

Abstract
As a model for molecular mimicry in neurological disease, we study people infected with human T-lymphotropic virus type 1 (HTLV-1) who develop HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP), an immune-mediated disease of the central nervous system (CNS). In HAM/TSP, data suggests molecular mimicry is the result of cross-reactive antibodies between HTLV-1-tax and heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1), a protein over-expressed in human CNS neurons. The hnRNP A1 epitope recognized by autoantibodies was unknown. In this study, we hypothesized that antibodies purified from HAM/TSP patients would react with functionally significant domains of hnRNP A1. Western blotting of functionally significant deletion mutants and overlapping fusion proteins using HAM/TSP IgG revealed two core epitopes within the C-terminal region of hnRNP A1. The first (aminoacids 191-SSQRGRSGSGNF-202), overlapped the RGG domain and the second (aminoacids 293-GQYFAKPRNQGG-304), with the M9 shuttling sequence, two functionally important regions of hnRNP A1. Monoclonal antibodies to HTLV-1-tax also reacted with the epitopes. These data fulfill an important criterion of molecular mimicry, namely that mimicking epitopes are not random, but include biologically significant regions of target proteins. This suggests an important role for the cross-reactive immune response between HTLV-1 and hnRNP A1 in the pathogenesis of immune-mediated neurological diseases via molecular mimicry.
Keywords: Human T-lymphotropic virus type 1, Heterogeneous nuclear ribonucleoprotein A1, Neurologic, HTLV-1 associated myelopathy/tropical spastic paraparesis, Multiple sclerosis, Autoimmune
A fundamental question concerning autoimmunity is the relationship between infection and the development of disease. One hypothesis that couples infection with autoimmune disease is molecular mimicry. Molecular mimicry is characterized by an immune response to an environmental agent that cross-reacts with a host antigen, which results in autoimmune disease [17]. As a model for molecular mimicry, we study patients infected with HTLV-1 who develop HAM/TSP, a neurological disease with important biologic similarities to multiple sclerosis (MS) [10,13]. Neurological disease in HAM/TSP patients is associated with cellular and antibody immune responses to HTLV-1-tax and HLA-DRB1*0101 [5,6,8]. Recently, we showed that HAM/TSP patients make antibodies to hnRNP A1, an autoantigen purified from CNS neurons [11]. Monoclonal antibodies to HTLV-1-tax (tax Mabs) cross-reacted with hnRNP A1 indicating molecular mimicry between the two proteins. Infusion of the tax Mabs and antibodies to hnRNP A1 completely inhibited neuronal firing indicative of their pathogenic nature [7,11]. These data establish a clear link between viral infection, autoimmunity and neurological disease and indicate that molecular mimicry plays a role in the pathogenesis of HAM/TSP [11]. Importantly, MS patients have also been found to develop antibodies to hnRNP A1, as well as to hnRNP A2/B1, all of which are derived from the same ancestral gene [19].
One requirement that establishes molecular mimicry is that immune responses to the mimicking proteins are not random, but must react with functionally significant regions of the proteins [17]. In HTLV-1 infected individuals, HTLV-1-tax is thought to play a critical role in the pathogenesis of HAM/TSP by regulating viral replication [10]. In addition, its C-terminus contains the immunodominant epitopes for antibody responses in humans [8]. This same region is coincident with the epitope recognized by the tax Mabs (tax346–353) [12]. hnRNP A1 is an RNA binding protein that binds mRNA and transports it in and out of the nucleus [18]. The N-terminal domain of hnRNP A1 contains two RNA binding domains (RBDs) [18]. The C-terminal is glycine-rich and interacts with other RNA binding proteins [4,16,18]. The antibody epitope for hnRNP A1 in HAM/TSP patients until now was unknown. Thus, we hypothesized that the antibody response to hnRNP A1 would recognize a functionally important region of the protein.
IgG from 5 HTLV-1 seronegative (‘normal IgG’) and 10 HAM/TSP patients were purified by absorption on protein A agarose (Bio-Rad, USA). Following isolation, biotinylation was performed using Biotin (Long Arm) NHS (Vector laboratories). E. coli BL21 and pGEX-6P-1 (Amersham Biosciences, Sweden) were used to express recombinant protein. Fragments of hnRNP A1 (GenBank accession NM 002136) were amplified from human brain mRNA by RT-PCR using the primer pairs in Table 1. The amplified fragments were subcloned into Bam HI and Hind III sites of pGEX-6P-1 to create recombinant expression vectors and transformed into E. coli BL21. Constructed plasmids were identified by restriction enzyme mapping and DNA sequencing. The recombinant expression vectors were transformed into E. coli BL 21 for expressing the fusion target protein. Overexpression and purification of transformed E. coli BL21 (GST-hnRNP A1 or GST-hnRNP A1 fragments) cells were performed according to the manufacturer’s instructions. For affinity chromatography, an AKTA FPLC (Amersham Biosciences) apparatus was used at 4 °C. The samples were loaded on a PBS-equilibrated 1 ml GSTrap FF column (Amersham Biosciences). GST-fusion proteins were eluted with elution buffer (15 mM reduced glutathione in 50 mM Tris–HCl, pH 8.0). Western blotting was performed as described previously [11]. Western blot analysis was performed using 1:25,000-diluted biotinylated HAM/TSP IgG (100 ng/ml), biotinylated HTLV-1 seronegative IgG (100 ng/ml), 1:5000 diluted rabbit anti-hnRNP A1 polyclonal antibody (an antibody to hnRNP A1 AA 293–304 developed with software designed to predict the immunogenicity of proteins – 200 ng/ml, Abcam, Cam-bridge, MA) followed by HRP-conjugated anti-rabbit IgG (1:25,000) or 1:5000 diluted biotinylated-mouse anti-tax Mab cocktail (200 ng/ml) [12] to detect hnRNP A1 and hnRNP A1 fragments. Absorption experiments were done by pre-incubating the HAM/TSP IgG with peptides representing the core epitopes overnight at 4 °C. Antibody staining was visualized with horseradish peroxidase-conjugated streptavidin (1:50,000) and enhanced chemiluminescence system (ECLplus, Amersham Biosciences). Protein concentration was determined by BCA method (Pierce) using bovine serum albumin as standard.
Table 1.
Nucleic acid and aminoacid sequences of recombinant proteins of hnRNP A1 epitopes
| Fragment no. | Fragment span | Sense primer cDNA/AA sequence | Anti-sense primer cDNA/AA sequence |
|---|---|---|---|
| 2A | AA 1–197 | (1)(cDNA 105–128) GGATCCATGTCTAAGTCAGAGTCTCCTAAA (AA 1–8) MSKSESPK |
(2)(cDNA 675–695) AAGCTTACTTCGACCTCTTTGGCTGGA (AA 191–197) SSQRGRS |
| 2B | AA 1–256 | (1)(cDNA 105–128) GGATCCATGTCTAAGTCAGAGTCTCCTAAA (AA 1–8) MSKSESPK |
(3)(cDNA 849–872) AAGCTTACCTCCAAAATTGCTTCCATCATT (AA 249–256) NDGSNFGG |
| 2C | AA 1–320 | (1)(cDNA 105–128) GGATCCATGTCTAAGTCAGAGTCTCCTAAA (AA 1–8) MSKSESPK |
(4)(cDNA 1044–1067) AAGCTTTTAAAATCTTCTGCCACTGCCATA (AA 314–320) YGSGRRF |
| 2D | AA 191–320 | (5)(cDNA 675–695) GGATCCTCCAGCCAAAGAGGTCGAAGT (AA 191–197) SRGRQSS |
(4)(cDNA 1044–1067) AAGCTTTTAAAATCTTCTGCCACTGCCATA (AA 314–320) YGSGRRF |
| 3–1 | AA 191–232 | (P1) (cDNA 675–692) GCTTGGATCCTCCAGCCAAAGAGGTCGA (AA 191–196) SSQRGR |
(P2) (cDNA 783–800) GTCAAAGCTTACGGCTGCCACCAAAGCC (AA 227–232) GFGGSR |
| 3–2 | AA 209–250 | (P3) (cDNA 729–746) GCTTGGATCCGGTTTCGGTGGGAATGAC (AA 209–214) GFGGND |
(P4) (cDNA 837–854) GTCAAAGCTTATCATTCTCAAATCCATT (AA 245–250) NGFGND |
| 3–3 | AA 227–268 | (P5) (cDNA 783–800) GCTTGGATCCGGCTTTGGTGGCAGCCGT (AA 227–232) GFGGSR |
(P6) (cDNA 891–908) GTCAAAGCTTATTGTTGTAATTCCCAAA (AA 263–268) FGNYNN |
| 3–4 | AA 245–286 | (P7) (cDNA 837–854) GCTTGGATCCAATGGATTTGAGAATGAT (AA 245–250) NGFGND |
(P8) (cDNA 945–962) GTCAAAGCTTAGAGCTTCTGCCTCCAAA (AA 281–286) FGGRSS |
| 3–5 | AA 263–304 | (P9) (cDNA 891–908) GCTTGGATCCTTTGGGAATTACAACAAT (AA 263–268) FGNYNN |
(P10) (cDNA 999–1016) GTCAAAGCTTGCCACCTTGGTTTCGTGG (AA 299–304) PRNQGG |
| 3–6 | AA 281–320 | (P11) (cDNA 945–962) GCTTGGATCCTTTGGAGGCAGAAGCTCT (AA 281–286) FGGRSS |
(P12) (cDNA 1050–1067) GTCAAAGCTTTTAAAATCTTCTGCCACT (AA 316–320) SGRRF |
| 3–7 | AA 303–320 | (P13) (cDNA 1011–1028) GCTTGGATCCGGTGGCTATGGCGGTTCC (AA 303–308) GGYGGS |
(P12) (cDNA 1050–1067) GTCAAAGCTTTTAAAATCTTCTGCCACT (AA 316–320) SGRRF |
| 4a(−) | AA 185–196 | (DS0) (cDNA 657–674) GCTTGGATCCGAGATGGCTAGTGCTTCA (AA 185–190) EMASAS |
(DAS0) (cDNA 675–692) GTCAAAGCTTTCGACCTCTTTGGCTGGA (AA 191–196) SSQRGR |
| 4a | AA 191–202 | (P1) (cDNA 675–692) GCTTGGATCCTCCAGCCAAAGAGGTCGA (AA 191–196) SSQRGR |
(DAS1) (cDNA 693–710) GTCAAAGCTTAAAGTTTCCAGAACCACT (AA 197–202) SGSGNF |
| 4b | AA 197–208 | (DS1) (cDNA 693–710) GCTTGGATCCAGTGGTTCTGGAAACTTT (AA 197–202) SGSGNF |
(DAS2) (cDNA 711–728) GTCAAAGCTTACCTCCACGACCACCACC (AA 203–208) GGGRGG |
| 4c | AA 203–214 | (DS2) (cDNA 711–728) GCTTGGATCCGGTGGTGGTCGTGGAGGT (AA 203–208) GGGRGG |
(DAS5) (cDNA 729–746) GTCAAAGCTTGTCATTCCCACCGAAACC (AA 209–214) GFGGND |
| 4d | AA 281–292 | (P11) (cDNA 945–962) GCTTGGATCCTTTGGAGGCAGAAGCTCT (AA 281–286) FGGRSS |
(DAS3) (cDNA 963–980) GTCAAAGCTTTCCACCGCCATAGGGGCC (AA 287–292) GPYGGG |
| 4e | AA 287–298 | (DS3) (cDNA 963–980) GCTTGGATCCGGCCCCTATGGCGGTGGA (AA 287–292) GPYGGG |
(DAS4) (cDNA 981–998) GTCAAAGCTTTTTTGCAAAGTATTGGCC (AA 293–298) GQYFAK |
| 4f | AA 293–304 | (DS4) (cDNA 981–998) GCTTGGATCCGGCCAATACTTTGCAAAA (AA 293–298) GQYFAK |
(P10) (cDNA 999–1016) GTCAAAGCTTGCCACCTTGGTTTCGTGG (AA 299–304) PRNQGG |
| 4g | AA 299–310 | (DS6) (cDNA 999–1016) GCTTGGATCCCCACGAAACCAAGGTGGC (AA 299–304) PRNQGG |
(DAS6) (cDNA 1017–1034) GTCAAAGCTTGCTGCTGGAACCGCCATA (AA 305–310) YGGSSS |
| 4h | AA 305–320 | (DS7) (cDNA 1017–1034) GCTTGGATCCTATGGCGGTTCCAGCAGC (AA 305–310) YGGSSS |
(P12) (cDNA 1050–1067) GTCAAAGCTTTTAAAATCTTCTGCCACT (AA 316–320) SGRRF |
cDNA begins at the first base following the restriction enzyme site and the cDNA number corresponds to the full-length cDNA (GenBank accession NM_002136) (restriction enzyme sites are underlined).
To identify epitopes recognized by antibodies derived from HAM/TSP patients, we designed primers representing biologically important regions of hnRNP A1 (Fig. 1A and Table 1) and these deletion mutants were cloned, expressed, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to membranes and used for Western blotting with HAM/TSP IgG. Full-length hnRNP A1 (Fig. 1A, fragment 2C), mutants containing the two RBDs (Fig. 1A, fragment 2A), as well as overlapping mutants containing the two RBDs plus the RGG domain (Fig. 1A, fragment 2B) and the RGG domain plus M9 (Fig. 1A, fragment 2D) were tested. All 10 HAM/TSP patients and the five ‘normal IgG’ controls were tested individually and representative Western blots are presented in Fig. 1C–E. As shown in Fig. 1C, ‘normal IgG’ showed no immunoreactivity. HAM/TSP IgG reacted with full-length hnRNP A1 (Fig. 1C, fragment 2C). There was no reactivity to the fragment containing the two RBDs alone (Fig. 1C, fragment 2A) and mild reactivity to the fragment containing the two RBDs plus the RGG domain (Fig. 1C, fragment 2B). There was intense immunoreactivity with the fragment containing the RGG domain plus M9 (Fig. 1C, fragment 2D). Taken together, these experiments suggest that immunoreactive epitopes recognized by HAM/TSP IgG do not coincide with the RBDs and may include AAs that overlap fragments B and D (AA 191–256) as well as those exclusive to fragment 2D (AA 257–320). The signal intensity of fragment 2D suggests that core epitopes recognized by HAM/TSP IgG are contained within this fragment. Importantly, the tax Mab showed the identical pattern (Fig. 1C).
Fig. 1.
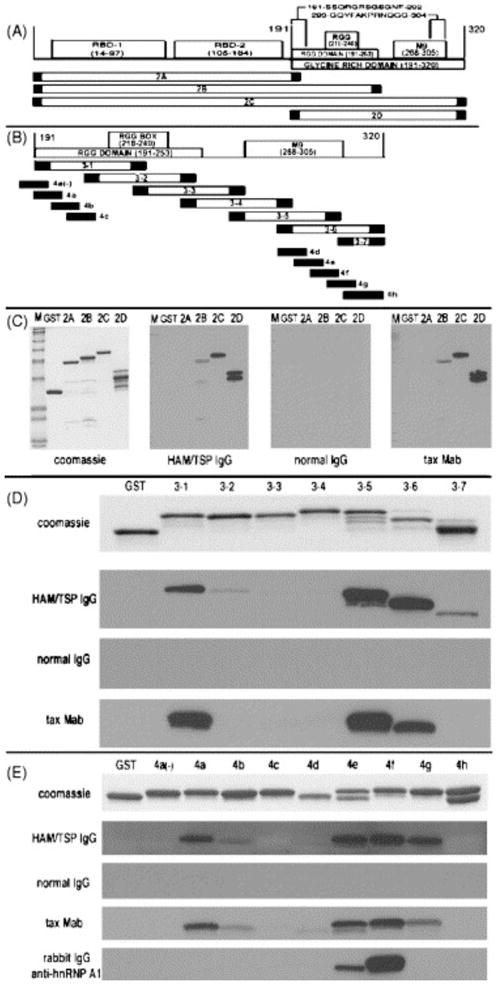
IgG immunoreactivity with hnRNP A1 protein fragments. (A) Schematic of hnRNP A1. The N-terminal region of hnRNP A1 contains two RBDs, RBD-1 (AA 14–97) and RBD-2 (AA 105–184). The C-terminal domain (AA 191–320) contains the RGG domain (AA 191–253), the RGG box (AA 218–240) and the M9 shuttling domain (AA 268–305). Four recombinant proteins representing hnRNP A1 epitopes were tested: fragments 2A–D. HAM/TSP IgG reacted with two core epitopes: AA 191–202 and AA 293–304 (see text). (B) Fragments 3–1 to 3–7 represent overlapping fusion proteins of fragment 2D. Fragments 4a(−) to 4h are coincident with areas of intense immunoreactivity defined by fragments 3–1 to 3–7. (C) HAM/TSP IgG showed intense immunoreactivity with fragments 2C and 2D and mild immunoreactivity with fragment 2B. The tax Mab parallels this pattern. There was no immunoreactivity using ‘normal IgG’. (D) HAM/TSP IgG showed intense immunoreactivity with fragments 3–1, 3–5 and 3–6 and mild immunoreactivity with fragments 3–2 and 3–7. The tax Mab closely paralleled this pattern and there was no immunoreactivity with ‘normal IgG’. (E) HAM/TSP IgG showed intense immunoreactivity with fragments 4a and 4e–g with minor immunoreactivity with fragment 4b. The tax Mab closely parallels this pattern. A polyclonal rabbit IgG to AA 293–304 reacted with only fragments 4e and 4f. ‘M’ is molecular weight (4–250 kDa).
Since these data suggested that the epitopes for HAM/TSP IgG were contained within fragment 2D, we designed overlapping primers representing this region (Fig. 1B, fragments 3–1 to 3–7, and Table 1) and each fragment prepared for Western blotting as described. As shown in Fig. 1D, there was no immunoreactivity using ‘normal IgG’. There was intense immunoreactivity to fragment 3–1, minimal immunoreactivity to fragment 3–2 and no immunoreactivity to fragment 3–3. Since fragment 3–1 was positive and fragment 3–3 was negative, this data suggests an epitope between the N-terminal of fragment 3–1 and the AA preceding the N-terminal of fragment 3–3 (corresponding to AA 191–226). The minimal immunoreactivity of fragment 3–2 suggests the contribution of this fragment (AA 209–226, that part that overlaps fragment 3–1 but does not include fragment 3–3) to the epitope is small compared to that of fragment 3–1. This epitope is contained within the RGG domain (AA 191–253). There was no immunoreactivity with fragment 3–4 and intense immunoreactivity with fragments 3–5 and 3–6. Fragments 3–4 (AA 245–286), 3–5 (AA 263–304) and 3–6 (AA 281–320) overlap with the M9 shuttling sequence (AA 268–305). Since fragment 3–4 (AA 245–286) was negative the epitope is not contained within its sequence as well as where it overlaps fragment 3–5 (AA 263–286). This suggests the epitope lies between AA 287 and the C-terminal of fragment 3–6 (AA 320). Fragment 3–7 (AA 303–320) was minimally positive compared to fragments 3–5 and 3–6. This suggests the core epitope is contained predominantly between AA 287–302 (AAs contained within fragments 3–5 and 3–6 that do not include AAs from fragments 3–4 and 3–7) and that the small region of overlap between fragment 3–7 in comparison to fragment 3–5 (AA 303–304) may contribute to the epitope, thus potentially extending the epitope to AA 287–304. The tax Mabs paralleled this immunoreactivity (Fig. 1D).
To further refine the epitopes recognized by HAM/TSP IgG, we designed overlapping primers representing the regions of fragment D where immunoreactivity was most intense (Fig. 1B, fragments 4a(−) to 4h, and Table 1). The N-terminal epitope of fragment D was tested using primers representing AA 191–202 (fragment 4a), AA 197–208 (fragment 4b) and AA 203–214 (fragment 4c). Since fragment 4a is positive and fragment 4c negative, and there is no overlap between these fragments, the epitope must lie within fragment 4a. Fragment 4b was minimally positive. This suggests that within fragment 4b, the region that overlaps fragment 4a (AA 197–202), but not fragment 4c (AA 203–208), contributes to the epitope. This suggests the epitope includes AA 191–202 (191-SSQRGRSGSGNF-202) with AA 191–196 making a greater contribution to the immunoreactivity with HAM/TSP IgG. To confirm that the core epitope is contained within the RGG domain, we added fragment 4a (−) (AA 185–196) to the analyses. Immunoreactivity to this fragment was negative, thus confirming that this core epitope is contained completely within the N-terminal region of the RGG domain. Similarly, we tested the M9 region. In this experiment we tested overlapping primers representing AA 281–292 (fragment 4d), AA 287–298 (fragment 4e), AA 293–304 (fragment 4f), AA 299–310 (fragment 4g) and AA 305–320 (fragment 4h). As shown in Fig. 1E, fragment 4d was negative and fragments 4e and 4f were strongly positive. Fragment 4g was also positive, although less intensely than fragments 4e and 4f. By excluding fragment 4d, it can be deduced that the epitope is contained predominantly between fragments 4e and 4f (AA 293–304) with a contribution from fragment 4g, which is believed to result from its overlap with fragment 4f (AA 299–304). Immunoreactivity with fragment 4h (AA 305–320) was negative. Thus, the second core epitope is AA 293–304 (293-GQYFAKPRNQGG-304), which is contained within the M9 shuttling domain. Importantly the epitope of the tax Mabs overlapped with that HAM/TSP IgG (Fig. 1E). Interestingly, a rabbit polyclonal antibody to hnRNP A1 AA 293–304 was found to overlap the M9 core epitope, but not the RGG domain epitope (Fig. 1E).
Next, we determined whether the core epitopes defined by the overlapping fusion proteins contributed to the immunoreactivity of HAM/TSP IgG for hnRNP A1. HAM/TSP IgG was tested for immunoreactivity using full-length hnRNP A1 and fragment 2D following pre-incubation with increasing concentrations of the core epitopes. As shown in Fig. 2A and B, increasing concentrations of either of the peptides representing the core epitopes abolished HAM/TSP IgG immunoreactivity for both full-length hnRNP A1 (fragment 2C) and fragment 2D. The control fusion protein (fragment 3–3) did not alter immunoreactivity of the HAM/TSP IgG for either protein. This confirms that these peptides are core epitopes and indicates that both epitopes are highly specific for the RGG domain and M9 shuttling domain of hnRNP A1. Importantly, all 10 HAM/TSP patients (Fig. 2E and G, lanes 1–10) immunoreacted with both epitopes in contrast to the 5 ‘normal IgG’ controls (2E and G, lanes 11–15).
Fig. 2.

Adsorption experiments using the core epitopes and screening of patient sera. HAM/TSP IgG was pre-incubated with either the recombinant peptide representing the RGG epitope (A) or the M9 shuttling epitope (B) prior to Western blot with fragments 2C and 2D. Increasing concentrations of each peptide resulted in dose dependent adsorption of immunoreactivity. Pre-incubation of HAM/TSP IgG with a control fusion protein (fragment 3–3) showed no change in immunoreactivity (C). Lanes 1: 0 μg; 2: 0.1 μg; 3: 1 μg; 4: 10 μg; 5: 100 μg protein. Patient sera was screened using the two core epitopes. (D and F) Coomassie stain of molecular weight markers (lane 1) and of fusion protein tested (lane 2). (E and G) HAM/TSP patients (lanes 1–10) showed intense immunoreactivity with each of the core epitopes (E, the RGG domain epitope, and G, the M9 shuttling epitope). There was no immunoreactivity shown by the ‘normal IgG’ controls (lanes 11–15).
Data in this study demonstrates that antibodies isolated from HAM/TSP patients reacted with functionally important regions of hnRNP A1 and further implicate hnRNP A1 as playing a key role in the pathogenesis of immune-mediated neurological disease via molecular mimicry. Specifically, we showed that IgG isolated from HAM/TSP patients reacted with two epitopes of hnRNP A1. Both epitopes were localized to the glycine-rich domain (AA 191–320) of hnRNP A1. This domain contains two important functional regions, the RGG domain (AA 191–253) and the M9 shuttling domain (AA 268–305) [4,16,18].
The first core epitope defined in this study (191-SSQRGRSGSGNF-202) localized to the RGG domain and overlaps its first motif (AA 194–196). The RGG domain interacts with RNA binding proteins and may assist in RNA binding [2,16]. A similar series of RGG sequences are also present in hnRNP A2 [16]. Deletion mutations in the RGG domain of hnRNP A2 results in its accumulation in the cell cytoplasm, suggesting this region plays a significant role in the import of hnRNP A2 into the nucleus [16].
The M9 sequence is the shuttling domain for hnRNP A proteins, including hnRNP A1 and A2, and thus, is required for the import and export of hnRNP A proteins between the nucleus and cytoplasm [4,16,18]. Recent reports suggest that the import and export functions of M9 are inseparable [14]. Deletion mutations of AA 268–272 or 301–305 interrupted both import and export of hnRNP A1 [14]. Our data indicate that the second epitope recognized by HAM/TSP IgG was localized to M9 at AA 293-GQYFAKPRNQGG-304, which overlaps one of the regions required for hnRNP A1 transport into and out of the nucleus.
Thus, HAM/TSP IgG reacts with epitopes of hnRNP A1, which when mutated result in disruption of protein function. Although the two epitopes may be related functionally, there is little primary sequence identity between them. Evaluation of the two epitopes using antigenicity and hydrophobicity models (https://peptideselect.invitrogen.com/peptide/) indicate that these regions are highly antigenic. Data presented here show that pre-incubation with one core epitope completely inhibited immunoreactivity between HAM/TSP IgG and hnRNP A1. Thus, pre-incubation with one epitope inhibited the immunoreactivity of both core epitopes. This suggests that these epitopes are not only related functionally, but also closely related antigenically. Importantly, both epitopes were also recognized by the tax Mabs, suggesting these functionally and antigenically related regions are targets for molecular mimicry between HTLV-1-tax and hnRNP A1. Database analyses of primary sequences showed little similarity between tax Mab epitope and the core epitopes of hnRNP A1 defined in this study. This suggests that molecular mimicry between these two proteins is based on immunological cross-reactivity rather than on the primary sequences of cross-reactive epitopes. Since epitope binding is based on three-dimensional structure, the data presented here (as well as that from other studies) suggest that molecular mimicry, defined by cellular or antibody mediated cross-reactivity, have increased biological significance compared to those defined by primary sequence analysis [17,20,21].
These data support the hypothesis that molecular mimicry contributes to the pathogenesis of HAM/TSP. HAM/TSP is an inflammatory disease of the CNS that results in progressive weakness associated with demyelination and axonal damage [9,10]. In HAM/TSP, there is no experimental evidence that direct infection of neural elements with HTLV-1 results in CNS damage [10]. Instead, data supports the hypothesis that immune-mediated mechanisms play a significant role. HAM/TSP patients have higher antibody and proinflammatory cytokine levels in sera and spinal fluid than do control patients [10]. It is the immune response to HTLV-1-tax and env, two immunodominant proteins, which differentiate patients with HAM/TSP from control populations [3,5,8]. Specifically, HAM/TSP patients develop a CD8+ cytotoxic T-lymphocyte (CTL) response specific for the HTLV-1-tax11–19 in association with HLA-A2 [5]. Some studies suggest that tax specific CTLs enter the CNS and cause parenchymal damage [5,10]. Other experiments indicate that tax specific CTLs in association with HLA-A2 protect against the development of disease [6]. In contrast to HLA-2, HLA-DRB1*0101 was found to increase the risk of developing HAM/TSP [6]. Antibodies also play a significant role in the pathogenesis of the disease [10]. HAM/TSP patients have elevated antibody titers to HTLV-1 in sera and spinal fluid [10]. People infected with HTLV-1 react with HTLV-env, which is used to diagnose infection [3,8]. The IgG response in HAM/TSP patients is to tax316–353 [8]. Data also indicate that viral load may stimulate immune responses in HAM/TSP since these patients have elevated viral loads of tax [15].
The present results along with our previous studies indicate that hnRNP A1 is a CNS autoantigens and immunoreactivity to it plays a role in the pathogenesis of HAM/TSP via molecular mimicry [7,11,12]. Specifically, we previously showed that HAM/TSP IgG reacts with CNS neurons [11]. Second, we determined that the CNS autoantigen is hnRNP A1 [11]. Third, we showed that Mabs to tax cross react with CNS neurons and hnRNP A1 [11,12]. Fourth, we showed that the immune response to both hnRNP A1 and tax involved biologically important epitopes. Finally, infusion of HAM/TSP IgG and the tax Mabs resulted in inhibition of neuronal firing [7,11]. Taken together these data indicate that molecular mimicry exists between HTLV-1-tax and hnRNP A1 and this cross-reactive immune response plays a role in the pathogenesis of HAM/TSP [9]. The contribution to this immune response by HTLV-1 infected, seropositive, asymptomatic individuals is not yet know, but will be addressed in future studies.
Autoimmunity to hnRNPs have been shown to play a significant role in the pathogenesis of other immune-mediated diseases such as MS, lupus, rheumatoid arthritis, and paraneoplastic diseases of the CNS [1,2,11]. Interestingly, recent data indicate that MS patients develop antibodies to hnRNP A1 as well as to hnRNP A2/B1 [19]. Thus, like HAM/TSP, immunoreactivity to hnRNP A1 may also play a role in the pathogenesis of MS as well as in other immune-mediated diseases of the CNS.
Acknowledgments
This material is based upon work supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs. This study was funded by a VA Merit Review Grant, NIH R01-NS-38876 (M.C.L.) and the NIH Medical Student Research Program Fellowship (T35 DK07405) (F.D.D.). Thanks to Dr. Mike Cremer for his review of the manuscript.
References
- 1.Buckanovich R, Posner J, Darnell R. Nova, the paraneoplastic Ri antigen, is homologous to an RNA binding protein and is specifically expressed in the developing motor system. Neuron. 1993;11:657–672. doi: 10.1016/0896-6273(93)90077-5. [DOI] [PubMed] [Google Scholar]
- 2.Cartegni L, Maconi M, Morandi E, Cobianchi F, Riva S, Biamonti G. hnRNP A1 selectively interacts through its Gly-rich domain with different RNA-binding proteins. J Mol Biol. 1996;259:337–348. doi: 10.1006/jmbi.1996.0324. [DOI] [PubMed] [Google Scholar]
- 3.Goon PK, Hanon E, Igakura T, Tanaka Y, Weber JN, Taylor GP, Bangham CR. High frequencies of Th1-type CD4(+) T cells specific to HTLV-1 Env and Tax proteins in patients with HTLV-1-associated myelopathy/tropical spastic paraparesis. Blood. 2002;99:3335–3341. doi: 10.1182/blood.v99.9.3335. [DOI] [PubMed] [Google Scholar]
- 4.Izaurralde E, Jarmolowski A, Beisel C, Mattaj IW, Dreyfuss G, Fischer U. A role for the M9 transport signal of hnRNP A1 in mRNA nuclear export. J Cell Biol. 1997;137:27–35. doi: 10.1083/jcb.137.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson S, Shida H, McFarlin DE, Fauci AS, Koenig S. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV-I pX in patients with HTLV-I associated neurological disease. Nature. 1990;348:245–248. doi: 10.1038/348245a0. [DOI] [PubMed] [Google Scholar]
- 6.Jeffery KJ, Usuku K, Hall SE, Matsumoto W, Taylor GP, Procter J, Bunce M, Ogg GS, Welsh KI, Weber JN, Lloyd AL, Nowak MA, Nagai M, Kodama D, Izumo S, Osame M, Bangham CR. HLA alleles determine human T-lymphotropic virus-I (HTLV-I) proviral load and the risk of HTLV-I-associated myelopathy. Proc Natl Acad Sci USA. 1999;96:3848–3853. doi: 10.1073/pnas.96.7.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalume F, Lee SM, Morcos Y, Callaway JC, Levin MC. Molecular mimicry: cross-reactive antibodies from patients with immune-mediated neurologic disease inhibit neuronal firing. J Neurosci Res. 2004;77:82–89. doi: 10.1002/jnr.20137. [DOI] [PubMed] [Google Scholar]
- 8.Lal RB, Giam CZ, Coligan JE, Rudolph DL. Differential immune responsiveness to the immunodominant epitopes of regulatory proteins (tax and rex) in human T cell lymphotropic virus type 1-associated myelopathy. J Infect Dis. 1994;169:496–503. doi: 10.1093/infdis/169.3.496. [DOI] [PubMed] [Google Scholar]
- 9.Lee SM, Morocos Y, Jang H, Stuart JM, Levin MC. HTLV-1 induced molecular mimicry in neurologic disease. In: Oldstone M, editor. Molecular Mimicry: Infection Inducing Autoimmune Disease. Springer; New York: 2005. [Google Scholar]
- 10.Levin MC, Jacobson S. HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP): a chronic progressive neurologic disease associated with immunologically mediated damage to the central nervous system. J Neurovirol. 1997;3:126–140. doi: 10.3109/13550289709015802. [DOI] [PubMed] [Google Scholar]
- 11.Levin MC, Lee SM, Kalume F, Morcos Y, Dohan FC, Jr, Hasty KA, Callaway JC, Zunt J, Desiderio D, Stuart JM. Autoimmunity due to molecular mimicry as a cause of neurological disease. Nat Med. 2002;8:509–513. doi: 10.1038/nm0502-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin MC, Lee SM, Morcos Y, Brady J, Stuart J. Cross-reactivity between immunodominant human T lymphotropic virus type I tax and neurons: implications for molecular mimicry. J Infect Dis. 2002;186:1514–1517. doi: 10.1086/344734. [DOI] [PubMed] [Google Scholar]
- 13.Levin M, Lehky T, Flerlage N, Katz D, Kingma D, Jaffe E, Heiss J, Patronas N, McFarland H, Jacobson S. Immunopathogenesis of HTLV-1 associated neurologic disease based on a spinal cord biopsy from a patient with HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP) New Eng J Med. 1997;336:839–845. doi: 10.1056/NEJM199703203361205. [DOI] [PubMed] [Google Scholar]
- 14.Michael WM, Choi M, Dreyfuss G. A nuclear export signal in hnRNP A1: a signal-mediated, temperature-dependent nuclear protein export pathway. Cell. 1995;83:415–422. doi: 10.1016/0092-8674(95)90119-1. [DOI] [PubMed] [Google Scholar]
- 15.Nagai M, Usuku K, Matsumoto W, Kodama D, Takenouchi N, Moritoyo T, Hashiguchi S, Ichinose M, Bangham CR, Izumo S, Osame M. Analysis of HTLV-1 proviral load in 202 HAM/TSP patients and 243 asymptomatic HTLV-1 carriers: high proviral load strongly predisposes to HAM/TSP. J Neurovirol. 1998;4:586–593. doi: 10.3109/13550289809114225. [DOI] [PubMed] [Google Scholar]
- 16.Nichols RC, Wang XW, Tang J, Hamilton BJ, High FA, Herschman HR, Rigby WF. The RGG domain in hnRNP A2 affects subcellular localization. Exp Cell Res. 2000;256:522–532. doi: 10.1006/excr.2000.4827. [DOI] [PubMed] [Google Scholar]
- 17.Oldstone MB. Molecular mimicry and immune-mediated diseases. FASEB J. 1998;12:1255–1265. doi: 10.1096/fasebj.12.13.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siomi H, Dreyfuss G. A nuclear localization domain in the hnRNP A1 protein. J Cell Biol. 1995;129:551–560. doi: 10.1083/jcb.129.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sueoka E, Yukitake M, Iwanaga K, Sueoka N, Aihara T, Kuroda Y. Autoantibodies against heterogeneous nuclear ribonucleoprotein B1 in CSF of MS patients. Ann Neurol. 2004;56:778–786. doi: 10.1002/ana.20276. [DOI] [PubMed] [Google Scholar]
- 20.Westall FC. Molecular mimicry or structural mimicry? Mol Immunol. 2006;43:1062–1064. doi: 10.1016/j.molimm.2005.06.039. [DOI] [PubMed] [Google Scholar]
- 21.Wucherpfennig K. Structural basis of molecular mimicry. J Autoimmun. 2001;16:293–302. doi: 10.1006/jaut.2000.0499. [DOI] [PubMed] [Google Scholar]