

Abstract
Direct acetonide protection of the catechol of dopamine has proven to be problematic due to formation of Pictet-Spengler isoquinolines. Here we report an efficient method for acetonide protection of dopamine, allowing preparation of a dopamine prodrug without complications from the Pictet-Spengler reaction. Acetonide-protected dopamine was first synthesized by pre-protecting the amino group with phthaloyl followed by refluxing with 2,2-dimethoxypropane in the presence of TsOH. Further work demonstrated that Fmoc or trifluoroacetyl were also suitable N-protective groups, while Boc-protected dopamine gave an isoquinoline product. Acetonide-protected dopamine was coupled to DHA (all cis-4,7,10,13,16,19-docosahexaenoic acid) to produce the N-DHA-dopamine prodrug in high purity.
In both vertebrate and invertebrate animals, 4-(2-aminoethyl)benzene-1,2-diol (dopamine) is the precursor of norepinephrine and epinephrine but also an important neurotransmitter itself, essential to the normal functioning of the central nervous system.1 Parkinson’s disease, affecting about 1% of the senior population, is characterized by a reduction of dopamine levels in the striatum.2 At physiological pH dopamine is almost completely ionized, resulting in low permeation across the blood brain barrier and precluding it as a direct treatment for Parkinson’s disease.
Prodrugs formed through conjugation of lipophilic molecules to dopamine display enhanced uptake into the brain. DHA (all cis-4,7,10,13,16,19-docosahexaenoic acid) is a major component of polyunsaturated fatty acids in retinal and neuronal membranes. Prodrug N-DHA-dopamine, a potentially useful appetite-suppressant, was found to have a Blood-Brain Barrier Index of 30%, comparable with that of D-glucose (33%).3,4 Unlike amphetamine-based suppressants, which usually have cardiovascular and neuropsychiatric side effects and induce tolerance, N-DHA-dopamine was inactive until it hydrolyzes to release dopamine in the brain and thus produced no harmful side effects.4 Its synthesis was previously reported by using symmetrical DHA anhydride and unprotected dopamine. However, carboxylic anhydride is highly reactive toward with both the amino group and the free hydroxyl groups of dopamine to give a mixture. After time-consuming purifications through mixed-bed ion exchange resins, a correct mass of N-DHA-dopamine remained elusive.4 Other methods using fatty acyl halogenides or imidazolides gave less satisfactory results.5 The target products were obtained in lower yields and were contaminated with the corresponding fatty acid imides.
One of the challenges in manipulating dopamine during reactions is that it is readily oxidized, especially under basic conditions, to dopamine quinone, which undergoes self-polymerization or nucleophilic addition reactions with amino or sulfhydryl groups.6 Proper protection of the catechol group is often required during chemical modification of dopamine. Various catechol protecting groups have been reported, including methyl ether,7 cyclic ethyl orthoformate,8 t-butyldimethylsilyl,9 and acetonide.10 Easy protection/deprotection together with good stability to strong bases and weak acids makes the acetonide protective group especially useful.11 Ideally, the only byproduct formed during deprotection is acetone which can be easily removed by evaporation. We report here a facile synthesis of N-DHA-dopamine prodrug with high purity and good yield by coupling reaction between DHA and acetonide-protected dopamine followed by removal of the acetonide group.
Direct protection of a catecholamine by acetonide is not easily accomplished by refluxing with acetone or its ketal 2,2-dimethoxypropane (DMP) in the presence of a catalyst, e.g., p-toluenesulfonic acid (TsOH). Like other beta phenylamines, dopamine readily undergoes Pictet-Spengler condensations with aldehydes and ketones to produce tetrahydroisoquinolines. Such acetonide compounds as 1-(2,2-dimethylbenzo[1,3]dioxol-5-yl)propan-2-amine are usually obtained by a complicated method involving construction of the target molecule from an acetonide protected catechol subunit, introduction of a nitro group by a nitration reagent such as nitroethane, and reduction of the -NO2 to -NH2 group with lithium aluminum hydride.12 Christian’s work demonstrated that when dopamine was blocked as a glucoamide, direct acetonide cyclization was readily accomplished by refluxing in acetone.13,14 Considering that cleavage of a glucoamide bond to free up the dopamine may require harsh conditions, e.g. heating in strongly basic solution,15 we decided to explore a strategy using an easily introducible and removable N-protecting group to temporarily mask the amino group of dopamine followed by refluxing with DMP in the presence of TsOH. A systematic study was also carried out to screen the amino protecting groups, including cyclic imide, carbamate, and amide.
One of the simplest methods to differentiate the reactivity of the amino and the catechol group is to form an ammonium salt by protonation with strong acids. For example, selective acetylation of the catechol of 3,4-dihydroxyphenylalanine (DOPA) was achieved by protonation of the amino group with hydrogen chloride or bromide.16,17 Unfortunately, refluxing dopamine hydrochloride with acetone gave a Pictet-Spengler isoquinoline instead of an acetonide product (Scheme 1).18
Scheme 1.
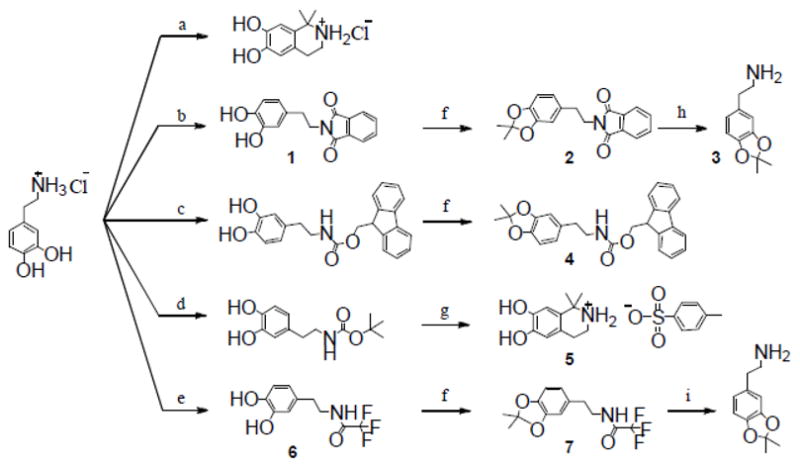
Reagents and conditions: (a), reference18; (b), N-carbethoxyphthalimide, Et3N, 74%; (c), reference19; (d), reference20; (e), CF3COOMe, Et3N in MeOH, 98%; (f), DMP, TsOH (4.5%), benzene, reflux, yield, 95% (2), 90% (4), 89% (7); (g), DMP, TsOH (1.05 equiv), benzene, reflux, >10%; (h), H2NNH2, DCM, 97%; (i), LiOH, THF/H2O, 87%.
A sufficient protecting method may be to completely remove both of the two hydrogen atoms of the amino group. For this purpose, the N-phthaloyl (Phth) protective group was introduced to dopamine by N-carbethoxyphthalimide in methanol. The resulting Phth-dopamine (1) was then refluxed with DMP and a catalytic amount of TsOH in benzene. To shift the reaction equilibrium, volatile byproducts were removed from the reaction system by distillation. The condensed liquid was recycled using a Soxhlet extractor, the thimble of which was filled with anhydrous CaCl2 to absorb water and methanol. In addition, anhydrous solvent and argon protection were used to prevent introduction of water from external sources, shortening the reaction time to ca. 1–2 h from 24 h sometimes required for traditional methods.21 The raw product was subjected to GC-MS analysis, which gave a major peak in the chromatogram and a molecular ion at 323 (calcd. 323.12) in the mass spectrum, indicating that full protection of the amino group results in acetonide cyclization rather than isoquinoline formation. The identification of Phth-dopamine(acetonide) (2) was further confirmed by high resolution mass spectrometry (HRMS) and nuclear magnetic resonance (NMR) spectroscopy, with the latter showing a peak at δppm 117.8 for the quaternary carbon of the acetonide group. Acetonide protected dopamine (3)22 was obtained by deprotection of the phthaloyl group of 2 with hydrazine in DCM. In a previous application of this strategy, Phth-protected DOPA methyl ester was converted to Phth-DOPA(acetonide)-OMe, leading to the synthesis of Fmoc-DOPA(acetonide)-OH, a useful intermediate to incorporate DOPA into synthetic peptides.10
The use of hazardous hydrazine23 and the difficulty to remove hydrazide byproduct24,25 during dephthalylation of Phth-DOPA(acetonide)-OMe prompted us to examine other N-protecting groups, such as the carbamates Boc and Fmoc and the amide trifluoroacetyl (Tfa), which remove only one of the two hydrogen atoms of the amino group. The introduction of a carbonyl group adjacent to the nitrogen atom followed by protonation of the amide nitrogen may form an N-acyliminium ion, which probably facilitates Pictet-Spengler type reactions due to increased eletrophilic reactivity.26 However, acetonide cyclization may still be favored if the reaction is carried out in an aprotic solvent with only catalytic amount of acids.
Fmoc- and Boc-protected dopamine behaved quite differently when refluxed with DMP and TsOH. Fmoc-dopamine, synthesized by reacting dopamine with fluorenylmethyloxycarbonyl chloride,19 readily underwent acetonide cyclization to form Fmoc-dopamine(acetonide)(4). The course of the reaction could be conveniently monitored by the FeCl3 test on a TLC plate, which produced a black spot for free catechols at room temperature. After acetonide protection, the spot was indistinguishable from yellow background, but turned black after heating to 105 °C. In the case of Boc-dopamine20, even after a 4 h reflux with 5.5% TsOH and DMP, ferric chloride test still produced a dark black dot on the TLC plate, indicating the presence of a significant amount of unprotected catechol. A second portion of TsOH (1 equiv) was added and the reaction mixture was stirred for another 1 h to give a precipitate, which was collected, purified, and subjected to intensive characterization. Positive mode LC-MS data revealed a monoisotopic molecular ion of m/z 194.10, corresponding to C11H16NO2 (MH+), suggesting the addition of a C2H6C group to dopamine. Negative mode LC-MS data showed a peak at 171.00, indicating the presence of p-toluenesulfonate anions ((M-H)−, Calcd. 171.01). Combining the data provided by HRMS and NMR spectra, the product was identified as a sulfonic salt of a tetrahydroisoquinoline (5). The result is not entirely unexpected because the acid-labile Boc protective group is readily removed when heated in the presence of TsOH, releasing dopamine that subsequently undergoes a Pictet-Spengler condensation.27
In contrast with the Boc protective group, Tfa is stable to acids, and it is smaller in size than the Fmoc group. Although Tfa-dopamine (6) could be isolated in low yield (19%) from the Bischler-Napieralski reaction of Tfa and methyl ether protected dopamine28, an improved procedure was developed by treating dopamine hydrochloride with methyl trifluoroacetate in methanol in the presence of triethylamine (Et3N), giving 6 in nearly quantitative yield. The acetonide protection of compound 6 ran smoothly and was completed in 1.5 h with a yield of ca. 89%. The advantage of using Tfa protective group is that its deprotection does not require hazardous hydrazine. Compound 3 was readily obtained by hydrolysis of Tfa-dopamine(acetonide) (7) in lithium hydroxide solution followed by simple extraction.
Compound 3 provides a convenient way to synthesize dopamine-containing molecules including highly pure N-DHA-dopamine prodrug. Commercially available DHA was activated with dicyclohexylcarbodiimide (DCC) and converted to an N-hydroxysuccinimide (NHS) ester, which was then stirred with dopamine(acetonide) in toluene and chloroform. The resulting N-DHA-dopamine(acetonide) (8) was quite stable to routine work-up and was readily purified by flash chromatography (hexane/EtOAc) to give a light yellow oil, yield 68% (90% pure by RP-HPLC). The molecular formula of this intermediate was established by HRMS: C33H45NO3, MH+, Calcd 504.34722, Found 504.34608. Further purification by semi-preparative RP-HPLC provides >98% pure compound 8, which was hydrolyzed in 30% trifluoroacetic acid (TFA) chloroform solution to quantitatively produce DHA-dopamine (9)29, a brown oil residue with a correct mass (HRMS): MH+, Calcd. 464.31592, Found 464.31560. Judging from analytical RP-HPLC chromatogram, the resultant compound 9 was more than 97% pure without requiring any chromatographic purification (see Supporting Information).
In conclusion, by masking the amino group with a proper N-protecting group, acetonide-protected dopamine was first synthesized in the presence of TsOH in anhydrous benzene. The resulting acetonide derivative should facilitate the preparation of highly pure catechol-containing compounds, as demonstrated by the synthesis a lipophilic prodrug N-DHA-dopamine. This novel strategy is easily generalized to acetonide protection of other catecholamines.
Supplementary Material
Scheme 2.
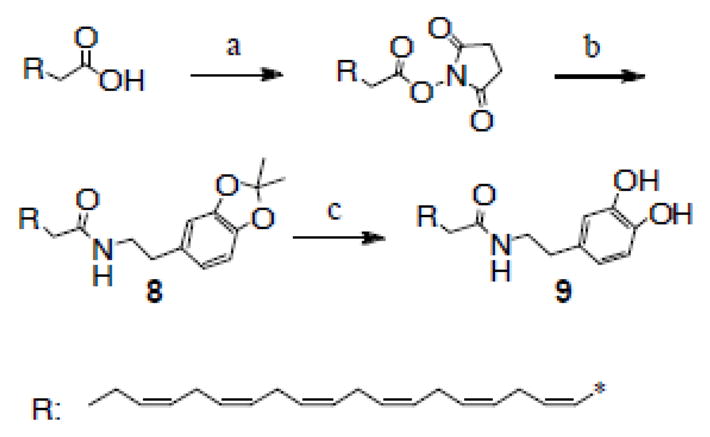
Reagents and conditions: (a), DCC/NHS; (b), dopamine(acetonide), 68% (two steps); (c), 25% TFA in CHCl3/H2O, 100%.
Acknowledgments
This research was supported by NIH Grants R37 DE 014193, UL1 RR025741 and U54 CA119341. NMR and Mass Spectra were completed at IMSERC, Northwestern University. The authors thank Dominic Fullenkamp for his comments on the manuscript.
Footnotes
Supplementary Materials
Supplementary materials associated with this article can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benes FM. Trends Pharmacol Sci. 2001;22:46–47. doi: 10.1016/s0165-6147(00)01607-2. [DOI] [PubMed] [Google Scholar]
- 2.Mendis T, Suchowersky O, Lang A, Gauthier S. Can J Neurol Sci. 1999;26:89–103. [PubMed] [Google Scholar]
- 3.Shashoua VE, Hesse GW. Life Sci. 1996;58:1347–1357. doi: 10.1016/0024-3205(96)00101-4. [DOI] [PubMed] [Google Scholar]
- 4.Shashoua VE. US2003050226A1. US Pat Appl Publ. 2003
- 5.Bezuglov V, Bobrov M, Gretskaya N, Gonchar A, Zinchenko G, Melck D, Bisogno T, Di Marzo V, Kuklev D, Rossi JC, Vidal JP, Durand T. Bioorg Med Chem Lett. 2001;11:447–449. doi: 10.1016/s0960-894x(00)00689-2. [DOI] [PubMed] [Google Scholar]
- 6.LaVoie MJ, Hastings TG. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Gancedo A, Gil C, Roldan CM, Perez S, Vilas P. Chemotherapy. 1979;25:83–90. doi: 10.1159/000237827. [DOI] [PubMed] [Google Scholar]
- 8.Hu BH, Messersmith PB. Tetrahedron Lett. 2000;41:5795–5798. [Google Scholar]
- 9.Li G, Cheng G, Xue H, Chen S, Zhang F, Jiang S. Biomaterials. 2008;29:4592–4597. doi: 10.1016/j.biomaterials.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Hu BH, Messersmith PB. Tetrahedron Lett. 2008;49:5519–5521. doi: 10.1016/j.tetlet.2008.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Statz AR, Meagher RJ, Barron AE, Messersmith PB. J Am Chem Soc. 2005;127:7972–7973. doi: 10.1021/ja0522534. [DOI] [PubMed] [Google Scholar]
- 12.Nichols DE, Kostuba LJ. J Med Chem. 1979;22:1264–1267. doi: 10.1021/jm00196a022. [DOI] [PubMed] [Google Scholar]
- 13.Christian ST. WO2001079244A1. PCT Int Appl. 2001
- 14.Christian ST, Sundsmo JS. US2005250739A1. US Pat Appl Publ. 2005
- 15.Mark C. WO2008006864. PCT Int Appl. 2008
- 16.Harwood HJ, Cassidy HG. J Am Chem Soc. 1957;79:4360–4365. [Google Scholar]
- 17.Fuller WD, Verlander MS, Goodman M. Biopolymers. 1978;17:2939–2943. doi: 10.1002/bip.1976.360150922. [DOI] [PubMed] [Google Scholar]
- 18.Morita S, Ito T, Tono T. Agric Biol Chem. 1975;39:547–549. [Google Scholar]
- 19.Felder Flesch D, Steibel J, Bertin A. WO2008043911. PCT Int Appl. 2008
- 20.Cai W, Kwok SW, Taulane JP, Goodman M. J Am Chem Soc. 2004;126:15030–15031. doi: 10.1021/ja0442062. [DOI] [PubMed] [Google Scholar]
- 21.Cole ER, Crank G, Minh HTH. Aust J Chem. 1980;33:675–680. [Google Scholar]
- 22.1H NMR (500 MHz, CDCl3): δppm 6.66-6.59 (m, 3H), 2.92 (t, 2H), 2.66 (t, 2H), 1.91 (br, 2H), 1.67 (s, 6H). 13C NMR (125 MHz, CDCl3): 147.6, 145.9, 132.1, 121.1, 117.6, 108.9, 108.0, 42.8, 38.0, 25.8 (2C). DEPT: CH3, 25.8; CH2, 42.8, 38.0; CH, 121.1, 108.9, 108.0. GC-MS: m/z 193 (17%), 164 (80.6%), 163 (32.4%), 149 (100%), 124 (18.1%), 123 (75%), 121 (18.1%), 106 (23.6%). HRMS (ESI): C11H15NO2, MH+, Calcd 194.11756, Found 194.11757.
- 23.Black TD, Briggs BS, Evans R, Muth WL, Vangala S, Zmijewski MJ. Biotechnol Lett. 1996;18:875–880. [Google Scholar]
- 24.Le Roy I, Mouysset D, Mignani S, Vuilhorgne M, Stella L. Tetrahedron. 2003;59:3719–3727. [Google Scholar]
- 25.Delon L, Laurent P, Blancou H. J Fluorine Chem. 2005;126:1487–1492. [Google Scholar]
- 26.Maryanoff BE, Zhang HC, Cohen JH, Turchi IJ, Maryanoff CA. Chem Rev. 2004;104:1431–1628. doi: 10.1021/cr0306182. [DOI] [PubMed] [Google Scholar]
- 27.Babu V, Patil B, Vasanthakumar GR. Synth Commun. 2005;35:1795–1802. [Google Scholar]
- 28.Niederstein Y, Peter MG. Liebigs Ann Chem. 1989:1189–1193. [Google Scholar]
- 29.1H NMR (500 MHz, CDCl3): δppm 6.79-6.52 (m, 3H), 5.99 (s, catecholic proton), 5.40-5.28 (m, 12H), 3.42 (m, 2H), 2.95-2.78(m, 10H), 2.64 (t, 2H, J = 6.8 Hz), 2.36 (q, 2H, J = 6.8 Hz), 2.21 (t, 2H, J = 6.8 Hz). 2.06 (m, 2H, J = 7.3 Hz). 0.96 (t, 3H, J = 7.3 Hz). 13C NMR (125 MHz, CDCl3): δ. 174.2, 144.1, 142.8, 131.8, 130.1, 129.6, 128.3, 128.18, 128.10 (2C), 127.77 (2C), 127.59, 127.54, 127.17, 126.7, 120.3, 115.3, 115.2, 41.2, 36.3, 34.7, 25.6 (4C), 25.5, 23.4, 20.6, 14.3. LC-MS: MH+, Calcd 464.31. Found 464.30; (2M+H)+, Calcd 927.63, Found 927.60. HRMS: C30H41NO3, MH+, Calcd 464.31592, Found 464.31560.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.