.19.5513-5522.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 66.Lobley A, Whitmore L, Wallace BA. Bioinformatics. 2002;18:211–212. doi: 10.1093/bioinformatics/18.1.211. [DOI] [PubMed] [Google Scholar]
- 67.Rej R. Anal Biochem. 1982;119:205–210. doi: 10.1016/0003-2697(82)90687-x. [DOI] [PubMed] [Google Scholar]
- 68.Lide DR. CRC Handbook of Chemistry and Physics. 85. CRC Press; Boca Raton, FL: 2004. pp. 9–77. [Google Scholar]
- 69.Wolfenden R. Annu Rev Biophys Bioeng. 1976;5:271–306. doi: 10.1146/annurev.bb.05.060176.001415. [DOI] [PubMed] [Google Scholar]
- 70.Glavas S, Tanner ME. Biochemistry. 1999;38:4106–4113. doi: 10.1021/bi982663n. [DOI] [PubMed] [Google Scholar]
- 71.Gerlt JA. Enzymatic catalysis of proton transfer at carbon atoms. In: Schowen RL, Klinman J, editors. Handbook of Hydrogen Transfer. Volume 2: Biological Aspects of Hydrogen Transfer. Wiley-VCH; Weinheim: 2006. [Google Scholar]
- 72.Gerlt JA, Gassman PG. J Am Chem Soc. 1993;115:11552–11568. [Google Scholar]
- 73.Cleland WW, Kreevoy MM. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 74.Shan SO, Herschlag D. Proc Natl Acad Sci USA. 1996;93:14474–14479. doi: 10.1073/pnas.93.25.14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shan SO, Loh S, Herschlag D. Science. 1996;272:97–101. doi: 10.1126/science.272.5258.97. [DOI] [PubMed] [Google Scholar]
- 76.Bell AF, Wu J, Feng Y, Tonge PJ. Biochemistry. 2001;40:1725–1733. doi: 10.1021/bi001733z. [DOI] [PubMed] [Google Scholar]
- 77.Fersht AR, Leatherbarrow RJ, Wells TN. Biochemistry. 1987;26:6030–6038. doi: 10.1021/bi00393a013. [DOI] [PubMed] [Google Scholar]
- 78.Fersht AR. Biochemistry. 1987;26:8031–8037. doi: 10.1021/bi00399a001. [DOI] [PubMed] [Google Scholar]
- 79.Wells TN, Fersht AR. Biochemistry. 1986;25:1881–1886. doi: 10.1021/bi00356a007. [DOI] [PubMed] [Google Scholar]
- 80.Wells TN, Ho CK, Fersht AR. Biochemistry. 1986;25:6603–6608. doi: 10.1021/bi00369a040. [DOI] [PubMed] [Google Scholar]
- 81.Breault GA, Comita-Prevoir J, Eyermann CJ, Geng B, Petrichko R, Doig P, Gorseth E, Noonan B. Bioorg Med Chem Lett. 2008;18:6100–6103. doi: 10.1016/j.bmcl.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 82.Geng B, Breault G, Comita-Prevoir J, Petrichko R, Eyermann C, Lundqvist T, Doig P, Gorseth E, Noonan B. Bioorg Med Chem Lett. 2008;18:4368–4372. doi: 10.1016/j.bmcl.2008.06.068. [DOI] [PubMed] [Google Scholar]
- 83.Basarab GS, Hill PJ, Rastagar A, Webborn PJ. Bioorg Med Chem Lett. 2008;18:4716–4722. doi: 10.1016/j.bmcl.2008.06.092. [DOI] [PubMed] [Google Scholar]
- 84.Dodd D, Reese JG, Louer CR, Ballard JD, Spies MA, Blanke SR. J Bacteriol. 2007;189:5265–5275. doi: 10.1128/JB.00352-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Molecular Operating Environment, version 2005–2009. Chemical Computing Group; Montreal, Quebec, Canada: [Google Scholar]
- 86.Spartan, version 04/1.0.3. Wavefunction Inc; Irvine, CA: [Google Scholar]
- 87.Q-site version 4.0. Schrodinger, LLC; New York, NY: 2005. [Google Scholar]
- 88.Cerius2 LigandFit 4.10. Accelrys, Inc; San Diego, CA: [Google Scholar]
- 89.Venkatachalam CM, Jiang X, Oldfield T, Waldman M. J Mol Graph Model. 2003;21:289–307. doi: 10.1016/s1093-3263(02)00164-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.