

Abstract
There is still an urgent need to develop nonnucleoside reverse transcriptase (RT) inhibitors (NNRTI) with a high-genetic barrier to resistance that facilitate patient adherence and allow durable suppression of HIV-1 replication. In this study, we describe the synthesis of a novel series of N-aminoimidazole (NAIM) analogs. Each of the NAIM analogs display potent activity against wild-type recombinant purified HIV-1 RT as well as RTs containing the K103N or Y181C resistance mutations. The analogs, however, do not exhibit significant antiviral activity in cell culture and were, in general, cytotoxic. Nevertheless, these data suggest that the NAIM backbone may provide a suitable scaffold from which inhibitors active against NNRTI-resistant HIV-1 could be developed.
Keywords: N-aminoimidazole, NNRTI, HIV, reverse transcriptase, synthesis, molecular modeling
Introduction
Reverse transcription of the single-stranded (+) RNA genome into double-stranded DNA is an essential step in the HIV-1 replication life-cycle. This process is complex and requires the concerted function of both the DNA polymerase and ribonuclease H (RNase H) active sites of HIV-1 reverse transcriptase (RT). Due to its essential role in HIV-1 replication, RT is a major target for antiviral drug development and two classes of inhibitors, (1) the nucleoside and nucleotide RT inhibitors and (2) the nonnucleoside RT inhibitors (NNRTIs) have been approved by the United States Food and Drug Administration (FDA) for the treatment of HIV-1 infection.
NNRTIs (e.g. nevirapine, delavirdine, efavirenz and etravirine) are routinely used in both first-line and salvage combination antiretroviral therapies, and to prevent mother-to-child transmission in resource poor settings. NNRTI interact with HIV-1 RT by binding to a single site in the p66 subunit of HIV-1 RT termed the NNRTI binding pocket (NNRTI-BP) that is situated approximately 10Å from the RT DNA polymerase active site [1]. Crystallographic analyses of HIV-1 RT in complex with NNRTI have suggested at least 3 possible mechanisms that explain NNRTI inhibition. (1) Esnouf et al reported that NNRTI binding distorts the precise geometry of the DNA polymerase catalytic site and proposed that this class of drugs inhibits DNA polymerization by locking the polymerase active site in an inactive conformation [2]. (2) Hsiou et al observed that NNRTI binding deformed the structural elements that comprise the “primer grip”, a region in RT that is involved in the precise positioning of the primer DNA strand in the polymerase active site. This change in “primer grip” conformation may alter the position and conformation of the template/primer (T/P) substrate thereby preventing the establishment of a catalytically competent ternary complex [3]. (3) Kohlstaedt et al proposed that the NNRTI-BP may normally function as a hinge between the palm and thumb domains. Since the mobility of the thumb may be important to facilitate T/P translocation, the binding of NNRTIs may restrict the mobility of the thumb domain thereby slowing down or preventing T/P translocation and/or elongation of nascent viral DNA [1]. The three mechanisms suggested above are not mutually exclusive, and NNRTIs may exert multiple inhibitory effects on RT catalyzed DNA synthesis. In fact, several kinetic and thermodynamic studies have illustrated the complexities of the RT-NNRTI interaction [4–7].
Although NNRTI containing regimens are effective and generally well-tolerated in the majority of patients, treatment durability is limited by drug-related side effects and the development of drug resistance. HIV-1 resistance to NNRTIs correlates directly with mutations of one or more RT residues in the NNRTI-BP. Mutations associated with resistance to NNRTIs include L100I, K101E, K103N, V106A, V108I, V179D, Y181C, Y188C/L/H, G190A/E/S, M230L, P236L and Y318F [8]. These mutations can affect inhibitor binding in a number of ways. (1) They can remove one or more favorable interactions between the inhibitor and NNRTI-binding pocket. For example, the Y181C mutation eliminates π-stacking interactions between this residue and the aromatic ring of the NNRTI pharmacophore [9]. (2) They can introduce steric barriers to NNRTI binding. For example, the G190E mutations introduce a bulky side-chain which may prevent NNRTI binding by sterically interfering with functional groups, such as the cyclopropyl ring of nevirapine [10]. (3) The mutations may introduce or eliminate inter-residue contacts in the NNRTI-binding pocket which interfere with the ability of other residues in the pocket to fold down over the NNRTI [11].
Accordingly, there is still an urgent need to develop NNRTI with a high-genetic barrier to resistance that facilitate patient adherence and allow durable suppression of viral replication. In this regard, previous studies have demonstrated that NNRTI which contain an imidazole nucleus exhibit activity against both wild-type (WT) and drug-resistant strains of HIV-1. For example capravirine (see Fig. (1)) retains activity against HIV-1 containing the single key NNRTI mutations K103N, V106A or L100I [12]. The N-aminoimidazoles (NAIMs) have also been reported to inhibit replication of the WT virus as well as an HIV-1 strain that contained both the K103N and Y181C mutations [13]. In light of these studies, we have synthesized and characterized the anti-HIV-1 activity of a novel series of NAIMs that contained a thiourea moiety (N-NH-CS-NH) with one aryl ring at the 5th position of the imidazole nucleus and the other aryl ring via an NHCSNH linker.
Fig. (1).

Chemical structures of capravirine, NAIM analogs described by Lagoja et al (13) and the novel NAIM analogs synthesized in this study.
Methods
Materials
The wild-type (WT), K103N, and Y181C HIV-1RTs (subtype B, LAI) were purified as described previously [14, 15]. The protein concentration of the purified enzymes was determined spectrophotometrically at 280 nm using an extinction coefficient (ε280) of 260450 M−1 cm−1, and by Bradford protein assays (Sigma-Aldrich, St. Louis, MO). The RNA- and DNA-dependent DNA polymerase activities of the purified WT and mutant enzymes were essentially identical (data not shown). [3H]TTP was acquired from PerkinElmer Life Sciences (Boston, MA). RNA and DNA oligonucleotides were synthesized by IDT (Coralville, IA). All the starting anilines were obtained from Central Drug House (CDH), Delhi, India. 85% hydrazine hydrate was procured from S.D. Fine Chemicals. p-nitrophenacylbromide and p-phenyl phenacylbromide were purchased from Fluka.
Inhibition of WT and mutant HIV-1 RT
Fixed time point assays were used to determine HIV-1 RT-associated RNA-dependent DNA polymerase activity, as reported previously [16]. Assays were carried out using poly(rA)-oligo(dT18) as the template/primer (T/P) substrate. The oligo (dT18) primers was synthesized with a biotin label on the 5′-terminus. DNA polymerase reactions were carried out in 50 mM Tris-HCl pH 7.5 (37 °C), 50 mM KCl, 10 mM MgCl2 containing 600 nM T/P, 1 μM of [3H]TTP, and variable concentrations of inhibitor. Reactions were initiated by the addition of 25 nM of RT, incubated for 20 min at 37 °C and then quenched with 0.5 M EDTA. Streptavidin Scintillation Proximity Assay beads (GE Healthcare, Piscataway, NJ) were then added to each reaction, and the extent of radionucleotide incorporation was determined by scintillation spectrometry using a 1450 Microbeta Liquid Scintillation Counter (Perkin Elmer, Waltham, MA).
Antiviral and cytotoxicity assays
Stock wild-type (WT) and mutant LAI HIV-1 were prepared as reported previously [17]. The 50% cell culture infective dose for the virus stock was determined for P4/R5 cells by 3-fold endpoint dilution assays and calculated using the Reed and Muench equation [18]. Drug susceptibility assays were performed using P4/R5 reporter cells as reported previously [19]. Inhibition of virus replication was calculated as the concentration of compound required to inhibit virus replication by 50% (EC50). Fold-resistance values were determined by dividing the EC50 for mutant HIV-1 by the EC50 for WT HIV-1. NNRTI toxicity was also evaluated in the P4/R5 cells. Briefly, log-phase cells were seeded at 5 × 103 cells/well in 96-well plates containing 2-fold serial dilutions of test compound and then incubated at 37°C with 5% CO2 for 48 hours. Cell Viability was assessed using CellTiter-Glo reagent (Promega, Madison, WI) according to the manufacturer’s instructions. The median 50% cytotoxic concentration (CC50) was determined from the concentration–response curve using the median effect method.
Molecular modeling
All computational studies were carried out using Glide v 5.0 [20] software package installed in a single machine running on a 3.4 GHz Pentium 4 processor with 1GB RAM and 160 GB Hard Disk with Red Hat Linux Enterprise version 5.0 as the Operating System.
Ligand Structure Preparation
Ligand structures were drawn and optimized using PRODRG online server [21] and saved in PDB format. By using the Ligprep utility of Glide, these structures were geometry optimized by using the Optimized Potentials for Liquid Simulations-2005 (OPLS-2005) force field [22, 23] with the steepest descent followed by truncated Newton conjugate gradient protocol. Partial atomic charges were computed using the OPLS-2005 force field.
Protein Structure Preparation
The geometry of the NNRTI-BP of the WT HIV-1 RT was taken from the structure of HIV-1 RT/TNK 651 complex filed in the RCSB Protein Data Bank [24] (PDB entry code 1rt2). Water molecules of crystallization were removed from the complex, and the protein was optimized for docking using the protein preparation wizard provided by Schrodinger Inc. Partial atomic charges were assigned according to the OPLS-AA force field.
Validation of Docking Protocol
The most suitable method of evaluating the accuracy of a docking procedure is to determine how closely the lowest energy pose predicted by the scoring function resembles an experimental binding mode as determined by X-ray crystallography. In the present study, Extra Precision Glide docking procedure was validated by removing TNK 651 from the binding site and re-docking it to the NNRTI-BP of HIV-1- RT. We found a very good agreement between the localization of the inhibitors upon docking and from the crystal structure, i.e. similar hydrogen bond interactions with Leu100 and Lys 102 (see Fig. (2A)). The root mean square deviations (RMSD) between the predicted conformation and the observed X-ray crystallographic conformation of compound TNK 651 equaled 2.445 Å (< 3 Å). This indicates the reliability of the docking method in reproducing the experimentally observed binding mode for 1rt2.
Fig. (2). Molecular modeling analyses.
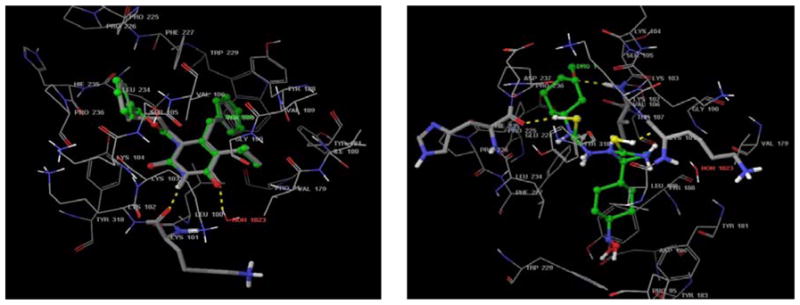
(A) Superimposition of experimental bound (co-crystallized) conformation of TNK651 (atom type) [22] and that predicted by Glide (ball and stick model). Active site amino acid residues are represented by lines. Hydrogen bond interactions are represented by dotted lines.
(B) Molecular model of 5 in the NNRTI-BP of HIV-1 RT (PDB entry 1RT2) Active site amino acid residues are represented as tubes while the inhibitor is shown as ball and stick model with the atoms colored as carbon: green, hydrogen: cyan, nitrogen: blue, and oxygen: red. Hydrogen bond interactions are represented by dotted lines.
Docking
The most active NAIM analog 5 was docked into the coordinates of the crystal structure of 1rt2. All docking calculations were performed using the “Extra Precision” (XP) mode of Glide Program v5.0. A grid was prepared with the center defined by the co-crystallized ligand TNK 651 for 1rt2. During the docking process, Glide initially performed a complete systematic search of the conformational, orientational and positional space of the docked ligand and eliminating unwanted conformations using scoring and followed by energy optimization. Finally the conformations were further refined via Monte Carlo sampling of pose conformation. Predicting the binding affinity and rank-ordering ligands in database screens was implemented by modified and expanded version of the Glide Score scoring function.
Results and Discussion
The primary objective of this study was to synthesize a series of novel NAIM analogs that exhibited potent activity against WT and drug-resistant HIV-1.
The synthesis of the novel NAIMs was carried out in two steps, as shown in Scheme 1. In the first step, a 1 ml solution of 0.025 mol 85 % hydrazine hydrate was added to a solution of 0.01 mol 1 dissolved in 15 ml of methanol, and was then stirred at 60 °C for 2 hr to obtain 2. Compounds 3 to 10 were then synthesized by treatment of 2 with an equivalent amount of 2-bromo-1-(4-nitrophenyl) ethanone or 1-(biphenyl-4-yl)-2-bromoethanone and potassium thiocyanate in glacial acetic acid at 60°C with stirring for 1 hr, then refluxed for 6 hr. The purity of the synthesized compounds was assessed by TLC and elemental analyses (see Table 1), and the desired compounds were identified by their spectral data. In general, the IR spectra showed a C=S peak at 719 cm−1 and a C-N peak at 1340 cm−1. The NMR spectra showed a singlet of 6.37 ppm and 2.05 ppm corresponding to the NH-Ar and imidazole-NH groups respectively. The elemental analysis results were within ±0.4% of the theoretical values (see Table 1).
Scheme 1. Scheme for the synthesis of the novel NAIM analogs.

Reagents and conditions: (a) NH2NH2, CH3OH, 60°C, 2 hr; (b) KSCN, CH3COOH, 60°C, 1 h, then reflux, 6 h
Table 1.
Elemental analyses of the synthesized NAIM analogs
| Compound | C | H | N | |||
|---|---|---|---|---|---|---|
| Theoretical | Determined | Theoretical | Determined | Theoretical | Determined | |
| 3 | 52.97 | 52.93 | 3.92 | 3.96 | 18.17 | 18.35 |
| 4 | 52.97 | 52.92 | 3.92 | 3.95 | 18.17 | 18.20 |
| 5 | 50.86 | 50.83 | 3.77 | 3.66 | 17.44 | 17.40 |
| 6 | 56.99 | 56.95 | 3.59 | 3.54 | 16.62 | 16.59 |
| 7 | 66.32 | 66.36 | 4.84 | 4.80 | 13.45 | 13.47 |
| 8 | 66.32 | 66.30 | 4.84 | 4.87 | 13.45 | 13.42 |
| 9 | 63.86 | 63.83 | 4.66 | 4.80 | 12.95 | 12.96 |
| 10 | 69.00 | 69.02 | 4.45 | 4.41 | 12.38 | 12.35 |
The anti-HIV-1 activity of compounds 3 to 10 was first assessed against recombinant purified HIV-1 RT using an RNA-dependent DNA polymerase activity assay. All of the compounds exhibited activity against WT HIV-1 RT that was comparable to that of the FDA approved NNRTI nevirapine (see Table 2). The most potent analog in the series was 5. Surprisingly, and in contrast to nevirapine, all of the NAIMs analogs retained good activity against HIV-1 RT containing either the K103N or Y181C mutations. For example, whereas the IC50 for nevirapine was increased >75-fold for both Y181C and K103N HIV-1 RT relative to the WT enzyme, the IC50 values for all of the NAIM analogs against the Y181C and K103N RTs was decreased less than 2-fold relative to the WT enzyme. We also assessed the anti-HIV-1 activity and cytotoxicity of each of the NAIMs in cell culture (see Table 2). Unfortunately, the NAIMs exhibited weak antiviral activity and were relatively toxic to the P4/R5 cells used in the study. In this regard, the weak antiviral activity might be due to poor cellular uptake or, alternatively, high protein binding in the cell.
Table 2.
Anti-HIV-1 activity of the synthesized NAIM analogs
| Compound | EC50(μM)a | CC50(μM)b | WT HIV-1 RT | Y181C HIV-1 RT | K103N HIV-1 RT | ||
|---|---|---|---|---|---|---|---|
| IC50(μM)c | IC50(μM)c | Fold-Rd | IC50(μM)c | Fold-Rd | |||
| 3 | 32.0 | 56.9 | 6.6±0.7 | 9.0±2.2 | 1.4 | 11.5±2.3 | 1.7 |
| 4 | 59.1 | 41.1 | 6.7±1.3 | 7.2±1.7 | 1.1 | 7.9±1.5 | 1.2 |
| 5 | 36.2 | 50.8 | 2.9±1.2 | 6.1±1.2 | 2.1 | 7.7±1.9 | 2.7 |
| 6 | >90 | >45 | 7.1±0.9 | 6.6±0.9 | 0.9 | 6.6±1.9 | 0.9 |
| 7 | 44.5 | 13.3 | 4.9±1.9 | 8.5±1.5 | 1.7 | 6.1±0.9 | 1.2 |
| 8 | 92.0 | 21.1 | 8.3±2.1 | 6.8±1.3 | 0.8 | 7.7±1.8 | 0.9 |
| 9 | >90 | 21.8 | NDe | NDe | NDe | NDe | NDe |
| 10 | 67.3 | 18 | 4.9±0.8 | 8.9±2.0 | 1.8 | 7.5±2.1 | 1.5 |
| Nevirapine | 0.048 | >45 | 7.2±1.4 | >75 | >10 | >75 | >10 |
| Efavirenz | 0.002 | >20 | 0.094±0.006 | 0.108±0.004 | 1.1 | 1.4±0.4 | 15.1 |
Antiviral activity as assessed in P4/R5 cells
Cytotxicity as assessed in P4/R5 cells
RDDP activity assessed using recombinant purified HIV-1 RT
Fold resistance (IC50Mutant/IC50WT)
Not determined due to insolubility of the compound.
To gain some structural insight into how the inhibitors interact with RT, we performed in silico docking studies to investigate the interaction of 5 with the NNRTI-binding pocket of the WT HIV-1 RT (PDB entry code 1rt2) using the Extra Precision (XP) mode of Glide software [20] (see Fig.(2)). To validate the Glide software, we first modeled the interaction between TNK651 and HIV-1 RT. Superimposition of the experimental bound (co-crystallized) conformation of TNK651 [22] and that predicted by Glide are shown in Fig. (2A). Glide successfully reproduced the experimental binding conformations of TNK 651 in the NNRTI-binding pocket of HIV-1 RT with an acceptable root-mean-square deviation (RMSD) of 2.4 Å. In our model of compound 5 with the NNRTI-binding pocket, the thiol group on the imidazole moiety forms a hydrogen bond with the main chain carbonyl oxygen of K101(SHimidazole ··· COK101 = 1.81 Å). The sulphur in the linker region forms a hydrogen bond with the C=O terminal of H235 (SHimidazole ··· COH235 = 2.11 Å). A hydrogen bond is also evident between the phenyl group attached to NHCSNH side chain making a hydrogen bond interaction with the NH terminal group of V106(SHimidazole ···COV106 = 1.96 Å). The same phenyl group also makes favorable interaction with the milder hydrophobic pocket formed by the side chain of V106, K102, K103 and P236. The imidazole nucleus made favorable interactions with the least hydrophobic portions of the NNRTI binding pocket. The NHCSNH side chain interacts with the side chain terminal of Y318. The other phenyl group attached to the fifth position of the imidazole nucleus makes favorable interactions with a bigger hydrophobic and aromatic rich pocket formed by the side chains of Y181, Y188, F227, W229 and L234. In this regard, the orientation of the phenyl groups as hydrophobic moieties into the hydrophobic pockets of NNRTI-BP and the imidazole nucleus as the hydrophilic moiety oriented towards the least hydrophobic portion may be responsible for the observed activity of this analog against RTs containing Y181C or K103N.
Conclusions
In this study we synthesized eight novel NAIM analogs and assessed their activity against both HIV-1 RT and virus. Although the NAIM analogs were found to be toxic and exhibited weak antiviral activity in cell culture, the compounds were remarkably potent against both WT and drug-resistant recombinant purified HIV-1 RTs. Taken together, these data suggest that the NAIM backbone may provide a suitable scaffold from which inhibitors active against NNRTI-resistant HIV-1 could be developed.
Acknowledgments
Research in the Sluis-Cremer laboratory was supported by a grant of the United States of America National Institutes of Health (2 R01 GM068406). Dr Ganguly acknowledges University Grants Commission for providing financial support in the form of a Minor Research Project.
References
- 1.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;26:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 2.Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- 3.Hsiou Y, Ding J, Das K, Clark AD, Jr, Hughes SH, Arnold E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996;15:853–860. doi: 10.1016/s0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- 4.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;17:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia Q, Radzio J, Anderson KS, Sluis-Cremer N. Probing nonnucleoside inhibitor-induced active-site distortion in HIV-1 reverse transcriptase by transient kinetic analyses. Protein Sci. 2007;16:1728–1737. doi: 10.1110/ps.072829007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S, Abbondanzieri EA, Rausch JW, Le Grice SF, Zhuang X. Slide into action: dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science. 2008;322:1092–1097. doi: 10.1126/science.1163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abbondanzieri EA, Bokinsky G, Rausch JW, Zhang JX, Le Grice SF, Zhuang X. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature. 2008;453:184–189. doi: 10.1038/nature06941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson VA, Brun-Vezinet F, Clotet B, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD. Update of the Drug Resistance Mutations in HIV-1. Top HIV Med. 2006;14:125–130. [PubMed] [Google Scholar]
- 9.Ren J, Nichols C, Bird L, Chamberlain P, Weaver K, Short S, Stuart DI, Stammers DK. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J Mol Biol. 2001;28:795–805. doi: 10.1006/jmbi.2001.4988. [DOI] [PubMed] [Google Scholar]
- 10.Huang W, Gamarnik A, Limoli K, Petropoulos CJ, Whitcomb JM. Amino acid substitutions at position 190 of human immunodeficiency virus type 1 reverse transcriptase increase susceptibility to delavirdine and impair virus replication. J Virol. 2003;77:15–23. doi: 10.1128/JVI.77.2.1512-1523.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren J, Stammers DK. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res. 2008;134:157–170. doi: 10.1016/j.virusres.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 12.Fujiwara T, Sato A, El-Farrash M, Miki S, Abe K, Isaka Y, Kodama M, Wu Y, Chen LB, Harada H, Sugimoto H, Hatanaka M, Hinuma Y. S-1153 inhibits replication of known drug-resistant strains of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 1998;42:1340–1345. doi: 10.1128/aac.42.6.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagoja IM, Pannecouque C, Van Aerschot A, Witvrouw M, Debyser Z, Balzarini J, Herdewijn P, De Clercq E. N-Aminoimidazole Derivatives. doi: 10.1021/jm0211117. [DOI] [PubMed] [Google Scholar]
- 14.Le Grice SF, Gruninger-Leitch F. Purification and characterization of human immunodeficiency virus type 1 reverse transcriptase. Eur J Biochem. 1990;26:307–314. doi: 10.1111/j.1432-1033.1990.tb15306.x. [DOI] [PubMed] [Google Scholar]
- 15.Le Grice SF, Cameron CE, Benkovic SJ. Rapid purification of homodimer and heterodimer HIV-1 reverse transcriptase by metal chelate affinity chromatography. Methods Enzymol. 1995;262:130–144. [Google Scholar]
- 16.Nissley DV, Radzio J, Ambrose Z, Sheen CW, Hamamouch N, Moore KL, Tachedjian G, Sluis-Cremer N. Characterization of novel non-nucleoside reverse transcriptase (RT) inhibitor resistance mutations at residues 132 and 135 in the 51 kDa subunit of HIV-1 RT. Biochem J. 2007;404:151–157. doi: 10.1042/BJ20061814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parikh UM, Koontz DL, Chu CK, Schinazi RF, Mellors JW. In vitro activity of structurally diverse nucleoside analogs against human immunodeficiency virus type 1 with the K65R mutation in reverse transcriptase. Antimicrob Agents Chemother. 2005;49:1139–1144. doi: 10.1128/AAC.49.3.1139-1144.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 19.Sluis-Cremer N, Koontz D, Bassit L, Hernandez-Santiago BI, Detorio M, Rapp KL, Amblard F, Bondada L, Grier J, Coats SJ, Schinazi RF, Mellors JW. Anti-human immunodeficiency virus activity, cross-resistance, cytotoxicity, and intracellular pharmacology of the 3′-azido-2′,3′-dideoxypurine nucleosides. Antimicrob Agents Chemother. 2009;53:3715–3719. doi: 10.1128/AAC.00392-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glide 5.0. Schrödinger, Inc; New York, U.S.A: 2008. www.schrodinger.com. [Google Scholar]
- 21.http://davapc1.bioch.dundee.ac.uk/prodrg/index.html
- 22.Hopkins AL, Ren J, Esnouf RM, Willcox BE, Jones EY, Ross C, Miyasaka T, Walker RT, Tanaka H, Stammers DK, Stuart DI. Complexes of HIV-1 Reverse Transcriptase with Inhibitors of the HEPT Series Reveal Conformational Changes Relevant to the Design of Potent Non-Nucleoside Inhibitors. J Med Chem. 1996;39(8):1589–1600. doi: 10.1021/jm960056x. [DOI] [PubMed] [Google Scholar]
- 23.William LJ, David SM, Julian T. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 24.http://www.rcsb.org/pdb/