

Abstract
Vesicular acetylcholine transporter (VAChT) is a member of the major facilitator superfamily (MFS). It contains conserved sequence motifs originally defined in the bacterial multidrug resistance transporter family of the MFS. Motif C (GSLV227A228PPFGGIL) is located at the C-terminal end of transmembrane helix 5 (TM 5) in VAChT. The motif is rich in glycine and proline residues that often have special roles in backbone conformations of TMs. The A228G mutant of VAChT transports >3-fold faster than wild-type does [Chandrasekaran et al. (2006) J. Neurochem. 1, 1551−1559.]. In the current study, the structure of Loop 4/5, TM 5, and motif C were taken from a three-dimensional homology model for human VAChT. The peptide was immersed in implicit membrane and energy-minimized, and molecular dynamics (MD) were simulated. Kinking and wobbling occur in otherwise helical peptide at the hinge residues L226 and V227. MD also were simulated for A228G single mutant and V227L−A228A double mutant peptides to investigate the structural roles of the A228G mutation and β-branching at V227. Mutant peptides exhibit increased wobbling at the hinge residues, but in the double mutant the increase is less. Because motif C participates in the interface that mediates hypothesized rocker-switch reorientation of the acetylcholine binding site during transport, dynamics in motif C might be an important contributor to transport rate.
Keywords: Vesicular acetylcholine transporter, molecular dynamics, acetylcholine, kink, wobble, transmembrane flexibility
The work reported here investigates an important transmembrane helix (TM) and motif in the vesicular acetylcholine transporter (VAChT), which is a member of the major facilitator superfamily (MFS) (1). VAChT takes up acetylcholine (ACh) synthesized in the cytoplasm of nerve terminals and stores it for evoked secretion (2). Most members of the MFS, including VAChT, contain 12 TMs that are predominantly α-helical.
There is no crystal structure of VAChT. However, nearly atomic resolution structures have been obtained for three bacterial members of the MFS, namely, lactose permease (LacY; PDB id 1PV6; 3.5 Å resolution) (3), glycerol-3-phosphate transporter (GlpT; PDB id 1PW4; 3.3 Å resolution) (4), and the bacterial multidrug resistance transporter EmrD (PDB id 2GFP; 3.5 Å resolution) (5). Despite low sequence identity for these proteins, the three-dimensional structures of the polypeptide backbones are highly similar. This observation led to the hypothesis that all members of the MFS containing 12 TMs share the same fold (6−12).
Accordingly, a homology model of VAChT has been proposed based on the GlpT structure (7). In the model, TMs 1−6 cluster together, as do TMs 7−12 (Figure 1). Because the template is distantly related to VAChT, the model is approximate. A transport channel for the substrate lies between the two clusters approximately perpendicular to the plane of the membrane. The binding site for ACh lies somewhere in the channel, and it is hypothesized to access each side of the membrane in alternating fashion when the two clusters of TMs rock back and forth against each other. This type of conformational change has been termed the “rocker-switch”.
Figure 1.
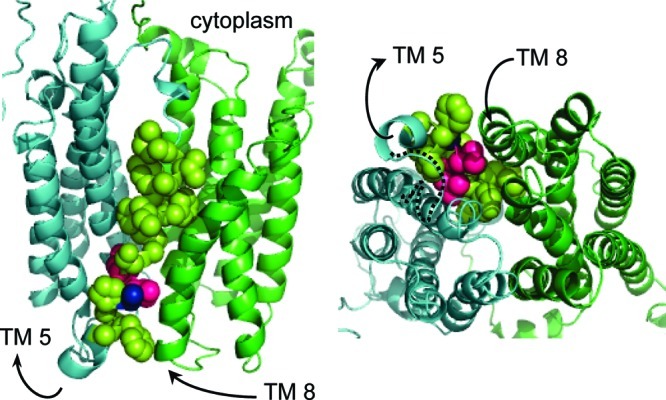
VAChT homology model. The left frame is a side view from inside the membrane, and the right frame is a view from inside the synaptic vesicle looking through the bottom of the transport channel toward cytoplasm. The first cluster of six TMs is cyan, and the second cluster is green. Not the entire homology model is visible. The arrows indicate the N- to C-terminal directions of TMs 5 and 8. Motif C is shown mostly in lemon-yellow as van der Waals spheres that cover the bottom two-thirds of TM 5. L226 and V227 are hot pink, and A228 is blue, even though they are part of motif C. The dotted black line in the right frame traces loop 5/6 from the end of TM 5 to the beginning of TM 6.
The MFS includes a family of bacterial multidrug resistance transporters containing six conserved sequence “motifs” 7−13 residues long (13−16). All of the motifs are present in VAChT (17). Four of them occur in TMs, and three of those are rich in proline and glycine residues. Proline and glycine promote formation of “defects,” such as hinges that mediate kinking and wobbling, and formation of short segments of 310- and π-helix in otherwise α-helical TMs (18,19). The defects often mediate special functions (20−24).
Motif C in TM 5 is especially interesting, because its consensus sequence is gxxxGPxxGGxl (capital ≥70% conservation, lower case ≥40% conservation, and x any residue). The consensus sequence for motif C in VAChT is G224SLVAPPFGGIL235 (boldface = invariant), where the numbering applies to human VAChT (hVAChT). This very highly conserved sequence contains invariant A228, which usually is G in the bacterial multidrug resistance transporter family.
In a previous study from this laboratory, A228 in motif C of VAChT was mutated to glycine. The change leads to >3-fold increase in the turnover rate for ACh transport (17). Equilibrium affinities for binding of ACh and an allosteric inhibitor called vesamicol are not affected, indicating that the resting conformation of VAChT is not affected by the mutation. The results suggest that the mutation lowers the energy barrier for the rocker-switch conformational change. Notably, motif C is located at an interface with the second bundle of TMs, and it probably participates in rocker-switch reorientation of the ACh binding site (Figure 1).
Molecular dynamics (MD) simulation of intact VAChT would be of interest to assess the conformational properties of TM 5 and motif C in situ, but as noted above, only an approximate homology model of VAChT is available. MD simulation of an approximate model would be of limited utility. However, analysis of a peptide having the TM 5 and motif C sequence should be useful. MD simulation of polyalanine mimicking a TM systematically and sparsely substituted with proline and glycine residues predicts that the flexibility of motif C is increased by the A228G mutation (17,25) Statistical correlations of sequence pattern descriptors with atomic-resolution structures of integral membrane proteins also do (26). In the study reported here, MD techniques were used to characterize the intrinsic conformational propensities for peptides corresponding to TM 5 and motif C in wild-type and mutant hVAChTs.
Results and Discussion
Simulated Peptides
The sequences of the peptides studied here are shown in Scheme 1. Residue numbers refer to the number in intact hVAChT, even though only 26-residue peptides have been characterized. The approach taken to investigate the conformational propensities of the peptides is similar to that reported by Brooks and colleagues for other TM-mimicking peptides (27,28). Although implicit membrane does not mimic the aqueous transport channel, most backbone atoms of TM 5 and motif C in the VAChT homology model do not contact channel water. CASTp software, which rolls a water sphere of 1.4 Å radius across the surface of a protein, was used to identify all VAChT atoms that contact channel water (29). Among backbone atoms in TM 5 and motif C, only some in A213, G215, and V216 do so (Table S1, Supporting Information). No backbone atoms in the region responsible for interesting structural behavior (L226 and V227, see below) do. Thus, implicit membrane should provide an adequate environment to test conformational propensities of peptides mimicking TM 5 and motif C in VAChT.
Scheme 1.

Dihedral phi (Φ) and psi (Ψ) angles flanking the α-carbon atoms for loop residues R210−R212 and G233−L235 were constrained to those in the VAChT homology model, but the side chains were free to move. The constraints mimic loop formation with flanking TMs 4 and 6 in intact VAChT and require the peptide to exhibit end effects approximating those in intact VAChT. Each peptide was immersed in implicit membrane, approximately parallel to the membrane perpendicular, and the structure was energy-minimized. Backbone structures changed little from that of the corresponding sequence in the VAChT homology model, suggesting that static structure in the flexible region of the backbone is determined primarily by intrinsic conformational propensity.
During the initial period of MD simulation, each peptide tilted relative to the membrane perpendicular, thereby increasing hydrophobic interactions between side chains and the membrane. The loop residues at each end of the peptides did not enter the membrane. Preliminary simulations demonstrated that a 27-Å thick membrane produces a 30° tilt away from the perpendicular and a 25-Å thick membrane produces a 35° tilt, but no significant differences in the internal dynamics of the peptides occur. The results reported here are for the 25-Å thick membrane, which produces tilt similar to that present for TM 5 in the intact model. To ensure that equilibrium was achieved, simulation during the first 6 ns was discarded.
Helical Hydrogen Bonding
Among TM helices, the α-helix with i + 4 to i hydrogen bonds (donor to acceptor) dominates because of favorable packing of atoms and nearly linear hydrogen bonds. Slightly less favorable energetically is the narrower 310-helix with i + 3 to i hydrogen bonds, and less favorable still is the wider π-helix with i + 5 to i hydrogen bonds. Hydrogen bonding in protein helices requires a distance ≤2.6 Å between the proton and oxygen. Thus, distance from the i + 4, i + 3, or i + 5 amide proton to the i carbonyl oxygen can be used to determine whether two residues interact in α-, 310-, or π-configuration.
At one moment, a particular amide proton might have α-interaction and at another moment 310-interaction. To quantitate the fraction of time that a donor residue spends in each type of interaction, a plot of the distance of the amide proton in the i + 4 or i + 3 residue to the carbonyl oxygen in the i residue versus cumulative percentage of saved structures is useful. The cumulative percentage that is ≤2.6 Å for each interaction estimates the fraction of time spent in that interaction. Similar plots demonstrate that π-interaction never occurs for the peptides investigated here.
Wild-Type Peptide
The following comments are organized in the order A213 to G232. Hydrogen bonding is analyzed from the amide proton donor to upstream carbonyl oxygen acceptor. A213−G215 exhibits distorted helical structure. Distortion probably is caused by the absence of hydrogen bonding to residues of loop 4/5. In contrast, V216−S225 forms good α-helix, with >90% of the sampled structures exhibiting α-interactions both upstream and downstream for all residues except S225, which has only the upstream α-interaction (not shown).
Beginning with L226, which is the third residue in motif C, α-helical structure begins to break down. Figure 2 displays cumulative distance analyses for the i + 3 to i (310-helix, top row) and i + 4 to i (α-helix, bottom row) interactions for L226, V227, and A228 in wild-type sequence. Each residue exhibits interaction with its potential upstream α-partner in only 63%, 62%, and 52% of the structures, respectively, for wild-type peptide (Table 1). It also exhibits interaction with its potential upstream 310-partner in 48%, 7%, and 27% of the structures, respectively. Some structures exhibit a bifurcated hydrogen bond, which explains why the sum for α- and 310-helical interactions is >100% for L226. Overall, L226 overwhelmingly samples α- and 310-distances, but >31% of V227 and >21% of A228 structures exhibit neither α- nor 310-interaction.
Figure 2.

Distance of amide protons for L226, V227, and A228 to the carbonyl oxygen of the indicated upstream residue versus cumulative percent of structures for wild-type peptide. The maximal distance of 2.6 Å for hydrogen bonding is indicated by the dark horizontal lines. The top row of graphs is for 310-interaction, and the bottom row is for α-interaction.
Table 1. The α-Helical Hydrogen Bonds to Upstream Carbonyl Oxygensa.
| residue | wild-type (5)b | fast transporter (4)b | double mutant (4)b |
|---|---|---|---|
| V216 through S225 | >90 | >90 | >90 |
| L226c | 63 | 72 | 78 |
| V227 or V227Lc | 62 | 94 | 98 |
| A228 or A228G | 52 | 68 | 75 |
| P229 | 0 | 0 | 0 |
| P230 | 0 | 0 | 0 |
| F231 | 80 | 90 | 90 |
| G232 | >90 | >90 | >90 |
Percentage of structures for which the distance of the amide proton in the i + 4 residue to the carbonyl oxygen in the i residue is ≤2.6 Å. Boldface residues are invariant in nature.
Number of α-hydrogen bonds not fully formed (that is, those <80%) relative to an idealized α-helix.
Hinge residue.
P229 and P230 have no amide protons. They cannot engage in hydrogen-bond donation to any potential acceptor. However, on the downstream side of the prolines, F231 engages in i + 4 to i hydrogen-bond donation to V227 upstream of the prolines 80% of the time (Figure 3 and Table 1), and G232 engages in i + 4 to i hydrogen-bond donation to A228 upstream of the prolines >90% of the time (Table 1). Thus, α-interactions reach around P229 and P230 in the wild-type peptide.
Figure 3.

Distance of amide proton for F231 to carbonyl oxygen of residue 227 versus cumulative percent of structures in the indicated peptide. The maximal distance of 2.6 Å for hydrogen bonding is indicated by the dark horizontal lines.
The presence of α-interaction between F231 and V227 is unexpected. In the case of a single proline in otherwise α-helix, steric hindrance between the side chain of the proline (here P230) and the carbonyl oxygen of the residue 3-positions upstream (here V227) disrupts donation of an α-helical proton by the residue following the proline (here F231) (30). Such hindrance does not occur here, presumably because of tandem prolines. Accepting ≥80% as a substantially established hydrogen bond, S225−F231 in wild-type peptide has completely or significantly lost five α-helical hydrogen bonds (Table 1). The deficit is expected a priori to result in kinked wild-type peptide.
Mutated Peptides
The A228G mutation is expected to introduce more flexibility into TM 5 and motif C of VAChT, because glycine residues can access a greater range of Φ and Ψ angles due to the absence of a β-carbon atom. But is the increased flexibility static or dynamic? The percent donations of α-hydrogen bonds to upstream residues of mutated peptides are listed in Table 1. In the fast-transporter peptide, L226, V227, and A228G become stronger donors than in wild-type peptide, as the fractions of α-distances to upstream acceptors increase to 72%, 94%, and 68%, respectively (Figure 4). The biggest increase occurs at V227, going from 62% to 94% α-helical. F231 increases its donation of a hydrogen bond to V227 to 90% of structures (Figure 3), and G232 still engages in donation of a hydrogen bond to A228 in >90% of structures. Overall, S225−F231 in the fast-transporter peptide has completely or significantly lost only four α-helical hydrogen bonds instead of the five for wild-type peptide. Nevertheless, fast-transporter peptide still is expected to be kinked.
Figure 4.

Distance of indicated amide protons to the carbonyl oxygen of the indicated residue versus cumulative percent of structures for mutant peptides. The maximal distance of 2.6 Å for hydrogen bonding is indicated by the dark horizontal lines. The top row of graphs is for α-helical interaction of L226, V227, and A228G in the fast-transporter peptide, and the bottom row is for α-helical interaction of L226, V227L, and A228G in the double mutant peptide.
β-Branched V227 is conserved except in Drosophila VAChT, in which the corresponding residue is F. Also, L226 and V227 are the major hinge residues in a kink caused by P229 and P230 (see below). To probe the importance of β-branching in this region, V227 was mutated in silico to V227L to create a double mutant that removes the β-branch and restores the same number of carbon atoms as in the wild-type peptide (Scheme 1; Figure 4). The percent donation of α-helical hydrogen bonds to upstream acceptors is listed in Table 1. L226, V227L, and A228G become even stronger donors than in the fast-transporter peptide, as the fractions of α-interaction with upstream acceptors increase further to 78%, 98% and 75%, respectively. Thus, β-branching makes V227 and the residues around it weaker donors for α-interactions. The A228G−V227L double mutation does not, of course, alter the inability of P229 and P230 to donate hydrogen bonds to upstream acceptors. The α-interactions between F231 to V227L (Figure 3) and G232 to A228G are unchanged. Overall, S225−F231 in the double mutant peptide has completely or significantly lost four α-helical hydrogen bonds. Double mutant peptide still is expected to be kinked.
Kink and Wobble
TMs containing a kink exhibit two types of motions between helical segments flanking the hinge residue that creates the kink. The first is a fluctuating kink angle (κ), and the second is a fluctuating wobble angle (ω). The angles are defined in Figure 5. The κ angle is 0° for a straight configuration. It can exceed 90° only for helical segments that bend back on themselves. The ω angle is 0° for a TM that kinks directly over Cα of the hinge residue, thereby putting Cα in the “arm pit.” It is positive for clockwise rotation of the axis for the N-terminal segment around the axis for the C-terminal segment when viewed from the C-terminal segment. These motions do not change winding around the helical axis, which is described by “phase change” between α-, 310-, and π-helices.
Figure 5.

Definitions of kink (κ) and wobble (ω) angles. H1 and H2 are the helical axes in vector notation before and after a hinge residue. Boldface is a vector. The identities of specific residues used here to calculate H1 and H2 are given in Methods. The axes do not have to intersect in order to calculate κ as the arccos(H1·H2). The ω is defined as follows. A plane normal to vector H2 is created by the vectors S and C, where C is the normalized distance vector between the Cα atom of the hinge residue and vector H2. The dark spot in the figure is the Cα atom of the hinge residue (either L226, V227, or V227L). S is defined as S = H2 × C. The vector H1 then is projected into the (S,C)-plane to give vector H1′. The angle between H1′ and C is defined as ω.
To carry out the analysis outlined in Figure 5 for the peptides, a hinge residue must be chosen. To do this, the Φ and Ψ angles and their rms deviations for all of the residues in all three peptides were examined (wild-type is listed in Table S2, Supporting Information). The largest differences in means from typical α-helical values [Φ ≈ −63° ± 6° and Ψ ≈ −43° ± 6° (mean ± rms deviation from the mean)] occur at residue 227 (either V227 or V227L) in all three peptides. For wild-type peptide, Φ = −86.2° ± 12.0° and Ψ = −15.8° ± 14.1°. The second largest deviations occur at L226 in all three peptides, and for wild-type Φ = −71.2° ± 11.8° and Ψ = −37.7° ± 10.4°. Smaller deviations occur at G224 and G232, but they are present also in control peptides (see next). The latter deviations are of less interest because they do not couple to P229 and P230.
An MD simulation was carried out for control peptides in which alanine or leucine residues replace P229 and P230 of the wild-type sequence. The Φ and Ψ angles and their rms deviations for all of the residues were examined (Table S3, Supporting Information). The values for L226 and V227 in the control peptides are typical for α-helix. Thus, the differences from α-helical structure in wild-type peptide, and presumably in fast-transporter and double mutant peptides as well, are due primarily to P229 and P230. Accordingly, residues 226 and 227 were chosen as hinges.
Figure 6 illustrates kink and wobble angles that L226 and residue 227 in wild-type, fast-transporter, and double mutant peptides access. Clusters of angle combinations that define unique structural populations occur. Approximate ranges for the clusters are listed in Table 2. Only notable populations will be discussed.
Figure 6.

Kink (κ) versus wobble (ω) angles. See Figure 5 for the definitions of kink and wobble. MD trajectories from 6 to 40 ns were analyzed and plotted as described in Methods (17 000 dots). The top row used L226 as the hinge residue. The bottom row used residue 227 (V227 or V227L) as the hinge residue.
Table 2. Kink and Wobble Centered at L226, V227, or V227La.
| peptide | residue | population (%) | kink (deg) | wobble (deg) |
|---|---|---|---|---|
| wild-type | L226 | 100 | 5 to 60 | 125 to 235 |
| V227 | 10 to 70 | 40 to 130 | ||
| fast transporter | L226 | 98 | 10 to 60 | 130 to 210 |
| V227 | 20 to 70 | 40 to 110 | ||
| L226 | 2 | 5 to 30 | −160 to 0 | |
| V227 | 0 to 25 | −180 to 180 | ||
| L226 | 0.1 | 50 to 70 | ∼−100 | |
| V227 | 30 to 60 | −170 to −125 | ||
| double mutant | L226 | 98 | 10 to 60 | 130 to 210 |
| V227L | 20 to 70 | 35 to 100 | ||
| L226 | 2 | 5 to 30 | −160 to 100 | |
| V227L | 0 to 25 | −180 to 180 |
A single population of wild-type peptide is present when analyzed at either L226 or V227. At L226, kink angles range from ∼5° to 60°, and wobble angles range from ∼125° to 235°. At V227, kink angles range over values similar to those at L226, but wobble angles range from ∼40° to 130°. This indicates that the average wobble angle at V227 points 95° away from that at L226. The plot of kink versus wobble angles for the control peptide that substituted alanine residues for P229 and P230 is completely different when analyzed at V227. Kink angles range from ∼0° to 30°, and wobble angles span the entire range from −180° to 180° with a concentration of structures at ∼110°. Thus, complex kink and wobble are produced by hinge residues L226 and V227 in wild-type peptide.
For mutant peptides analyzed at L226 or residue 227, large populations exhibit kink and wobble ranges similar to those of wild-type, but the ranges are somewhat narrower. The narrowing probably is the result of moderately increased α-helical character. Overall, there are only small structural differences in the major populations for all three peptides.
Mutant peptides also access minor, ∼2%, populations having structures very different from that of wild-type peptide when analyzed at either L226 or residue 227. At L226 in either mutant, the kink angles range from ∼5° to 30°, and wobble angles range from approximately −160° to 0°. This means more nearly straight peptide (small kink) allows the N-terminal segment to wobble away from the average structure of the major population, but only in the less negative direction. At residue 227 in either mutant, the kink angles are similar to those at L226, but wobble angles range from approximately −180° to 180°. This wobble range extends much beyond that at L226 and demonstrates that V227 is more flexible than is L226. More nearly straight peptide (small kink) allows the N-terminal segment to undergo very large wobbles in positive and negative directions away from the average structure.
Fast-transporter peptide also accesses a ∼0.1% population in which (a) L226 exhibits kink angles that range from ∼50° to 70° and wobble angles are approximately −100° and (b) V227 exhibits kink angles that range from ∼30° to 60° and wobble angles that range from −170° to −125°. Double mutant peptide does not access a corresponding 0.1% population, demonstrating that the A228G single mutant peptide is more flexible than is the A228G−V227L double mutant peptide.
Overall, deviation of these peptides from α-helical structure occurs primarily at hinge residues L226 and V227 or V227L. Kinking and wobbling are similar in mutant and wild-type peptides for 98−100% of the structures in each case. Thus, static structures determined by energy minimization using molecular mechanics are very similar to each other for all three peptides. However, MD reveals that the mutations produce small dynamic populations having substantially altered structures. The answer to the earlier question about whether A228G induces static or dynamic flexibility is that it induces both. A228G increases α-helical character in the helical segments flanking a complex hinge region, thus making the flanking regions stiffer. It also increases kink and wobble dynamics in small populations.
Figure 7 shows superposition of ribbon cartoons for 10 representative structures of wild-type peptide. Except for the three N-terminal residues corresponding to loop 4/5, alignment of the rest of the N-terminal residues is excellent until residue G224 is reached, after which the hinge region causes fraying of the C-terminal end of the bundle. Nevertheless, all of the wild-type structures are similar to each other.
Figure 7.
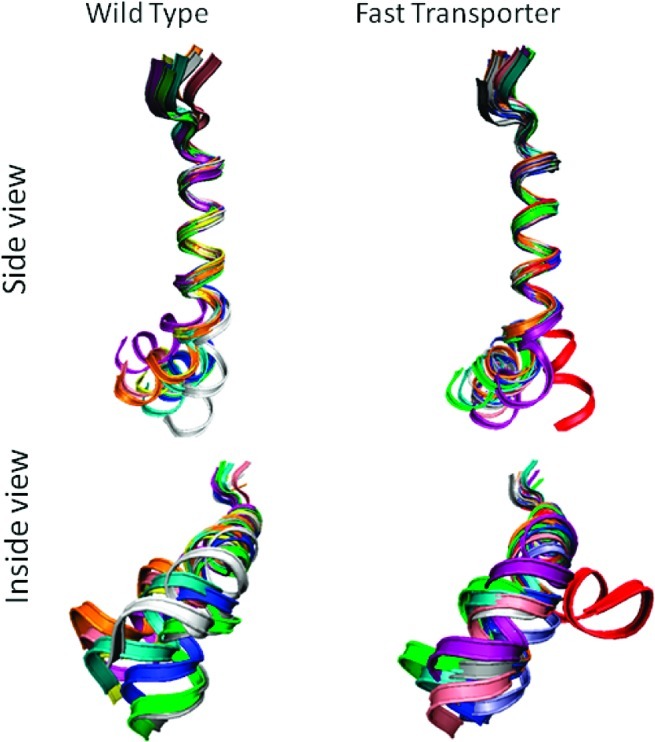
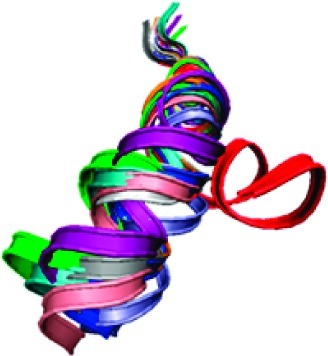
Ribbon drawings of 10 representative structures for wild-type and fast transporter peptides. The region of the peptides corresponding to the end of loop 4/5 and the start of the TM 5 in VAChT are at the top. The root-mean-square distances between corresponding α-carbon atoms of residues V216−G224 were minimized to obtain the superposition shown. Left is wild-type peptide. Right is fast transporter peptide. Drawings on top are viewed from the side of the peptide inside the membrane, and drawings on bottom are viewed from what would be the inside of a synaptic vesicle looking toward cytoplasm.
Superposition of ribbon cartoons for 10 representative structures of fast transporter peptide is revealing. Nine of the structures superimpose similarly to the wild-type, except that fraying of the C-terminal end is reduced slightly. In the tenth structure (red), the helical axes before and after the hinge region are more nearly colinear than are the axes in the other nine structures. Wobble-like rotational displacement of the C-terminus around the N-terminal axis also is much greater than those for the other structures.
Relationship of Peptide Dynamics to Intact VAChT
We return to the VAChT homology model (Figure 1). The mouth of the transport channel inside the synaptic vesicle is closed in the conformation modeled. The seal is formed by three pairs of interactions, namely, the end of TM 1 meeting the end of TM 7, the beginning of TM 2 meeting the end of TM 11, and the end of TM 5 meeting the beginning of TM 8. TMs 5 and 8 are bent but approximately parallel to each other with the cytoplasmic ends splayed. The bend in TM 5 occurs in motif C at approximately L226 and V227. TMs 2 and 11 have a similar relationship to each other. TMs 5 and 8 form one pair of rocking surfaces, TMs 2 and 11 form the other pair, and together they mediate the hypothesized rocker-switch.
The dynamics revealed in the current study are the intrinsic propensities of peptides mimicking TM 5 and motif C in VAChT. Such propensities will be dampened when a polypeptide is packed against adjacent protein. Nevertheless, non-α-helical structures found in TMs archived by the Protein Data Bank correlate with particular sequence descriptors (26). The correlation means that conformational propensities arising from amino acid sequences of peptides often persist in proteins. Partial persistence has been confirmed by comparative MD simulations of intact glycerol-3-phosphate transporter and its constituent TMs (31). To at least a significant extent, the dynamics propensities of the peptides mimicking TM 5 and motif C could persist in VAChT.
We hypothesize that (a) the complex hinge region at L226 and V227 persists in VAChT, (b) the increased kink dynamics and helical stiffness produced by the A228G mutation persist in VAChT, and (c) the rocker-switch in the A228G mutant of VAChT occurs faster. The latter effect could result in faster transport. It might happen as follows. In the VAChT homology model, loop 5/6 is long, unstructured, and slack (Figure 1, right). It probably allows the C-terminal segment of TM 5 after the hinge more freedom of motion than the N-terminal segment before the hinge. The protein context merely exchanges and inverts H1 and H2 in Figure 5, but it does not affect the ability of TM 5 and motif C to kink and wobble. Large-amplitude kinking and wobbling of the C-terminal segment would push it against the beginning of TM 8, which lies next to motif C. Increased wobbling and stiffness in TM 5 and motif C caused by the A228G mutation could enhance the effectiveness of pushing. Such a push would help initiate the rocker-switch by promoting a split of the closed interface between TM 5 and TM 8. It would be of interest to make the double mutant of VAChT mimicking the double mutant of the peptide studied here. The prediction is that smaller stimulation of the transport rate will be obtained because there is less propensity to wobble, particularly at high kink angles.
Methods
MD simulations of peptides corresponding to TM 5 and motif C in hVAChT and its mutants A228G and V227L/A228G were carried out using a membrane implicit generalized Born model with a simple switching function (the GBSW module) (27,28). The CHARMM biomolecular simulation program (32,33) and the PARAM22/CMAP (34) force field were used. Physical parameters representing the membrane were 0.03 kcal/(mol·Å2) for the surface tension coefficient, 25 or 27 Å for the thickness, 1.0 for the dielectric constant, and 0.4 Å for a smoothing length. Temperature was set to 298 K, and water dielectric constant was 80. The planar membrane was perpendicular to the z axis and centered at z = 0.
The structures of loop 4/5, TM 5, and motif C (Scheme 1) were taken from a three-dimensional model of VAChT done by Vardy et al. (7). A228 in the simulated wild-type peptide was mutated to A228G to mimic the mutant of VAChT, which shows >3-fold faster transport when expressed in a neurosecretory cell line (17). In the double mutant (which has not been created in VAChT), V227 was mutated in silico to V227L in addition to mutation A228G. The N-terminus was acetylated, and the C-terminus was blocked with N-methyl amine to mimic contiguous polypeptide in intact VAChT. The Φ and Ψ angles flanking the α-carbon atoms for loop residues R210−R212 and G233−L235 were constrained to those in the VAChT homology model, but the side chains were free to move. The side chains and Φ and Ψ values of residues A213−G232 were free to move. The resulting structures were immersed in the implicit membrane approximately parallel to the z-axis. This procedure placed the loop residues outside of the membrane. A chloride counterion was placed 5 Å from each arginine residue outside the membrane to establish a charge-neutral system. The structures then were energy-minimized by molecular mechanics to relieve close contacts in preparation for the MD runs. The integration step size was 0.001 ps. Total time simulated was 40 ns for each system, except for control peptides in Supporting Information. Coordinates were saved every 200 steps, with 200 000 structures collected. Every tenth structure was extracted.
Structural visualization and manipulation were performed using the VMD program (35). Calculations of hydrogen-bond distances and backbone torsion angles were performed using the CHARMM program. Helical axes for hinge residue L226 were calculated for F220−S225 (H1) and residue 227−G232 (H2), helical axes for hinge residue 227 (V227 or V227L) were calculated for I221−L226 (H1) and residue 228 (A228 or A228G)−G233 (H2), and the kink (κ) and wobble (ω) angles were calculated using the Simulaid program (36). The histogram and parameter correlation diagrams were plotted using the program KaleidaGraph.
Acknowledgments
We thank Eyal Vardy and Shimon Schuldiner for providing coordinates for the VAChT homology model. We thank Professor Mihaly Mezei for generously allowing access to the SIMULAID program.
Abbreviations
In addition to those of the IUPAC-IUB Commission on Biochemical Nomenclature (J. Biol. Chem. 1985, 1, 14−42), this paper uses the following symbols and abbreviations: ACh, acetylcholine; GlpT, glycerol-3-phosphate transporter; hVAChT, human VAChT; MD, molecular dynamics; MFS, major facilitator superfamily; TM, transmembrane helix; rms, root-mean-square; VAChT, vesicular acetylcholine transporter.
Supporting Information Available
Tables providing backbone atoms in TM 5 touching water in the channel and dihedral angles ± rms deviation in wild-type and control peptides and figure showing kink (κ) versus wobble (ω) angles at residue 227 in control peptide. This material is available free of charge via the Internet at http://pubs.acs.org.
J.L. carried out simulations, and J.L. and S.M.P. wrote the manuscript.
This research was supported by Grant NS15047 from the National Institute of Neurological Disorders and Stroke.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Saier M. H. , Jr.; Beatty J. T.; Goffeau A.; Harley K. T.; Heijne W. H. M.; Huang S.-C.; Jack D. L.; Jahn P. S.; Lew K.; Liu J.; Pao S. S.; Paulsen I. T.; Tseng T.-T.; Virk P. S. (1999) The major facilitator superfamily. J. Mol. Microbiol. Biotechnol. 1, 257–279. [PubMed] [Google Scholar]
- Parsons S. M. (2000) Transport mechanism in acetylcholine and monoamine storage. FASEB J. 14, 2423–2434. [DOI] [PubMed] [Google Scholar]
- Abramson J.; Smirnova I.; Kasho V.; Verner G.; Kaback H. R.; Iwata S. (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Lemieux M. J.; Song J.; Auer M.; Wang D. N. (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301, 616–620. [DOI] [PubMed] [Google Scholar]
- Yin Y.; He X.; Szewczyk P.; Nguyen T.; Chang G. (2006) Structure of the multidrug transporter EmrD from Escherichia coli. Science 312, 741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai T.; Subbramaniam S. (2004) Structure and transport mechanism of the bacterial oxalate transporter OxlT. Biophys. J. 87, 3600–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardy E.; Arkin I. T.; Gottsschalk K. E.; Kaback H. R.; Schuldiner S. (2004) Structural conservation in the major facilitator superfamily as revealed by comparative modeling. Protein Sci. 13, 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramson J.; Kaback H. R.; Iwata S. (2004) Structural comparison of lactose permease and the glycerol-3-phosphate antiporter: Members of the major facilitator superfamily. Curr. Opin. Struct. Biol. 14, 413–419. [DOI] [PubMed] [Google Scholar]
- Abramson J.; Iwata S.; Kaback H. R. (2004) Lactose permease as a paradigm for membrane transport proteins. Mol. Membr. Biol. 21, 227–236. [DOI] [PubMed] [Google Scholar]
- Defelice L. J. (2004) Transporter structure and mechanism. Trends Neurosci. 27, 352–359. [DOI] [PubMed] [Google Scholar]
- Lemieux M. J.; Huang Y.; Wang D.-N. (2004) The structural basis of substrate translocation by the Escherichia coli glycerol-3-phosphate transporter: A member of the major facilitator superfamily. Curr. Opin. Struct. Biol. 14, 405–412. [DOI] [PubMed] [Google Scholar]
- Hirai T.; Heymann J. A. W.; Maloney P. C.; Subramanian (2003) Structural model for 12-helix transporters belonging to the major facilitator superfamily. J. Bacteriol. 185, 1712–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouch D. A.; Cram D. S.; DiBerrardino D.; Littlejohn T. G.; Skurray R. A. (1990) Efflux-mediated antiseptic resistance gene qacA from Staphylococcus aureus: Common ancestry with tetracycline and sugar-transport proteins. Mol. Microbiol. 4, 2051–2062. [DOI] [PubMed] [Google Scholar]
- Paulsen I. T.; Brown M. H.; Skurray R. A. (1996) Proton-dependent multidrug efflux systems. Microbiol. Rev. 60, 575–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putman M.; van Veen H. W.; Konings W. N. (2000) Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 64, 672–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardy E.; Steiner-Mordoch S.; Schuldiner S. (2005) Characterization of bacterial drug antiporter homologous to mammalian neurotransmitter transporters. J. Bacteriol. 187, 7518–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran A.; Ojeda A. M.; Kolmakova N. G.; Parsons S. M. (2006) Mutational and bioinformatics analysis of proline- and glycine-rich motifs in vesicular acetylcholine transporter. J. Neurochem. 98, 1551–1559. [DOI] [PubMed] [Google Scholar]
- Cordes F. S.; Bright J. N.; Sansom M. S. P. (2002) Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323, 951–960. [DOI] [PubMed] [Google Scholar]
- Senes A.; Engel D. E.; DeGrado W. F. (2004) Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 14, 465–479. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Takeshima K.; Park C. B.; Kim S. C.; Matsuzaki K. (2000) Interactions of the novel anti-microbial peptide buforin 2 with lipid bilayers: Proline as a translocation promoting factor. Biochemistry 39, 8648–8654. [DOI] [PubMed] [Google Scholar]
- Chia B. C. S.; Carver J. A.; Mulhern J. A.; Bowie J. H. (2000) Maculatin 1.1, an anti-microbial peptide from the Australian tree frog, Litoria genimaculata - solution structure and biological activity. Eur. J. Biochem. 267, 1894–1908. [DOI] [PubMed] [Google Scholar]
- Lu H.; Marti T.; Booth P. J. (2001) Proline residues in transmembrane alpha helices affect the folding of bacteriorhodopsin. J. Mol. Biol. 308, 437–446. [DOI] [PubMed] [Google Scholar]
- Sansom M. S. P.; Weinstein H. (2000) Hinges, swivels & switches: The role of prolines in signaling via transmembrane α-helices. Trends Pharmacol. Sci. 21, 445–451. [DOI] [PubMed] [Google Scholar]
- Shelden M. C.; Loughlin P.; Tierney M. L.; Howitt S. M. (2001) Proline residues in two tightly coupled helices of the sulphate transporter, SHST1, are important for sulphate transport. Biochem. J. 356, 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright J. N.; Sansom M. S. P. (2003) The flexing/twirling helix: Exploring the flexibility about molecular hinges formed by proline and glycine motifs in transmembrane helices. J. Phys. Chem. B 107, 627–636. [Google Scholar]
- Rigoutsos I.; Riek P.; Graham R. M.; Novotny J. (2003) Structural details (kinks and non-α conformations) in transmembrane helices are intrahelically determined and can be predicted by sequence pattern descriptors. Nucleic Acids Res. 31, 4625–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im W.; Feig M.; Brooks C. L., III (2003) An implicit membrane generalized Born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 85, 2900–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu L.; Im W.; Brooks C. L., III (2007) Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys. J. 92, 854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundas J.; Ouyang Z.; Tseng J.; Binkowski A.; Turpaz Y.; Liang J. (2006) CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]; CASTp URL: http://sts.bioengr.uic.edu/castp/.
- Visiers I.; Braunheim B. B.; Weinstein H. (2000) Prokink: A protocol for numerical evaluation of helix distortions by proline. Protein Eng. 13, 603–606.11054453 [Google Scholar]
- D'rozario R. S. G.; Sansom M. S. P. (2008) Helix dynamics in a membrane transport protein: comparative simulations of the glycerol-3-phosphate transporter and its constituent helices. Mol. Membr. Biol. 25, 571–583. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. (1983) CHARMM: A program for macromolecular energy minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217. [Google Scholar]
- MacKerell A. D., Jr.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E. III; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Feig M.; Mackerell A. D.; Brooks C. L., III (2003) Force field influence on the observation of α-helical protein structures in molecular dynamics simulations. J. Phys. Chem. B 107, 2831–2836. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Mezei M. (2009) Simulaid: Simulation Setup Utilities. URL: http://atlas.physbio.mssm.edu/∼mezei/simulaid/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.