

Abstract
Nanomaterials for targeted delivery are uniquely capable of localizing delivery of therapeutics and diagnostics to diseased tissues. The ability to achieve high, local concentrations of drugs or image contrast agents at a target site provides the opportunity for improved system performance and patient outcomes along with reduced systemic dosing. In this review, the design of targeted nanodelivery systems is discussed with an emphasis on in vivo performance, the physicochemical properties that affect localization at the target site, and the incorporation of therapeutic drugs into these systems.
Keywords: Targeted drug delivery, active targeting, biodistribution, drug loading
Introduction
Successful targeted delivery systems are designed to allow delivery of therapeutic or diagnostic agents to a preferential site. As targeted nanodelivery involves local delivery of therapeutics and diagnostics at disease sites, this method has received considerable attention over the last 15 years and is poised to have a significant impact on medicine. Efficient targeted delivery systems allow for a reduced systemic dosage while resulting in relatively higher or more efficient dosing at the target site. The promising benefits of reduced systemic side effects and simultaneously improved efficacy have fueled the development of this field. To date, targeted delivery has become a rich field of drug delivery and nanomaterials.
Nanoscale materials are a necessity for most targeted delivery systems as these systems must be allowed to transport through different tissue spaces in order to localize at the target site. For tumor delivery, intravenously administered particles must circulate in the bloodstream and be small enough to escape circulation through tumor microvasculature which typically requires the system to have a diameter less than 100 nm to 2 μm [1]. In other applications such as the treatment and diagnosis of atherosclerotic plaques, targeted delivery systems have been developed on the order of 10 nm to greater than 1 μm.
The ability of nanoparticles to localize at a target site is dependent on a number of factors. In general, it is unclear whether chemical properties, the presence of a targeting ligand, or size is the primary determinant for nanoparticle biodistribution. In many cases, it is a combination of these properties that shapes biodistribution. One of the advantages of nanoparticle delivery systems is that the circulation time of drugs and diagnostic agents can be prolonged, and delivery can be controlled through targeting.
Even with targeted delivery, only a fraction of the administered dose localizes at the target site while the remaining nanoparticles distribute throughout the body. At this point, pharmacokinetics pertaining to the nanodelivery system determine the dose in nontargeted tissues. A furthered understanding of nanoparticle biodistribution and pharmacokinetics will be a significant contribution to the successful development and translation of targeted delivery systems.
An often overlooked design parameter in targeted delivery systems is the method and amount of incorporation of the drug or diagnostic agent. There are a variety of methods for incorporating these functions into a delivery system which depend on the nanomaterial and the agent of interest. The small volumes of nanomaterials inherently limit their maximum payloads, and their sizes present challenges to traditional purification and measurement techniques.
From current work in the field, it is evident that the design of optimized targeted delivery systems will be based on the drug or agent of interest, the nanoparticle type that allows sufficient loading of the drug, and the physicochemical properties that allow for targeting. We will highlight some of this recent work on targeted delivery systems and focus on in vivo performance, localization, and the incorporation of diagnostic and therapeutic agents in targeted delivery systems.
In vivo studies on targeted delivery: localization, uptake, and performance
One of the key features of targeted delivery systems is the addition of targeting ligands to the surface of the nanoparticle either through physical adsorption or more commonly chemical attachment. Various in vitro and in vivo studies have shown that the addition of these targeting ligands improves the therapeutic effects of the delivery systems compared to control systems without the targeting ligands. Nanodelivery systems with targeting ligands are often referred to as actively targeted systems but will be referred to as targeted delivery systems throughout this paper. Nanodelivery systems without targeting ligands can be passively targeted systems but will be referred to as nontargeted systems in this paper. Recent in vivo studies have shed light on some of the mechanisms behind this improvement in therapeutic efficacy. Findings from these studies suggest that the targeting ligands act less like a homing device seeking out the target site but more like sticky shoes that improve nanoparticle association with the target cells’ membranes or even uptake by the target cells. In this section, we will cite recent studies on targeted delivery systems focusing on comparisons against nontargeted delivery systems for the two most common applications of targeted delivery: targeting of tumors and atherosclerotic plaques.
Tumor targeting
Nanoparticles are well-suited materials for targeted tumor delivery due to their ability to circulate in the bloodstream for relatively long time periods and their ability to accumulate in tumor spaces. Often, actively targeted nanodelivery systems are modifications of passively targeted delivery systems. A number of studies have shown improved treatment in vivo and in vitro with targeted systems. However, there is growing evidence to suggest that for many nanoparticle delivery systems accumulation at the tumor site is independent of the presence or absence of a targeting ligand. In these cases, the enhanced permeability and retention (EPR) effect is the dominating mechanism for localization at the tumor site. Exploitation of the EPR effect requires careful size selection of nanoparticles that allows nanoparticles to extravasate leaky tumor vasculature and to be retained in the tumor due to insufficient lymphatic drainage. However, in a number of studies, nanodelivery systems with targeting ligands have performed better than nontargeted systems. This improved performance or more effective treatment has been attributed to improved nanoparticle association with target cell membranes and target cell internalization. We will discuss a variety of recent in vivo studies which have compared targeted and nontargeted nanodelivery systems towards understanding the relationship between active and passive tumor targeting.
Active Tumor Targeting
This relationship between cell uptake and performance of targeted nanodelivery systems has been demonstrated in several studies with folate targeted-nanoparticles. To target tumor cells overexpressing the folate receptor, folate was incorporated into the nanoparticle delivery system as a targeting ligand.
In a study by Wang et al [2], the biodistribution and performance of targeted nanoparticles composed of heparin-folate-paclitaxel conjugates loaded with paclitaxel (HFT-T) were compared to nontargeted nanoparticles of heparin-paclitaxel conjugates loaded with paclitaxel (HT-T). HFT-T targeted systems significantly reduced tumor volume over nanoparticle and paclitaxel controls in a KB-3-1 human nasopharyngeal carcinoma xenograft-bearing mouse model. Despite this, biodistribution studies revealed that the difference between accumulation of targeted and nontargeted systems in the tumor was not statistically significant. However, fluorescence microscopy of the tumor cells showed that the targeted systems were more commonly internalized. These results were confirmed by flow cytometry which showed that the targeted nanoparticles were found in three times more tumor cells than nontargeted nanoparticles (21.8 ± 2.9% cells compared to 5.6 ± 1.8% cells). This phenomenon was further evidenced by in vitro studies which showed the uptake of targeted nanoparticles was higher than nontargeted nanoparticles and was folate-dependent. In this study, the improved performance of targeted nanoparticles was due to enhanced cell uptake, rather than enhanced localization compared to nontargeted nanoparticles.
Likewise, comparable biodistributions and tumor accumulation between folate receptor-targeted and nontargeted gadolinium (Gd) nanoparticles has been observed in a KB xenograft-bearing athymic mouse model [3]. The results showed that the targeted and nontargeted systems had similar biodistributions and that there was no statistical difference in tumor localization at either 5 or 8 hours. Furthermore, there was a higher tumor cell uptake of targeted nanoparticles in vitro and an enhanced retention following intratumor injection of nanoparticles in vivo. While this study did not examine the tumor microdistribution of nanoparticles in vivo, it relied on in vitro data to support the assertion that enhanced internalization was a feature of folate-coated Gd nanoparticles.
Tumor delivery via folate receptor targeting has also been evaluated using micelles with differing amounts of the targeting ligand [4]. Targeted micelles were formed with varying amounts of folate by increasing the molar ratio of folate-PEG-poly(aspartate-hydrazone-adriamycin) [Fol-PEG-p(Asp-Hyd-ADR)] to methoxy-PEG-poly(aspartate-hydrazone-adriamycin) [PEG-p(Asp-Hyd-ADR)] while nontargeted control micelles were formed entirely from PEG-p(Asp-Hyd-ADR). The effective dose for the targeted system improved upon the nontargeted system, which was also lower than the effective dosage for the free drug in in vivo studies with tumor-bearing CD-1 nude mice. These results indicated that targeted micelles (containing 10% of the folate-conjugated polymer) performed more effectively than nontargeted micelles despite similar pharmacokinetic profiles of micelle accumulation in the tumor and relatively close area under the curves (AUC) for the drug accumulation in tumors (132.67 and 143.60 percent initial dose (%ID)/g organ × h, respectively). While the internalization of targeted and nontargeted nanoparticles was not monitored in vivo in this study, the preferential internalization of targeted nanoparticles is hypothesized to be the reason for the improved effective dose.
Another common targeting ligand, transferrin (Tf), has been conjugated to a variety of targeted delivery systems to target overexpressed Tf receptors common to many cancers. As observed in folate receptor targeted systems, nanoparticles with Tf targeting ligands improve delivery but have a biodistribution and tumor accumulation similar to nontargeted systems [5]. For example, Bartlett et al focused on delivery of siRNA using cyclodextrin-containing polycations and used positron emission tomography (PET) and bioluminescence imaging to simultaneously monitor the nanoparticle location and siRNA activity as shown in Figure 2. In this study, siRNA activity was higher for the targeted delivery systems (Figure 2c). The improved siRNA activity was attributed to a greater tumor cell internalization of the targeted nanoparticles versus the nontargeted nanoparticles, because the tumor accumulation of targeted and nontargeted nanoparticles was similar. Approximately 1% initial dose (ID) accumulated per tumor volume one day after injection. Additionally, Bartlett et al found that nanoparticle accumulation in tumors was independent of the Tf-receptor level.
Figure 2.
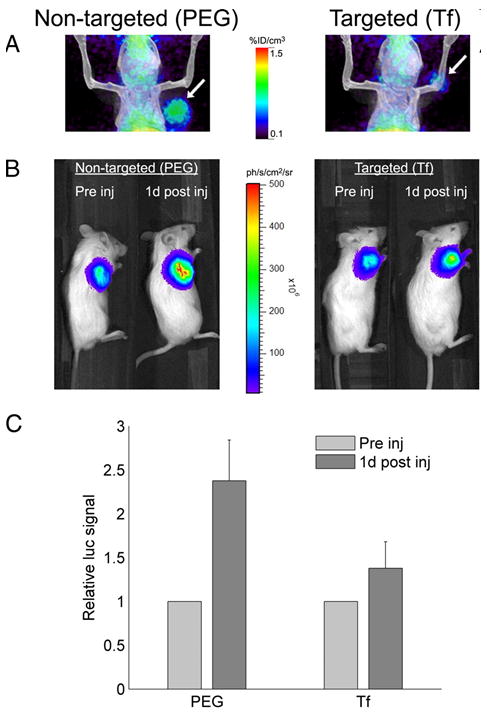
The in vivo performance of siRNA administered via targeted and nontargeted nanodelivery systems. (a) Micro-PET/CT images of mice show the distribution of targeted and nontargeted nanoparticles in tumors (arrow) 1 day after administration. (b) Bioluminescence imaging of mice shows luciferase expression in the tumor before and 1 day after administration of nontargeted and targeted nanoparticles. (c) A decrease in luciferase activity was associated with siRNA activity 1 day after administration of targeted (Tf) nanoparticles and nontargeted (PEG) nanoparticles. Error bars represent standard error. Reproduced from [5]. Copyright 2007 National Academy of Sciences, U.S.A.
In addition to the improvements in delivery via folate and transferrin targeting, enhanced performances due to nanoparticle internalization have been seen for multiple systems with antibody-directed targeting. In a study focused on spectroscopic detection of cancer using surface-enhanced Raman (SERS) via colloidal gold Raman nanotags, single-chain variable fragment antibodies for epidermal growth factor receptor (EGFR) were harnessed for targeting of 80 nm nanotags [6]. Transmission electron micrographs of tumor cells revealed that the targeted nanotags were more readily internalized. The enhanced internalization of targeted nanotags paralleled an increase in Raman signal intensity from tumors treated with the targeted nanotags. Biodistribution measurements showed that the accumulation and retention of EGFR targeted nanotags in the tumor was about 10 times greater than that of nontargeted controls.
Similar findings have been reported for antibody targeting of liposomes. For example, liposomes possessing an anti-HER2 monoclonal antibody (MAb) composed of phosphatidylcholine and polyethylene glycol (PEG)-modified distearoylphoshphatidylethanolamine (DSPE) (PEG-DSPE) with an average particle diameter of 100 nm were shown to improve antitumor efficacy of doxorubicin over various control formulations [7]. Kirpotin et al further evaluated the biodistribution and uptake of these targeted liposomes containing the anti-HER2 MAb compared to nontargeted liposomes [8]. They found that the biodistribution of targeted and nontargeted liposomes was similar. In fact, localization of targeted and nontargeted liposomes in HER-2 overexpressing xenografts (BT-474) and non-HER-2-overexpressing xenografts (MCF-7) were not statistically different with a range of 7–8% of the injected dose per gram of tumor tissue (ID/g tumor). Targeted and nontargeted liposomes were shown to extravasate tumor blood vessels in both xenograft models, yet a significant difference between the targeted and nontargeted liposomes was observed in the microdistribution within HER-2-overexpressing tumor xenografts. Targeted liposomes were found predominantly in tumor cells while nontargeted liposomes were found in the extracellular spaces or in macrophages associated with tumor tissue. For targeted liposomes, there was a four-fold greater preference for nanoparticle localization in xenograft tumor cells rather than host cells while there was no statistical preference for tumor cells over host cells in the case of nontargeted liposomes.
Likewise, ElBayoumi and Torchilin used antibody targeting to enhance delivery of doxorubicin loaded PEGylated liposomes [9]. The targeting ligand, monoclonal antibody 2C5 (mAb 2C5) with nucleosome-restricted activity, increased accumulation of liposomes in Lewis lung carcinoma tumor models over nontargeted systems. Following administration of liposomes, a greater reduction in tumor volume was achieved by doxorubicin delivery from targeted liposomes.
Passive Targeting of Tumors
While the active targeting schemes described above involve the use of molecules designed to specifically interact with a physiological target, passive targeting takes advantage of natural properties and processes in tissues to localize a delivery agent at a desired target site. The most common type of passive targeting is tumor targeting by the EPR effect. To exploit this effect, the size of nanoparticles is designed to allow nanoparticles to extravasate from defects in tumor vasculature. Nanoparticles between 100–200 nm have been shown to accumulate in tumors by the EPR effect. Larger molecules such as nanoparticles can diffuse out of defective tumor vasculature but are retained in the tumor space due to insufficient venous and lymphatic clearance. The retention of large molecules such as nanoparticles is typically higher than small molecules which can more easily diffuse back into vasculature as shown in Figure 3 [10]. In addition to appropriate size, nanoparticles must have properties that allow for a long circulation time. Passive tumor targeting by the EPR effect requires a driving concentration gradient of nanoparticles for a sustained time period of at least 6 hours [10, 11]. As a result, PEGylation and surface charge are important design parameters due to their known effect on circulation times. In vivo targeting delivery studies suggested that the presence of a targeting ligand does not appear to significantly affect extravasation, but inefficient extravasation could significantly affect targeted delivery.
Figure 3.
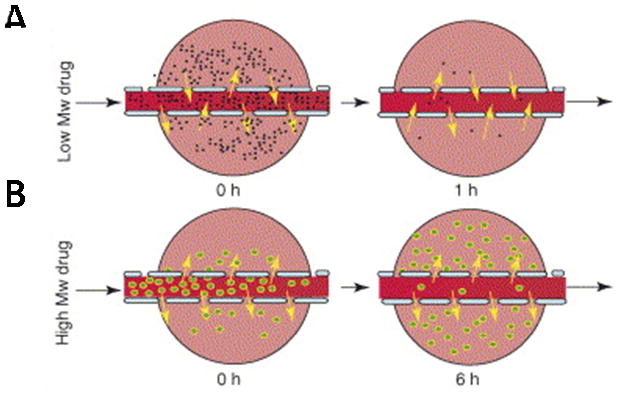
Transport of small and large molecular weight drugs and nanoparticles from defective tumor vasculature into tumor space. (a) Small molecular weight drugs (black) can diffuse out of tumor vasculature but can also diffuse back into tumor vasculature which allows clearance of these compounds from the tumor space. (b) Larger molecular weight drugs or nanoparticles (green) can diffuse into the tumor space but are retained due to the EPR effect. Reprinted from [10], Copyright 2006, with permission from Elsevier.
Challenges to Tumor Delivery
One of the challenges to developing a successful tumor targeting delivery platform, is that even with passive and active targeting only a fraction of the administered dose accumulates in the tumor. This brings two questions to light: is there enough accumulation at the target site and what is the systemic dose?
In short, effective delivery can be achieved at the tumor site. Whether the dose administered is an effective dose is easily gauged by a sufficient suppression in tumor growth. The effective dose is dependent on nanoparticle localization at the tumor site but also on the maximum payload that each nanoparticle can deliver. To put this into perspective, nanoparticles localize in tumors at much lower levels than the administered dose (Table 1). So, if the amount of drug or image contrast agent is increased in each nanoparticle, a higher dose arrives at the tumor site. However, there is a balance between the payload and the number of nanoparticles. The fraction of the administered dose that localizes in the tumor relative to the mass of the tumor (%ID/g tumor) is also lower than other in organs (the % ID/g tissue) such as the liver [12]. What this tells us is that other organs have an increased exposure per gram of tissue to the nanoparticles and their payloads than the tumor often does. So, an increase in the drug or contrast agent could be detrimental.
Table 1.
Summary of studies on tumor accumulation of nanodelivery systems
| Formulation | Tumor Accumulation | Dose | Tumor and Animal Model | Study | |
|---|---|---|---|---|---|
| PEGylated Gadolinium nanoparticles | Folate and PEG-coated PEGylated |
5% ID at 5 h, 7% ID at 8 h 4% ID at 5 h, 9% ID at 8 h |
10 mg NPs/kg mouse weight (1.6 mg Gd/kg) | KB, athymic mice | [3] |
| PEG-p(ASP-Hyd-ADR) nanoparticles | Folate PEGylated |
6–8% ID at 4 h, 4–7 % ID at 24 h ~6% ID at 4 h, ~4% ID at 24 h |
10 mg/kg ADR | KB, CD-1 nude mice | [4] |
| 64Cu labeled shell-crosslinked nanoparticles of PAA-b-PMA | Folate Nontargeted |
5.9 ± 2.8% ID/g, 0.04 ± 0.02 %ID 6.0 ± 1.9% ID/g, 0.13 ± 0.04 %ID at 4 h |
370–440 kBq NPs or 3–5 mg/kg mouse weight | KB, folate-deficient athymic nude mice | [12] |
| 64Cu labeled nanoparticles with cyclo-dextrin containing polycations | Tf PEGylated |
< 2% ID/cm3 over 60 min < 2% ID/cm3 |
100–300 μCi 64Cu | Neuro2A-Luc, NOD/SCID mice | [5] |
| Colloidal gold nanoparticles | EGFR ScFv nanotags PEGylated nanotags |
~ 8 ppm (gold/g tissue) at 5 h <1 ppm |
45 femtomoles | Tu686, nude mice | [6] |
| Immunoliposomes | Anti-HER2 liposomes | 8.34% ID/g tissue at 24 h 7.18% ID/g tissue |
40 μmol/kg mouse weight | BT-474, mice MCF-7, mice |
[8] |
| Nontargeted liposomes | 7.32% ID/g tissue 8.59% ID/g tissue |
BT-474 MCF-7 |
|||
| PEGylated gold nanoparticles | 20 nm 80 nm |
6.63 % ID at 48 hours 0.30% ID |
2.6 × 1011 NPs/mouse | A431, mouse | [24] |
| Hollow gold nanospheres with α–melanocyte-stimulating hormone analog (NDP-MSH) | NDP-MSH-PEG-HauNS PEG-HAuNS |
4.3 +−1.2% ID/g at 4 hours 12.6 +− 3.1% ID/g |
2 × 1012 NPs/mouse | B16/F10, mouse | [75] |
List of abbreviations for Table 1: initial dose (ID), nanoparticles (NPs), poly(ethylene glycol)-poly(aspartate-hydrazone-adriamycin) (PEG-p(ASP-Hyd-ADR)), Gadolinium (Gd), poly(acrylic acid-b-methyl acrylate) (PAA-b-PMA), epidermal growth factor receptor (EGFR), single-chain variable fragment antibody (ScFv), transferrin (Tf), α-melanocyte-stimulating hormone analog (NDP-MSH), PEGylated-hollow gold nanospheres (PEG-HauNS)
A greater cell internalization accompanied by a simultaneous improvement in performance of targeted nanodelivery systems is a recurring theme in cancer delivery. Clearly, the selection of the targeting ligand is important, but the nanoparticle properties that influence accumulation in the tumor are important design parameters as well.
Targeting of atherosclerotic plaques
Second to tumor targeting, the most common application of targeted imaging and treatment is atherosclerosis. In targeting atherosclerosis, the delivery system circulates in the bloodstream and must adhere to a specific region of the vasculature. The small size and relatively large surface area of nanoparticles makes them attractive candidates for this application. Because circulating nanodelivery systems must adhere to atherosclerotic lesions, understanding nanoparticle adhesion and behavior in flow conditions is extremely important, although it is beyond the scope of this paper. To date, the majority of in vivo studies have focused on the delivery of image contrast agents. As a result, the preferential accumulation of targeted nanoparticles at atherosclerotic lesions has often been confirmed by improved image contrast or increases in the signal at the target site relative to the background.
In one such study, an improved contrast in fluorescence imaging and delivery of a heparin hirulog therapeutic was achieved using micelles with a clot-binding peptide, cysteine-arginine-glutamic acid-lysine-alanine (CREKA) to target fibrin deposits on atherosclerotic plaques [13]. The micelles were formed from PEG-DSPE polymers with variable head groups labeled with carboxyfluorescein (FAM-labeled) for imaging, CREKA for targeting, or a heparin hirulog for treatment. Results showed that the targeted micelles improved the fluorescence intensity of atherosclerotic plaques in the aortic tree over nontargeted micelles as well as the delivery of the heparin hirulog as seen in Figure 4. The CREKA targeting ligand was shown to be responsible for this improved contrast because through competitive inhibition studies. The contrast provided by targeted FAM-labeled micelles was depleted with the addition of excess unlabeled targeted micelles but remained the same with the addition of excess unlabeled nontargeted micelles. Additionally, CREKA targeted micelles were more effective than nontargeted control micelles in delivering hirulog to plaques in an ApoE-null mouse model. The ability to target delivery to diseased tissue using the CREKA targeted micelles by showing that hirulog delivery was significantly lower in nonatherosclerotic (wild-type) mice (Figure 3c).
Figure 4.

Delivery of image contrast agent and a heparin hirulog to atherosclerotic plaques in the aortic tree of ApoE-null mice using micelles. (a) Fluorescence images of excised aortic trees show that image contrast with fluorescently labeled (FAM-labeled) nontargeted micelles is improved upon by targeted (FAM-CREKA) micelles. The contrast provided by FAM-CREKA micelles disappears with the addition of excess targeted micelles lacking a fluorescent label. Contrast is not significantly decreased when FAM-CREKA targeted micelles are administered along with nontargeted micelles without a fluorescent label. (b) The average fluorescence intensity of image pixels for the various studies. (c) The delivery of the anticoagulant peptide hirulog-2 to atherosclerotic plaques from micelles was quantified with a thrombin activity assay. In an ApoE null mouse model, hirulog delivery was significantly improved using targeted micelles compared to nontargeted micelles. The delivery of hirulog via targeted micelles in wild-type mice was significantly lower than targeted delivery in the ApoE atherosclerotic model. Reproduced from [13]. Copyright 2009 National Academy of Sciences, U.S.A.
Enhanced contrast of magnetic resonance imaging (MRI) of atherosclerotic plaques has also been achieved using targeted nanodelivery of contrast agents. For example, magnetofluorescent nanoparticles targeted to vascular adhesion molecule-1 (VCAM-1), a biomarker for atherosclerosis, enhanced MRI and fluorescence image contrast agent delivery compared to nontargeted controls in a cholesterol-fed apoE−/−mouse model [14]. In another study, Neubauer et al examined targeted delivery of paramagnetic liquid perfluorocarbon nanoparticles which incorporated a peptidomimetic victronectin antagonist for targeting the αvβ3 integrins [15]. Targeted systems enhanced MRI images compared to the nontargeted systems with double the concentration at the aortic wall (0.38% ID compared to 0.18% ID for nontargeted, or 1.64 compared to 0.84 nmol Gd/g tissue/h, respectively) in a cholesterol-fed rabbit model.
Several other studies have focused on targeted delivery systems as platforms for image contrast agents in atherosclerosis and have shown significant image enhancement over controls. For example, Park et al. demonstrated that hydrophobically modified chitosan nanoparticles with a peptide targeting ligand selectively accumulate at atherosclerotic plaques [16]. In addition, magnetofluorescent nanoparticles were targeted to macrophages associated with atheroma using dextran [17]. Administration of the targeted nanoparticles resulted in significant image enhancement over saline controls in the blood-plaque contrast-to-noise ratio of MRI. Improvements were also observed in near infrared reflectance fluorescence images in the plaque-to-background ratio. While these studies and others have not compared targeted delivery with nontargeted delivery to atheroma, these results indicate that targeted delivery is a promising approach to treatment and diagnosis of cardiovascular diseases.
The “fate” of the nanoparticle delivery system
The systemic biodistribution of nanoparticle delivery systems after administration is one of the most important issues and design elements to consider in targeted delivery. There is an innate relationship between the physicochemical properties of the nanoparticle and what happens to the particle after administration. This section will focus on the biodistribution, opsonization, and clearance of nanoparticles related to targeted delivery.
Opsonization and Clearance
For targeted delivery applications, there is a delicate balance between the circulation time necessary for particle localization at the target site and a suitable time frame for elimination. The eventual elimination (or whole body elimination) of nanomaterials is particularly important due to concerns over long-term exposure and interference with other diagnostics and therapies.
Both renal filtration and opsonization result in removal of nanomaterials from the bloodstream. Nanoparticles smaller than the renal filtration cut-off of 50 kDa or 5–6 nm are rapidly cleared from the bloodstream and excreted as demonstrated by Choi et al and summarized in Figure 5 [18]. Larger nanoparticles can be removed from the bloodstream by the reticuloendothelial system (RES) through opsonization resulting in nanoparticle accumulation in the liver, spleen, and lungs. Both the size and charge of nanoparticles is known to affect opsonization. In general, smaller particles tend to circulate longer [19]. Surface engineering such as charge shielding and the reduction of the adsorption of opsonin proteins on the nanomaterial has been shown to slow opsonization by imparting ‘stealth’ properties to the nanoparticle. For example, the addition of hydrophilic molecules such as PEG has been used in a variety of studies to slow opsonization. While we have alluded to the effect that PEGylation has on nanoparticle opsonization, there have been several extensive reviews that give a more in depth discussion of the factors affecting opsonization and strategies for slowing opsonization [19, 20].
Figure 5.

CdSe core and ZnS shell quantum dot clearance from blood, biodistribution, and renal clearance. (a) The concentration of radiolabeled quantum dots in blood after intravenous injection in a CD-1 mouse model decreases with nanoparticle size. (b) For quantum dots between 4 and 9 nm, the blood half-life of quantum dots increases with size. (c) Color video (left) and gamma-ray images taken with an Anger camera (middle) to show the biodistribution of 4.36 nm quantum dots in animals immediately after sacrifice (top row) and in organs (bottom row). The merged color image and gamma-ray image show nanoparticles in the bladder (Bl) (top right). The overall biodistribution of quantum dots in each organ is also quantified (bottom right). Abbreviations are: skin (Sk), adipose (Ad), muscle (Mu), bone (Bo), heart (He), lungs (Lu), spleen (Sp), liver (Li), kidneys (Ki), stomach (St), intestine (In), and brain (Br). (d) Biodistribution of 8.65 nm quantum dots following intravenous injection. Data corresponds to Figure 5c. (3) Quantum dot excretion via urine 4 h post-injection (blue) decreases with size and retention in the body (red) increases with size. Data is mean ± standard deviation. Reprinted by permission from Macmillan Publishers Ltd: [18], Copyright 2007.
In general, the selection of nanoparticle clearance rate is dependent on the application. For tumor targeting, the blood half-life in combination with the administered dose should afford a high blood concentration of nanoparticles for several hours [11]. Shorter blood half-lives may be desirable in other applications like image contrast agent delivery. Rapid localization and short half-lives would allow administration of contrast agent and imaging to be performed on a patient during a single appointment. Because nanoparticles are highly tunable, shorter or longer blood half-lives can be achieved on the order of minutes to hours. For example, quantum dots have been shown to have half-lives that range from 48 min to 20 h as demonstrated in Figure 5 [18].
An emerging issue in the field of nanoparticle delivery is the overall elimination of the nanoparticles. Clearance from the bloodstream is different than clearance or elimination from the body. Biodegradable polymeric systems do address the issue of elimination as long as they degrade to eliminable byproducts, but eventual elimination is an issue for organic and inorganic systems that are not degradable. One recent study has highlighted this issue by demonstrating that quantum dots can remain fluorescent in vivo for two years [21]. This nonelimination could cause significant problems if the quantum dots interfere with other treatments or diagnostics.
Biodistribution
Because understanding biodistribution is fundamental to the development of successful targeted nanodelivery systems, it is important to understand some of the factors affecting biodistribution both from an experimental perspective and material design perspective. Currently, there are not standardized practices for evaluating the performance of targeted delivery systems. As a result, it is difficult to compare results across studies. For example, some of the experimental factors affecting the measured biodistribution of nanomaterials are the initial dose [4, 22] the method of administration [23], the experimental model (including the tumor mass in the case of tumor delivery) [12], and the experimental time range. As targeted nanodelivery systems continue to be developed, standardized practices and the effect of experimental conditions on results will become key experimental considerations.
Despite the fact that there are not standardized procedures in measuring biodistribution, there are some commonalities and trends in the development of new experimental techniques. Perhaps, the use of autoradiography in monitoring nanoparticle biodistribution of radiolabeled nanomaterials is one of the most common elements across studies [19]. The widespread usage of autoradiography is due to the ability to easily and quantitatively measure small concentrations, but there are several drawbacks to radiography. Autoradiography requires tissue excision so only one time point can be measured per sample, and there are safety considerations in practicing this technique. Because of this, there is a growing body of literature that has used alternative methods to quantify biodistribution (Table 2).
Table 2.
Measurement techniques for quantifying biodistribution
| Technique | Agent | Study |
|---|---|---|
| Computed tomography (CT) | Iohexol | [76] |
| Fluorescence imaging | Cy5.5 | [2] |
| Inductively coupled atomic emission spectroscopy (ICP-AES) | Gd | [3] |
| Inductively coupled plasma-mass spectrometry (ICP-MS) | Au Fe |
[6, 22, 25] [77] |
| Micro-positron emission tomography (PET)/computed tomography (CT) | 64Cu | [5] |
| MRI imaging | Gd | [15] |
| Radiography |
111In 67Ga 125I-CA 64Cu Tc-O complex C14 |
[3, 9, 24, 32] [8] [30] [12,29] [78] [79] |
| Superconducting quantum interference device (SQUID) magnetometer | Iron | [28] |
| Two-photon luminescence | Gold nanorods | [80] |
Similar biodistributions for systems with and without targeting ligands is a recurring theme in targeted delivery. In only a few studies, has the presence of targeting ligands been shown to affect biodistribution in tissues other than the target site [4, 6]. To the best of our knowledge, there have not been any studies to determine whether internalization is improved by a higher accumulation at the target site. However, it is evident that a higher concentration of nanodelivery systems at the target site allows a greater availability for cell uptake. What this underscores is that the physicochemical properties affecting biodistribution are equally important for active and passive targeting. Size, shape, surface charge, and mechanical properties can be selected based on their effect on biodistribution.
It has been shown that the biodistribution of nanoparticles is size-dependent, as demonstrated by two recent studies on gold nanoparticles and PEGylated gold nanoparticles [24, 25]. For example, smaller gold nanoparticles (10 nm in diameter) were distributed in more tissues than larger particles (50, 100, or 250 nm) [25]. In all of the samples, nanoparticle accumulation occurred primarily in the liver, spleen, and the blood as measured by the concentration (ng/g tissue) and percent of the initial dose. Like gold nanoparticles, PEGylated gold nanoparticles exhibited a size-dependent biodistribution [24]. Accumulation of 20 nm gold nanoparticles in the tumor was significantly higher than that of 80 nm particles in an A341 xenograft-bearing mouse model. Outside of the tumor, nanoparticles of both sizes accumulated predominantly in the liver and spleen. As expected from a slow uptake by reticuloendothelial system (RES) organs, the 20 nm particles had a longer blood half-life than 80 nm particles and a lower accumulation in the liver and spleen.
Many studies have predicted that the shape of nanoparticles affects in vivo performance [26, 27], but the influence of shape on nanoparticle biodistribution is only beginning to be explored. To the best of our knowledge, only one study has compared the in vivo biodistributions of nanoparticles with different shapes. Park et al. evaluated the biodistribution of magnetic iron oxide nanoworms, elongated iron oxide nanoparticles, and compared them to spherical iron oxide nanoparticles [28]. They observed that nanoworms more readily accumulated in tumors in an MDA-MB-435 xenograft-bearing mouse model than nanospheres. After 48 h, nanoworms still remained in the tumor while nanospheres did not. The overall biodistributions of the two systems after 24 h were similar, where each showed a lower accumulation in the kidney relative to the liver and a higher accumulation in the spleen relative to the liver. The circulation time for both systems was comparable with blood circulation half-lives between 15 and 18 h.
In conjunction with size, the composition of nanoparticles has been shown to affect biodistribution by determining the “flexibility or shape adaptability” of the nanoparticle [29]. When comparing the biodistribution of two relative sizes of shell cross-linked nanoparticles having diameters of approximately 20 nm or 37 nm and core compositions of either glassy polystyrene or fluid-like poly(methyl acrylate), Sun et al found that small particles with polystyrene cores (18 ± 3 nm) had a greater retention in the blood compared to small particles with poly(methyl acrylate) cores (24 ± 3 nm). The initial accumulation of nanoparticles in the liver, lung, and spleen was higher for small particles with a poly(methyl acrylate) core composition than particles with polystyrene core compositions at 10 min and 1 h. The authors suggested that increased shape adaptability imparted by the more flexible poly(methyl acrylate) core contributed to the observed differences.
Surface properties, such as surface charge and PEGylation, also affect the biodistribution of nanoparticles. Generally, the addition of neutral hydrophilic polymers to the surface of nanoparticles neutralizes the surface charge. Neutralization of surface charge has been associated with improved blood retention time and decreased opsonization by the RES giving rise to the term ‘stealth properties’ [30]. For example, the addition of PEG to poly(lactide-co-glycolic acid) (PLGA) liposomes (PLGA-mPEG liposomes) affects biodistribution in a manner dependent on the ratio of PLGA to PEG [31]. A comparison between similarly sized PLGA nanoparticles and PLGA-mPEG(256) nanoparticles revealed that they had different surface charges (−54.2 mV and −5.9 mV, respectively). The neutralized surface charge of the PLGA-mPEG system increased nanoparticle retention in the blood after 3 hours to 42.6% ID in comparison to 4.1% ID of the PLGA dose. Meanwhile, the accumulation of PLGA-mPEG nanoparticles in the liver was generally lower than PLGA nanoparticles. Similar effects have been achieved with the PEGylation of poly(caprolactone) (PCL) nanoparticles [31]. For example, the surface charge of the PCL nanoparticles was increased from −27.17 mV to −6.046 mV by the addition of PEG, and a significant improvement in the blood retention time was accompanied by a drop in nanoparticle accumulation in the liver and lungs for PEGylated PCL nanoparticles.
The change in surface charge imparted by a polymer surface coating is not the only factor affecting biodistribution. The composition of the polymer is a determining factor as well. This phenomenon was explored with the adsorption of poloxamer 407, poloxamine 904, and poloxamine 908 onto the surface of polystyrene and PLGA nanoparticles [32]. Following administration in a rabbit model, the poloxamer 407 coating was the most effective in reducing nanoparticle clearance from the blood and liver accumulation after 3 hours even though the poloxamine 908 formulation had a more neutral surface charge than those with a poloxamer 407 coating.
For much smaller PEGylated nanoparticles, the length of PEG chains and end group functionality plays a greater role in determining biodistribution as demonstrated with 10 nm diameter Gadolinium nanoparticles [33]. The biodistribution of polymer coated Gadolinium nanoparticles was affected by the length of the PEG chains. Differences in the biodistribution of nanoparticles coated with similar lengths of PEG revealed that the end group (carboxylic acid, methoxy, or amine) of the PEG chain affected biodistribution.
Given that lowering systemic toxicity is a goal of targeted delivery and only a fraction of the total dose administered accumulates at the target site, the biodistribution in other tissues and clearance rate are extremely important.
Incorporation and Release of Therapeutics from Nanodelivery Systems
The importance of controlling nanoparticle localization for targeted delivery is inconsequential if a particle cannot carry a relevant amount of drug and release it at the target site. The ability to efficiently incorporate a drug into nanoparticle delivery systems is dependent on the drug itself, the design of the nanoparticle, and the method of loading. Countless strategies have been used to synthesize organic nanoparticles with an intended application in drug delivery, however, only a fraction of these systems have demonstrated a definitive ability to entrap a drug or model molecule, and most of these studies have focused on a relatively small number of nanodelivery systems, most commonly biodegradable PLGA. This section will focus specifically on nanoparticle systems designed to successfully entrap a therapeutic, the strategies used for loading, and data on drug release when available.
Selection of a general nanodelivery system, be it targeted or nontargeted, should reflect the properties of the drug and the intended application. For example, the nanodelivery system should maintain drug stability and should be hydrophilic or hydrophobic to ensure compatibility with the drug. Properties of the drug, such as size, charge, and hydrophilicity or hydrophobicity, typically determine whether the carrier can effectively entrap, protect, and release the drug. Mechanisms for drug entrapment vary depending on the type of nanoparticle and the drug itself. In many cases, the method of drug loading determines the efficiency of the loading process and ultimately whether a nanodelivery system is suitable for the intended application.
Most nanodelivery systems can be placed into one or more of three large groups: biodegradable polymer nanoparticles; self-assembled carriers including micelles, liposomes, and polymersomes; or hydrophilic crosslinked systems such as nanoscale hydrogels, also called nanogels. While in the case of self-assembled carriers, therapeutics can be simply loaded during micelle or vesicle assembly, biodegradable nanoparticle matrices are loaded during post-polymerization nanoparticle formation using emulsion and solvent evaporation techniques. On the other hand, nanogel systems are typically loaded after nanoparticle formation by incubating the particles in a solution containing the drug. The drug becomes entrapped in the crosslinked network by simple partitioning effects, which allow the drug to diffuse into the nanoparticle matrix. The loading of hydrophilic and hydrophobic drugs in some of these types of nanodelivery systems is shown in Figure 6.
Figure 6.
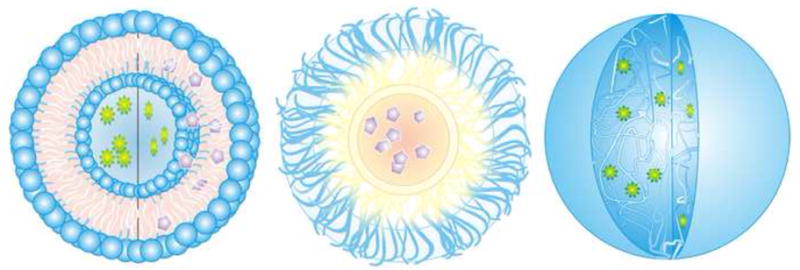
Drug loading of various nanodelivery systems. (a) Liposome loaded with hydrophilic (green) and hydrophobic drugs (purple). (b) Encapsulation of hydrophobic drugs in a micelle. (c) Entrapment of hydrophilic drugs in nanogels.
The amount of a drug that has been successfully loaded into a nanodelivery system is typically quantified in one of two ways. The most common measure cited is the entrapment efficiency, also referred to as the encapsulation or incorporation efficiency, which is defined as the fraction of the drug loaded per the total amount of drug used (or available for loading). The other number that is often reported is the loading capacity or loading efficiency, which refers simply to the ratio of the weight of drug incorporated to the weight of the carrier. These quantifications will be cited here as reported in the studies that are discussed.
Biodegradable Nanoparticles
Biodegradable nanoparticles have been widely investigated for use as drug delivery systems, and are versatile platforms for successful encapsulation of a range of therapeutics. These biodegradable polymer nanoparticles are formed most commonly using emulsion and solvent evaporation techniques, where the polymer is first dissolved in an organic phase that is subsequently dropped into a continuous aqueous phase and stirred until the solvent evaporates. Hydrophobic drugs can easily be incorporated into particles during this process by solvating the drug along with the polymer in the organic phase. These oil-in-water emulsions have been used to load a variety of hydrophobic drugs, such as anti-cancer therapeutics [34–36]. In addition to hydrophobic drugs, encapsulation of water-soluble drugs and biomolecules, including proteins and certain chemotherapeutics, has been achieved with water-in-oil-in-water double emulsion techniques [37–39]. Other synthesis techniques, such as nanoprecipitation and spray drying have also been used effectively to produce PLGA nanoparticles loaded with therapeutics [40].
Recent studies have carefully evaluated the effects of PLGA nanoparticle properties on drug loading. For example, smaller nanodelivery systems have a smaller volume, and therefore, would be expected to have a smaller maximum loading capacity. In the majority of studies, the maximum loading capacity is not determined for a carrier. The achieved loading capacity of a delivery system is dependent on factors beyond volume alone. For example, drug loading and release from PLGA micro- and nanospheres have been shown to be inversely correlated with size. In a study that compared two sizes of PLGA particles, dexamethasone was first loaded into PLGA microspheres using a typical oil-in-water solvent evaporation method to produce 20 μm particles. Smaller 1 μm particles loaded with dexamethasone were produced via emulsion polymerization. The emulsion polymerization scheme for the smaller particles led to a dexamethasone encapsulation efficiency of 11.2% compared to 1% for the larger microparticles when incubating 100 mg of dexamethasone with 500 mg of polymer [41].
Drug delivery systems composed of polyanhydrides have been widely investigated because of their surface eroding mechanism which can lead to near zero-order release profiles [42, 43]. In general, drugs have been successfully incorporated in polyanhydride disks such as the Gliadel® system or microparticles [44]. However, there have been limited studies demonstrating loading and release from polyanhydride carriers on the nanoscale. In one study, paclitaxel or 2-hydroxypropyl-β-cyclodextrin (HPCD)-complexed paclitaxel was encapsulated in polyanhydride nanoparticles composed of poly(methyl vinyl ether-co-maleic anhydride) during particle formation using a solvent displacement method which simultaneously allowed nanoparticle formation and loading. Up to a 167 μg/mg loading capacity was obtained for nanoparticles with an average diameter of 302 nm. The controlled release of HPCD-paclitaxel was then demonstrated in different simulated biological fluids. No release was observed from particles in simulated gastric fluid (pH 1.2) whereas there was complete release over a 24 hour period in simulated intestinal fluid (pH 7.5) [45].
Like synthetic biodegradable systems, nanodelivery systems composed of naturally occurring biodegradable polymers have been investigated for drug delivery. Some of the most widely studied are systems based on the biodegradable and biocompatible polysaccharide chitosan. The potential use of chitosan carriers has been demonstrated for a vast number of drugs and applications. For example, chitosan carriers have received considerable attention for delivery of nucleic acids due to chitosan’s net positive charge. One of the contributing factors to chitosan’s versatility is that a number of techniques have been developed to synthesize chitosan nanoparticles including ionic gelation, precipitation, reverse micelle formation, self-assembly, and spray drying [46, 47].
In one study examining the use of chitosan for delivery of biomolecules, a model protein, bovine serum albumin (BSA), was loaded into chitosan nanoparticles prepared by two different methods using tripolyphosphate (TPP) as an ionic crosslinker. In the first method termed “incorporation”, BSA was incorporated into the polymer matrix during particle formation by adding BSA to a chitosan solution before the addition of the TPP crosslinker at which point particles spontaneously formed during mixing. Using the “incubation” method, BSA was simply adsorbed to the surface of nanoparticles during incubation following particle coacervation. Encapsulation efficiencies up to 88% were obtained and BSA release was primarily shown to be a fast release over a 6 hour period [48]. In another study, chitosan nanoparticles crosslinked with TPP were loaded with methotrexate disodium (MTX) using a post-polymerization incubation method. Loading capacities up to 52% and loading efficiencies up to 78% were obtained when an MTX concentration of 1.26 mg/mL was incubated with a 0.16 wt% nanogel solution [49].
Chitosan nanoparticles have also been formulated to deliver anticancer drugs. For example, a water-in-oil microemulsion method was used to entrap a doxorubicin-dextran conjugate in chitosan nanoparticles having a hydrodynamic diameter of 100 nm. Entrapment efficiencies were measured between 60–65%. In a mouse model, treatment with the loaded nanoparticles led to faster and more complete tumor regression than free drug or empty nanoparticles [50].
Another natural biodegradable material used to synthesize nanoparticles for drug delivery applications is the anionic biopolymer alginate. Most often calcium chloride is added to a solution of alginate to induce ionic crosslinking, resulting in micro- and nanoparticle polymer networks. In one case, alginate gelation with calcium ions followed by coating with chitosan produced nanoparticles intended for oral insulin delivery applications. A maximum insulin encapsulation efficiency was measured at 92% with loading capacities up to 14.3% [51]. Isoniazid, rifampicin, and pyrazinamide were encapsulated in alginate particles using a cation gelation method in an effort to design aerosolized nanoparticles for treatment of tuberculosis. Calcium chloride was added to sodium alginate solution containing varying amounts of the three drugs to induce the gelation and encapsulation of the drug. Drug encapsulation efficiencies ranged from 70–90% in particles at the upper limit of so-called nanoparticles with an average size just around 1 μm [52].
Self-Assembled Nanoparticles
Self-assembled nanoparticles, including liposomes, micelles, and polymersomes, can be formed using various amphiphiles such as natural lipids or block copolymers. Liposomes are vesicles that consist of a spherical shaped lipid bilayer, or multilayers, containing an inner aqueous void space. Liposomal encapsulation is a versatile method to load therapeutics, because hydrophilic drugs and biomolecules can be loaded in the inner aqueous void, while hydrophobic molecules can be entrapped in the lipid bilayer. Liposomal delivery of chemotherapeutics has been established as an effective method to increase efficacy and decrease toxicity over free drug, evidenced by the success of liposomal doxorubicin, Doxil®, which has been approved for human use in cancer treatment [53].
In addition to the lipid structures, synthetic block copolymers built with one hydrophilic block and one hydrophobic block have been shown to exhibit a variety of self-assembled structures including spherical or cylindrical micelles and vesicles with architectures that resemble liposomes known as polymersomes [54]. Micelles, nanoparticles with a hydrophobic core and hydrophilic outer layer, self-assembled from amphiphilic block copolymers in aqueous media, are frequently used to entrap hydrophobic drugs during micelle formation. During spontaneous micelle formation in an aqueous environment, hydrophobic drugs are localized in and around the hydrophobic core [55]. This loading method has been used to entrap a variety of therapeutics, particularly anticancer agents, in block copolymers of poloxamers, which consist of blocks of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO), and NK105, which consists of blocks of PEG and a modified polyaspartate [56–58].
Using this method of drug loading, micelle nanoparticles have been used to entrap hydrophobic anticancer therapeutics. Biodegradable, cationic micelle nanoparticles were self-assembled using amphiphilic poly{(N-methyldietheneamine sebacate)-co-[(cholesteryl oxocarbonylamido ethyl) methyl bis(ethylene) ammonium bromide] sebacate}, P(MDS-co-CES). In this study, Herceptin®, which is a monoclonal antibody, was attached to the surface of the particles while paclitaxel was loaded into the micelles by dissolving the polymer and drug in DMF and dialyzing against a sodium acetate/acetic acid buffer. This method let to encapsulation efficiencies of paclitaxel of 58.1% and a loading capacity of 14.3%. Release studies demonstrated the that the drug could be released from the nanoparticles over a 69 hour period [59].
While traditional micelle systems have been used primarily to incorporate hydrophobic drugs, adapted systems have been used to entrap more hydrophilic species such as biomolecules. For example, drug carrier systems called polyion complex (PIC) micelles have been synthesized from PEG block copolymers. The PIC micelles are formed using block copolymers consisting of a hydrophilic block and a polyionic block. In this case, electrostatic interactions between the ionic polymer and an oppositely charged species are a driving force for the formation of the micelles. Poly(ethylene glycol-grafted-chitosan), PEG-g-chitosan, block copolymers were used to entrap diammonium glycyrrhizinate (DG) in micelles assembled in acetate buffer after addition of TPP to induce chitosan aggregation. Loading efficiencies of DG over 96% have been obtained using these systems and the average diameter of micelles was between 20 and 30 nm. Release profiles varied based on the free ions in solution. In ionic solution, a burst release was observed, but there was limited release in DI water [60].
Polymer vesicles called polymersomes or polymerosomes have been used to entrap a variety of therapeutics and model drugs. For example, polymersomes with diameters around 100 nm were prepared from triblock copolymers, poly(caprolactone)-poly(ethylene glycol)-poly(caprolactone), PCL-PEG-PCL, using a double emulsion method. Insulin was incorporated during self-assembly of 122 nm particles [61]. In another study, triblock copolymers, poly(ethylene oxide)-b-poly(acrylic acid)-b-poly(N-isopropylacrylamide) (PEO-PAA-PNIPAM), were synthesized via RAFT polymerization. The resulting polymers were water soluble at room temperature, but formed polymer vesicles with diameters between 170 and 250 nm above 32° C. These polymersomes were loaded with FITC-dextran during self-assembly at elevated temperatures and then crosslinked using cystamine via carbodiimide chemistry. The crosslinks were shown to degrade in the presence of dithiothreitol, and therefore, it was hypothesized that these systems are degradable in vitro and in vivo. Loading capacity of these nanoparticles was measured at levels exceeding 85% by weight [62].
Recently, there has been interest in creating systems that are hybrid particles combining properties of liposomes and synthetic polymer systems for nanodelivery applications. Cationic lipids have been combined with peptide-lipid amphiphiles and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carboxy(polyethylene glycol) to design liposome nanoparticles targeted to the urokinase plasminogen activator receptor (uPAR) overexpressed on many tumors. The particles have an ABCD structure, an inner aqueous (A) layer of loaded anionic nucleic acids, a cationic lipid bilayer (B), a PEG layer for stealth characteristics (C), and a peptide sequence layer for targeting of uPAR (D). Loading efficiencies as high as 60% were achieved when 10 wt% docetaxel was added during the particle self-assembly process [63]. In another example, hybrid systems were formed through self-assembly to form nanoparticles with a lipid monolayer surrounding a PLGA core loaded with a hydrophobic drug. This unique system relied on PEG-conjugated lecithin to form a hydrophilic outer shell of PEG as the lipid monolayer formed around the hydrophobic PLGA polymer. Drug loading of docetaxel into the PLGA core was accomplished during this self-assembly process by first dissolving polymer and drug in an organic solvent and then dripping this solution into an aqueous solution containing PEG-lecithin. This process led to the formation of the particles via self-assembly and the solvent was allowed to evaporate. Encapsulation efficiencies of 62% were obtained using this method when adding 10 wt% docetaxel to the organic polymer solution. Controlled release was observed over a 100 h period and shown to be a function of lipid coverage, where the release rate could be slowed by increasing the lipid to polymer ratio [64].
Nanogels
Nanoscale hydrogels, or nanogels, are hydrophilic crosslinked polymer networks that imbibe water, but are insoluble in aqueous environments. Drugs can be loaded into the polymer matrix of these materials and controlled release is dependent on the diffusion coefficient of the drug through the hydrogel network. Additionally, hydrogel nanoparticle systems have been developed in a class of drug carriers know as “intelligent” or “smart” delivery systems. Nanogels can swell in aqueous medium and also can respond to a variety of environmental stimuli such as pH, temperature, ionic strength, or electric field through an increase or decrease in volume [65, 66]. Typically, nanogels are used to carry a drug when the matrix is in a more collapsed state. Following a change in stimulus the nanogel swells, thereby increasing the mesh size and the rate of diffusion of the drug out of the polymer matrix. Alternatively, a method in which the drug is squeezed out of the nanogel has been used to obtain a fast release, where a drug is entrapped in a hydrogel in the swollen state, but upon deswelling the drug is squeezed out of the polymer matrix [67, 68].
While most biodegradable or self-assembled nanoparticle systems are formed from previously synthesized polymers, hydrogel nanoparticles are most commonly synthesized using heterogeneous polymerization techniques. Because the hydrogel nanoparticles are formed during polymerization, incorporating a therapeutic at this stage requires exposing the drug to polymerization conditions that could potentially damage or modify the drug. However, the porous characteristics of nanogel matrices allows for drug loading following polymerization by partitioning methods, which eliminates the need to subject the drug to harsh polymerization conditions. In most situations, the drug is loaded into the nanogel by incubating the nanoparticles with the drug in aqueous conditions such that the particles are in their most swollen state. After a period of time to allow the therapeutic to diffuse into the polymer matrix, a condition (most commonly pH or temperature) is altered, leading to a particle size transition to a more collapsed state with a smaller mesh size, physically entrapping the therapeutic in the polymer matrix.
The hydrophilic nature of hydrogel systems makes them an ideal candidate for the loading and delivery of water soluble therapeutics, of particular interest biomacromolecules such as proteins or nucleic acids. Several nanogel systems have been investigated for the delivery of insulin to the small intestine. In addition to their ability to entrap the water soluble insulin, some of these systems have advantageous pH-sensitive properties allowing them to protect insulin in the acidic environment of the stomach and release it in the more neutral pH of the small intestine [69, 70]. Polybasic nanoparticles are cationic pH responsive hydrogels with PEG grafts. A UV-initiated free radical emulsion polymerization was used to synthesize poly[2-(diethylamino) ethyl methacrylate] surface grafted with PEG (PDGP) nanogels. Insulin as a model drug was added to a solution of the nanogels and the pH was adjusted to 6.5 to swell the nanoparticles. After a loading period the pH was raised quickly to 7.4 to collapse the particles and entrap the loaded protein. The loaded nanogels were dialyzed against water for 5 days to remove any excess protein not loaded. Encapsulation efficiencies were measured up to 92% for insulin in PDGP particles with low crosslinking densities when equal weights of particles and protein were added to the loading solution [71].
Controlled drug loading and release has also been shown in several studies using hydrogels based on the temperature-sensitive polymer poly(N-isopropylacrylamide) (PNIPAAm). PNIPAAm exhibits a negative swelling transition at 34 °C, which makes it an attractive system from a physiological standpoint for applications in drug delivery. For example, poly(NIPAAm) and poly(NIPAAm-co-AA) nanoparticles were loaded with 5-fluorouracil and release was shown to be a function of pH and temperature [72].
Nanogels of poly(vinyl alcohol), PVA, and poly(vinyl pyrrolidone), PVP have also been investigated for drug delivery applications. For example, composite systems of PVA-crosslinked PVP nanogels were used to entrap ferromagnetic particles as well as bleomycin A5 hydrochloride. Nanogels were prepared using a water-in-oil emulsion polymerization initiated by gamma ray irradiation. For this emulsion polymerization, the aqueous phase contained the PVA, PVP, and ferromagnetic particles. The resulting nanogels entrapped the ferromagnetic particles and were then used to load bleomycin A5 hydrochloride. Dry nanoparticles were allowed to swell in an aqueous solution containing the drug, sonicated, and then placed on a shaker plate for 12 h of storage until no apparent liquid was visible. Encapsulation efficiency was not reported, because it was assumed that all of the drug had been immobilized, but in vitro release occurred over a minimum period of 8 hours [73].
While most hydrogel nanodelivery systems have been used to entrap hydrophilic molecules, recent work on modified hydrogels has demonstrated the potential to load hydrophobic drugs as well. In order to do so, the hydrophobicity of the nanogels has been increased through the incorporation of amphiphiles into the hydrogel matrix. An acrylated PEG-PPG-PEG triblock copolymer with both hydrophobic and hydrophilic groups was used to synthesize crosslinked nanoparticles via an inverse microemulsion using a PEG crosslinker. Properties of the emulsion could be altered to control for particle sizes ranging from 50 to 500 nm. These nanogels were then loaded with doxorubicin to determine their ability to carry hydrophobic drugs. Doxorubicin, solubilized in CHCl3 with triethylamine, was added to an aqueous solution of nanoparticles and the CHCl3 was allowed to evaporate overnight. The results indicated that the formation of nanogels from amphiphilic macromers was a successful strategy for loading hydrophobic drugs in a hydrogel with up to 9.8% loading capacity of doxorubicin [74].
While the control and optimization of drug loading and release are key challenges in the field of nanodelivery, there is not a single carrier or loading mechanism that is ideal for any drug and any application. Each nanodelivery system and drug pairing is unique. Future studies on the interplay between the properties of the carrier, properties of the drug, and the mechanisms of loading will to significantly improve nanodelivery from a drug loading and release vantage point.
Conclusion
As the field of targeted drug delivery continues to move forward, it will be increasingly important to design nanoscale systems with tailorable properties for efficient delivery and improved therapeutic efficacy. Important design considerations will include the physicochemical properties that govern targeting, biodistribution, and clearance as well as the system’s effectiveness in carrying, protecting, and even releasing active therapeutic and diagnostic agents.
The selection and addition of targeting ligands to the surface of nanoparticles has already been shown to improve therapeutic efficacy over systems without targeting ligands. We are only beginning to understand the role that targeting ligands play in interfacing interactions of nanoparticles and cells. The few studies that have compared targeted and nontargeted systems have demonstrated that the role of targeting ligands in localization at the target site is application dependent. Targeted delivery to atherosclerotic lesions is greatly enhanced by targeting ligands which impart an improved ability to accumulate at the target site. However, in the case of tumor targeting, improved performances of targeted systems over nontargeted systems has been accomplished by improved cellular uptake as well as improved accumulation at the target site. Some of these studies have suggested that the primary benefit of incorporating targeting ligands is to mediate cell internalization. Only a small number of studies have focused on comparing the in vivo performance and biodistribution of targeted and nontargeted delivery systems. Further study is necessary to truly determine how the presence of a targeting ligand affects targeted delivery.
Design of the physicochemical properties, including size, shape, surface charge, and mechanical characteristics, of the nanoparticle itself will continue to be key design parameters due to their effect on biodistribution. It is important to account for all of these factors in concert with targeting strategies to understand how to predict and control nanoparticle distribution. Many of the same properties that govern biodistribution also determine the clearance rate. The challenge in designing materials for targeted delivery is that the material properties should allow an appropriate residence time that is long enough for particle localization and delivery of the drug or diagnostic but does not prevent the system from being fully eliminated in an acceptable amount of time. The desired particle residence time will vary with the intended application, and this time frame can be tuned. Ultimately, the challenge lies in designing a system that has the perfect combination of properties to allow for sufficient interaction of the targeted nanoparticles with the target site, desirable biodistribution, and efficient removal.
The material properties that contribute to targeting, biodistribution, and elimination must also be balanced with the properties that affect the ability to entrap a therapeutic or diagnostic. The primary consideration in determining the ability of a nanoparticle system to entrap a molecule is the interaction between the polymer and the therapeutic. This relationship is often a balance of intermolecular interactions including hydrophobic/hydrophilic and electrostatic interactions. Another consideration is the method in which the drug can be incorporated into the material, and whether the available methods will allow an acceptable amount of the therapeutic to be loaded into the carrier. Both the loading strategy and stability of the entrapped drug must also be carefully considered in order to design systems that are able to carry a sufficient amount of drug to the target site.
As we move towards developing optimally effective, high performance nanodelivery systems, it will be necessary to design new materials that incorporate the best properties for targeting, biodistribution, clearance, drug incorporation, and release at the target site.
Figure 1.

The in vivo biodistribution of intravenously administered targeted HFT-T nanodelivery systems and nontargeted HT-T was investigated in a KB-3-1 xenograft-bearing mouse model. (a) Near-infrared fluorescence imaging of mice at 1, 24, and 48 h showed the biodistribution of nanodelivery systems. (b) At 48 h, the biodistribution of nanoparticles in various tissues was quantified by fluorescence intensity. (c) Nanoparticle (red) uptake by xenograft cells was shown by colocalization with EpCAM-positive (green) xenograft cells. Targeted systems were found predominantly in cells while nontargeted systems were less likely to be internalized remaining in the extracellular matrix. (d) Flow cytometry of disaggregated xenograft cells stained with a FITC-conjugated EpCAM antibody distinguished nanoparticle uptake in human xenograft tumor cells from nanoparticle uptake in host non-tumor cells for targeted and control nanoparticles. Human tumor cells were EpCAM positive in Q4-2 (green in lower right quadrant) and Q2-2 (yellow). Nanoparticle uptake by tumor cells are shown in Q2-2 (yellow), and nanoparticle uptake in host cells are shown in Q1-2 (red). Reprinted with permission from [2]. Copyright 2009 American Chemical Society.
Acknowledgments
This work was supported in part by the National Institutes of Health (grant EB000246 and a Physical Science-Oncology Centers U54 grant) and the National Science Foundation (grant DGE-03-33080).
Biographies

Margaret A. Phillips received her Honors B.S. in biomedical engineering and Mathematics) from Saint Louis University in 2006. She is currently a PhD student with Nicholas Peppas in biomedical engineering at the University of Texas at Austin where she is a Thrust 2000 fellow and an NSF-IGERT trainee. Her thesis focuses on the surface modification of anionic complexation hydrogels with polysaccharides for drug delivery applications. She is currently finishing a three year term as the National Student Chapter President of the Society For Biomaterials.

Martin L. Gran received his B.S. in chemical engineering from Iowa State University in 2006. He is currently pursuing a Ph.D. at the University of Texas at Austin in chemical engineering under the guidance of Dr. Nicholas A. Peppas. At the University of Texas, he has received a Thrust 2000 Fellowship in Engineering and is a trainee in the NSF-IGERT program in Cellular and Molecular Imaging for Diagnostics and Therapeutics. His current research focuses on the development of temperature-sensitive polymer nanoparticle systems for externally-controlled drug delivery.

Nicholas A. Peppas is the Fletcher S. Pratt Chair in Chemical Engineering, Biomedical Engineering, and Pharmacy, and is the director of the Center for Biomaterials, Drug Delivery and Bionanotechnology at the University of Texas at Austin. He is a member of the National Academy of Engineering, the Institute of Medicine of the National Academy of Sciences, the National Academy of Pharmacy of France and the Texas Academy of Engineering, Sciences and Medicine. He received his Diploma in Engineering (D. Eng.) from the National Technical University of Athens, Greece in 1971 and his Sc.D. from MIT in 1973, both in chemical engineering.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Byrne JD, Betancourt T, Brannon-Peppas L. Adv Drug Deliv Rev. 2008;60:1615. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Wang X, Li J, Wang Y, Cho KJ, Kim G, Gjyrezi A, et al. ACS Nano. 2009;3:3165. doi: 10.1021/nn900649v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oyewumi MO, Yokel RA, Jay M, Coakley T, Mumper RJ. J Control Release. 2004;95:613. doi: 10.1016/j.jconrel.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Bae Y, Nishiyama N, Kataoka K. Bioconjugate Chem. 2007;18:1131. doi: 10.1021/bc060401p. [DOI] [PubMed] [Google Scholar]
- 5.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Proc Natl Acad Sci U S A. 2007;104:15549. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian X, Peng XH, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, et al. Nat Biotechnol. 2008;26:83. doi: 10.1038/nbt1377. [DOI] [PubMed] [Google Scholar]
- 7.Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, et al. Clin Cancer Res. 2002;8:1172. [PubMed] [Google Scholar]
- 8.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, et al. Cancer Res. 2006;66:6732. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 9.ElBayoumi TA, Torchilin VP. Mol Pharm. 2009;6:246. doi: 10.1021/mp8001528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer AK, Khaled G, Fang J, Maeda H. Drug Discov Today. 2006;11:812. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Pirollo KF, Chang EH. Trends Biotechnol. 2008;26:552. doi: 10.1016/j.tibtech.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Rossin R, Pan D, Qi K, Turner JL, Sun X, Wooley KL, et al. J Nucl Med. 2005;46:1210. [PubMed] [Google Scholar]
- 13.Peters D, Kastantin M, Kotamraju VR, Karmali PP, Gujraty K, Tirrell M, et al. Proc Natl Acad Sci U S A. 2009;106 doi: 10.1073/pnas.0903369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly KA, Allport JR, Tsourkas A, Shinde-Patil VR, Josephson L, Weissleder R. Circ Res. 2005;96:327. doi: 10.1161/01.RES.0000155722.17881.dd. [DOI] [PubMed] [Google Scholar]
- 15.Neubauer AM, Sim H, Winter PM, Caruthers SD, Williams TA, Robertson JD, et al. Magn Reson Med. 2008;60:1353. doi: 10.1002/mrm.21795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park Y, Hong HY, Moon HJ, Lee BH, Kim IS, Kwon IC, et al. J Control Release. 2008;128:217. doi: 10.1016/j.jconrel.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 17.Jaffer FA, Nahrendorf M, Sosnovik D, Kelly KA, Aikawa E, Weissleder R. Mol Imaging. 2006;5:85. [PubMed] [Google Scholar]
- 18.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, et al. Nat Biotechnol. 2007;25:1165. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owens DE, Peppas NA. Int J Pharm. 2006;307:93. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Moghimi SM, Szebeni J. Prog Lipid Res. 2003;42:463. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 21.Fitzpatrick JA, Andreko SK, Ernst LA, Waggoner AS, Ballou B, Bruchez MP. Nano Lett. 2009;9:2736. doi: 10.1021/nl901534q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akiyama Y, Mori T, Katayama Y, Niidome T. J Control Release. 2009;139:81. doi: 10.1016/j.jconrel.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Sun W, Zou W, Huang G, Li A, Zhang N. J Drug Target. 2008;16:357. doi: 10.1080/10611860801927598. [DOI] [PubMed] [Google Scholar]
- 24.Zhang G, Yang Z, Lu W, Zhang R, Huang Q, Tian M, et al. Biomaterials. 2009;30:1928. doi: 10.1016/j.biomaterials.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJ, Geertsma RE. Biomaterials. 2008;29:1912. doi: 10.1016/j.biomaterials.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 26.Glangchai LC, Caldorera-Moore M, Shi L, Roy K. J Control Release. 2008;125:263. doi: 10.1016/j.jconrel.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 27.Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, et al. Proc Natl Acad Sci U S A. 2008;105:11613. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JH, von Maltzahn G, Zhang L, Schwartz MP, Ruoslahti E, Bhatia SN, et al. Adv Mater. 2008;20:1630. doi: 10.1002/adma.200800004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun X, Rossin R, Turner JL, Becker ML, Joralemon MJ, Welch MJ, et al. Biomacromolecules. 2005;6:2541. doi: 10.1021/bm050260e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Livaniou E, Evangelatos G, et al. Int J Pharm. 2003;259:115. doi: 10.1016/s0378-5173(03)00224-2. [DOI] [PubMed] [Google Scholar]
- 31.Shan X, Yuan Y, Liu C, Tao X, Sheng Y, Xu F. Biomed Microdevices. 2009;11:1187. doi: 10.1007/s10544-009-9336-2. [DOI] [PubMed] [Google Scholar]
- 32.Dunn SE, Coombes AGA, Garnett MC, Davis SS, Davies MC, Illum L. J Control Release. 1997;44:65. [Google Scholar]
- 33.Faure AC, Dufort S, Josserand V, Perriat P, Coll JL, Roux S, et al. Small. 2009;5:2565. doi: 10.1002/smll.200900563. [DOI] [PubMed] [Google Scholar]
- 34.Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. J Control Release. 1999;57:171. doi: 10.1016/s0168-3659(98)00116-3. [DOI] [PubMed] [Google Scholar]
- 35.Mu L, Feng SS. J Control Release. 2003;86:33. doi: 10.1016/s0168-3659(02)00320-6. [DOI] [PubMed] [Google Scholar]
- 36.Fonseca C, Simões S, Gaspar R. 2002;83:273. doi: 10.1016/s0168-3659(02)00212-2. [DOI] [PubMed] [Google Scholar]
- 37.Bilati U, Allemann E, Doelker E. J Microencapsul. 2005;22:205. doi: 10.1080/02652040400026442. [DOI] [PubMed] [Google Scholar]
- 38.Li YP, Pei YY, Zhang XY, Gu ZH, Zhou ZH, Yuan WF, et al. J Control Release. 2001;71:203. doi: 10.1016/s0168-3659(01)00235-8. [DOI] [PubMed] [Google Scholar]
- 39.Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas AG, Ithakissios DS. J Control Release. 2002;79:123. doi: 10.1016/s0168-3659(01)00530-2. [DOI] [PubMed] [Google Scholar]
- 40.Astete CE, Sabliov CM. J Biomater Sci Polym Ed. 2006;17:247. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- 41.Dawes GJ, Fratila-Apachitei LE, Mulia K, Apachitei I, Witkamp GJ, Duszczyk J. J Mater Sci Mater M. 2009;20:1089. doi: 10.1007/s10856-008-3666-0. [DOI] [PubMed] [Google Scholar]
- 42.Tabata Y, Gutta S, Langer R. Pharm Res. 1993;10:487. doi: 10.1023/a:1018929531410. [DOI] [PubMed] [Google Scholar]
- 43.Lopac SK, Torres MP, Wilson-Welder JH, Wannemuehler MJ, Narasimhan B. J Biomed Mater Res B. 2009;91:938. doi: 10.1002/jbm.b.31478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langer R. J Control Release. 1991;16:53. [Google Scholar]
- 45.Agüeros M, Ruiz-Gatón L, Vauthier C, Bouchemal K, Espuelas S, Ponchel G, et al. Eur J Pharm Sci. 2009;38:405. doi: 10.1016/j.ejps.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 46.Agnihotri SA, Mallikarjuna NN, Aminabhavi TM. J Control Release. 2004;100:5. doi: 10.1016/j.jconrel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 47.Berger J, Reist M, Mayer JM, Felt O, Peppas NA, Gurny R. Eur J Pharm Biopharm. 2004;57:19. doi: 10.1016/s0939-6411(03)00161-9. [DOI] [PubMed] [Google Scholar]
- 48.Gan Q, Wang T. Colloid Surface B. 2007;59:24. doi: 10.1016/j.colsurfb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 49.Zhang H, Mardyani S, Chan WC, Kumacheva E. Biomacromolecules. 2006;7:1568. doi: 10.1021/bm050912z. [DOI] [PubMed] [Google Scholar]
- 50.Mitra S, Gaur U, Ghosh PC, Maitra AN. J Control Release. 2001;74:317. doi: 10.1016/s0168-3659(01)00342-x. [DOI] [PubMed] [Google Scholar]
- 51.Sarmento B, Ribeiro AJ, Veiga F, Ferreira DC, Neufeld RJ. J Nanosci Nanotechno. 2007;7:2833. doi: 10.1166/jnn.2007.609. [DOI] [PubMed] [Google Scholar]
- 52.Zahoor A, Sharma S, Khuller GK. Int J Antimicrob Ag. 2005;26:298. doi: 10.1016/j.ijantimicag.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 53.Malam Y, Loizidou M, Seifalian AM. Trends Pharmacol Sci. 2009;30:592. doi: 10.1016/j.tips.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 54.Discher DE, Ortiz V, Srinivas G, Klein ML, Kim Y, Christian D, et al. Prog Polym Sci. 2007;32:838. doi: 10.1016/j.progpolymsci.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Torchilin VP. Pharm Res. 2007;24:1. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 56.Kabanov AV, Batrakova EV, Alakhov VY. J Control Release. 2002;82:189. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 57.Hamaguchi T, Matsumura Y, Suzuki M, Shimizu K, Goda R, Nakamura I, et al. Brit J Cancer. 2005;92:1240. doi: 10.1038/sj.bjc.6602479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kataoka K, Harada A, Nagasaki Y. Adv Drug Deliv Rev. 2001;47:113. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 59.Lee AL, Wang Y, Cheng HY, Pervaiz S, Yang YY. Biomaterials. 2009;30:919. doi: 10.1016/j.biomaterials.2008.10.062. [DOI] [PubMed] [Google Scholar]
- 60.Yang KW, Li XR, Yang ZL, Li PZ, Wang F, Liu Y. J Biomed Mater Res A. 2009;88:140. doi: 10.1002/jbm.a.31866. [DOI] [PubMed] [Google Scholar]
- 61.Rastogi R, Anand S, Koul V. Colloid Surf B. 2009;72:161. doi: 10.1016/j.colsurfb.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 62.Xu H, Meng F, Zhong Z. J Mater Chem. 2009;19:4183. [Google Scholar]
- 63.Wang M, Lowik DW, Miller AD, Thanou M. Bioconjugate Chem. 2009;20:32. doi: 10.1021/bc8001908. [DOI] [PubMed] [Google Scholar]
- 64.Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, et al. Biomaterials. 2009;30:1627. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 65.Ganta S, Devalapally H, Shahiwala A, Amiji M. J Control Release. 2008;126:187. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 66.Qiu Y, Park K. Adv Drug Deliv Rev. 2001;53:321. doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 67.Sershen SR, Westcott SL, Halas NJ, West JL. J Biomed Mater Res. 2000;51:293. doi: 10.1002/1097-4636(20000905)51:3<293::aid-jbm1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 68.Peppas NA, Bures P, Leobandung W, Ichikawa H. Eur J Pharm Biopharm. 2000;50:27. doi: 10.1016/s0939-6411(00)00090-4. [DOI] [PubMed] [Google Scholar]
- 69.Yamagata T, Morishita M, Kavimandan NJ, Nakamura K, Fukuoka Y, Takayama K, et al. J Control Release. 2006;112:343. doi: 10.1016/j.jconrel.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 70.Morishita M, Goto T, Nakamura K, Lowman AM, Takayama K, Peppas NA. J Control Release. 2006;110:587. doi: 10.1016/j.jconrel.2005.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fisher OZ, Peppas NA. Macromolecules. 2009;42:3391. doi: 10.1021/ma801966r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen H, Gu Y, Hu Y, Qian Z. PDA J Pharm Sci Tech. 2007;61:303. [PubMed] [Google Scholar]
- 73.Guowei D, Adriane K, Chen X, Jie C, Yinfeng L. Int J Pharm. 2007;328:78. doi: 10.1016/j.ijpharm.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 74.Missirlis D, Kawamura R, Tirelli N, Hubbell JA. Eur J Pharm Sci. 2006;29:120. doi: 10.1016/j.ejps.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 75.Lu W, Xiong C, Zhang G, Huang Q, Zhang R, Zhang JZ, et al. Clin Cancer Res. 2009;15:876. doi: 10.1158/1078-0432.CCR-08-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zheng J, Jaffray D, Allien C. Mol Pharm. 2009;6:571. doi: 10.1021/mp800234r. [DOI] [PubMed] [Google Scholar]
- 77.Jain TK, Reddy MK, Morales MA, Leslie-Pelecky DL, Labhasetwar V. Mol Pharm. 2008;5:316. doi: 10.1021/mp7001285. [DOI] [PubMed] [Google Scholar]
- 78.Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, Heuser JE, et al. J Clin Invest. 2009;119:2830. doi: 10.1172/JCI38842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bazile DV, Ropert C, Huve P, Verrecchia T, Marlard M, Frydman A, et al. Biomaterials. 1992;13:1093. doi: 10.1016/0142-9612(92)90142-b. [DOI] [PubMed] [Google Scholar]
- 80.Tong L, He W, Zhang Y, Zheng W, Cheng JX. Langmuir. 2009;25:12454. doi: 10.1021/la902992w. [DOI] [PMC free article] [PubMed] [Google Scholar]