

Abstract
Activating transcription factor 5 (ATF5) is highly expressed in malignant glioma and plays an important role in promoting cell survival. Here we perform a genome-wide RNA interference (RNAi) screen to identify transcriptional regulators of ATF5. Our results reveal an essential survival pathway in malignant glioma, whereby activation of a RAS/MAPK or PI3K signaling cascade leads to induction of the transcription factor CREB3L2, which directly activates ATF5 expression. ATF5, in turn, promotes survival by stimulating transcription of MCL1, an anti-apoptotic BCL2 family member. Analysis of human malignant glioma samples indicates that ATF5 expression inversely correlates with disease prognosis. The RAF inhibitor sorafenib suppresses ATF5 expression in glioma stem cells and inhibits malignant glioma growth in cell culture and mouse xenografts. Our results demonstrate that ATF5 plays an essential role in malignant glioma genesis, and reveal that the ATF5-mediated survival pathway described here provides potential therapeutic targets for treatment of malignant glioma.
A hallmark of cancer is uncontrolled cellular proliferation, which can arise as a result of over-expression of genes promoting cell survival or down-regulation of genes inducing cell death1. Genes that are highly expressed in cancer cells and are essential for their survival are appealing targets for the development of novel cancer therapeutics2.
ATF5 (also known as ATFx) is a member of the ATF/camp responsive element-binding (CREB) family of transcription factors, which comprises a large group of basic leucine zipper proteins whose members mediate diverse transcriptional regulatory functions3. ATF5 is an anti-apoptotic factor that plays an important role in promoting survival in a variety of cell types4. Increased levels of ATF5 have been observed in primary brain tumors, and ATF5 expression is particularly high in glioblastoma, an aggressive form of malignant glioma5. Previous studies have shown that interference with ATF5 function results in glioma cell death in primary tumors but does not affect normal cells surrounding the tumor6. The high level of ATF5 in brain tumors, coupled with the absence of ATF5 expression in mature neurons, suggests that ATF5 may be an attractive therapeutic target for the treatment of malignant glioma.
The upstream regulators of ATF5 expression and the downstream effectors that mediate the anti-apoptotic effects of ATF5 remain to be determined. Here we describe a novel genome-wide RNAi negative-selection screening strategy that has enabled us to elucidate a pathway by which ATF5 is expressed and prevents apoptosis. Our results reveal an essential survival pathway in malignant glioma, the inhibition of which has therapeutic implications.
RESULTS
A signaling pathway required for ATF5 expression
To identify factors that regulate ATF5 transcription, we performed a genome-wide small hairpin RNA (shRNA) screen to isolate genes that, when knocked down, greatly reduce ATF5 expression. Because loss of ATF5 function would induce apoptosis5,7 and prevent the subsequent identification of candidate shRNAs, we developed a negative-selection strategy (Fig. 1a) based on the ability of diphtheria toxin (DT) to kill cells expressing the DT receptor (DTR). Mouse cells lack a functional DTR and are resistant to DT8. We generated a mouse malignant glioma GL261 cell line stably expressing the human DTR driven by the mouse Atf5 promoter (pATF5-DTR). To prevent cell death due to loss of endogenous ATF5, we also engineered the cell line to stably express ATF5 driven by the constitutive cytomegalovirus (CMV) promoter (pCMV-ATF5). The resulting cell line was termed GL261/pATF5-DTR/pCMV-ATF5; as expected, these cells were efficiently killed by addition of DT (Supplementary Fig. 1a).
Figure 1.
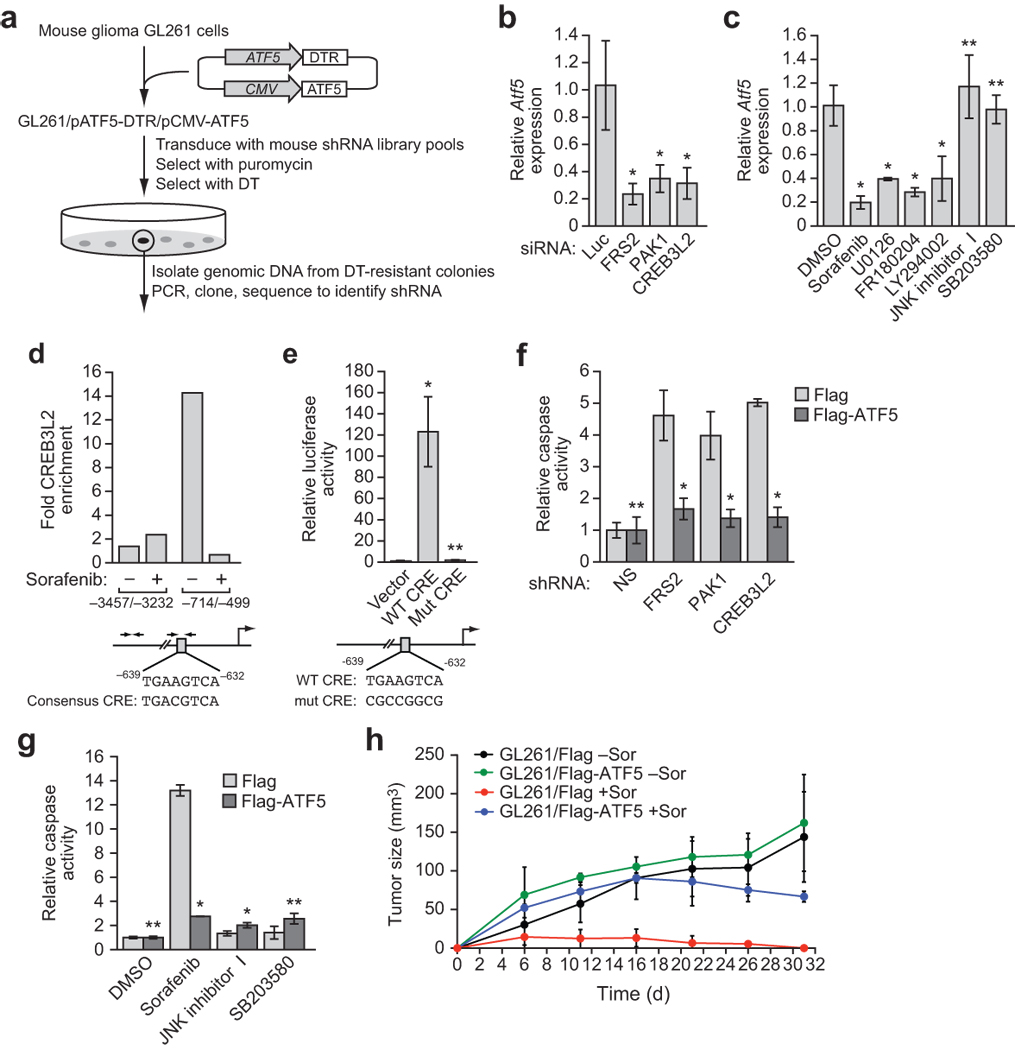
A genome-wide RNAi screen reveals a signaling pathway required for ATF5 expression. (a) Schematic summary of the genome-wide RNAi screen. (b) qRT-PCR analysis of endogenous Atf5 expression in GL261 cells transiently transfected with a luciferase (Luc), FRS2, PAK1 or CREB3L2 siRNA. (c) qRT-PCR analysis of endogenous Atf5 expression in GL261 cells treated with sorafenib (10 µM), U0126, FR180204, LY294002, JNK inhibitor I, SB203580 or, as a control, DMSO. (d) ChIP analysis monitoring binding of CREB3L2 to two regions of the Atf5 promoter in GL261 cells treated with or without sorafenib (10 µM). A consensus CRE motif is present at −632 to −639 bp upstream of the transcription start-site (bottom). Enrichment of CREB3L2 is expressed relative to a no antibody control. (e) Luciferase reporter assay in GL261 cells transiently transfected with an Atf5 promoter reporter construct carrying a wild-type (WT) or mutant (Mut) CRE. (f) Caspase 3/7 activity assay in GL261 cells stably expressing Flag-ATF5 or, as a control, Flag and transiently transfected with a non-silencing (NS), FRS2, PAK1 or CREB3L2 shRNA. (g) Caspase 3/7 activity assay in GL261 cells expressing Flag or Flag-ATF5 and treated with either DMSO, sorafenib (20 µM), JNK inhibitor I or SB203580. (h) Mouse tumorigenensis assays. Sorafenib treatment decreased tumor volume significantly in the absence of ATF5 (P = 0.0045) but not in the presence of ATF5 (P = 0.62). All error bars shown, ±s.d.; *P < 0.05; **P > 0.05.
A mouse shRNA library comprising ~62,400 shRNAs directed against ~28,000 genes9 was divided into 10 pools, which were packaged into retrovirus particles and used to stably transduce GL261/pATF5-DTR/pCMV-ATF5 cells. We treated cells with DT, and isolated DT-resistant clones. We identified positive shRNAs by sequence analysis and tested their ability to inhibit endogenous Atf5 gene expression (Supplementary Fig. 1b). Using this approach, we identified and validated 12 genes that were required for endogenous Atf5 expression in mouse malignant glioma GL261 cells (Supplementary Table 1).
We elected to focus on three factors: fibroblast growth factor receptor (FGFR) substrate 2 (FRS2), p21 protein-activated kinase 1 (PAK1) and CREB protein 3-like 2 (CREB3L2). FRS2 is a docking protein that binds to receptor tyrosine kinases, such as FGFR or epidermal growth factor receptor (EGFR), and leads to activation of RAS signaling10; notably, the RAS signaling pathway is frequently activated in malignant gliomas due to aberrant expression of receptor tyrosine kinases including FGFR and EGFR11. PAK1 is a kinase that functions as a positive regulator of mitogen-activated protein kinase (MAPK) signaling10,12. CREB3L2 is a member of the CREB family of transcription factors, which bind to DNA sequences known as cAMP responsive elements (CRE)13. CREB3L2 is homologous to CREB3, whose activity and expression is regulated by MAPK signaling14. We therefore hypothesized that FRS2, PAK1 and CREB3L2 comprise a signaling pathway that culminates in increased Atf5 expression. To test this hypothesis, we first confirmed that FRS2, PAK1 and CREB3L2 were required for Atf5 expression. We designed small interfering RNAs (siRNAs) against FRS2, PAK1 and CREB3L2 whose sequences were unrelated to the shRNAs isolated in the primary screen; siRNA-mediated knockdown of FRS2, PAK1 or CREB3L2 (Supplementary Fig. 1c) resulted in decreased Atf5 expression (Fig. 1b), confirming that these three factors are upstream activators of Atf5 transcription.
To determine whether Atf5 expression is modulated by signaling through FGFR, EGFR or RAS, we treated GL261 cells with an inhibitor of FGFR (PD173074)15, EGFR (CL-387,785)16, or RAS (manumycin A)17, and monitored Atf5 expression by quantitative real-time RT-PCR (qRT-PCR) and immunoblot analysis. All three inhibitors reduced Atf5 expression at both the mRNA and protein level (Supplementary Fig. 1d,e).
To determine whether downstream signaling pathways of RAS, such as MAPK and phosphoinositide-3-kinase (PI3K), are required for Atf5 expression, we treated GL261 cells with the RAF kinase inhibitor sorafenib18, the MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK) inhibitor U0126 (ref. 19), the ERK inhibitor FR180204 (ref. 20), or the PI3K inhibitor LY294002 (ref. 21). All four inhibitors substantially reduced Atf5 expression, whereas no effect was observed upon treatment with the p38MAPK inhibitor SB203580 (ref. 22) or c-Jun N-terminal kinase (JNK) inhibitor I (ref. 23) relative to the vehicle dimethyl sulfoxide (DMSO) (Fig. 1c).
We next investigated whether CREB3L2 directly regulates Atf5 expression. Bioinformatic analysis of the Atf5 promoter revealed a consensus CRE motif at −632 to −639 basepairs upstream of the transcription start-site (Fig. 1d, bottom). Chromatin immunoprecipitation (ChIP) analysis demonstrated that CREB3L2 directly bound to a region of the Atf5 promoter containing a consensus CRE motif, but not to a region further upstream (Fig. 1d, top). Moreover, binding of CREB3L2 to the Atf5 promoter was inhibited by sorafenib. To confirm the functional role of CREB3L2 in Atf5 transcription, we constructed a reporter gene in which ~4 kb of the Atf5 promoter was fused upstream of the luciferase gene. Figure 1e shows that the wild-type Atf5 promoter, but not a derivative in which several nucleotides in the CRE motif were mutated (see Fig. 1e, bottom), substantially stimulated luciferase production. Lastly, we found that endogenous Creb3l2 expression was substantially reduced by treatment of GL261 cells with an siRNA against FRS2 or PAK1, or an inhibitor of MAPK or PI3K (Supplementary Fig. 1f–h). Taken together, these results indicate that FRS2, PAK1 and CREB3L2 are components of RAS/MAPK- and PI3K-directed pathways that regulate ATF5 expression.
Next, we performed several experiments to confirm that the ATF5-mediated survival pathway is required for viability of malignant glioma cells. First, shRNA-mediated knockdown of FRS2, PAK1 or CREB3L2 induced apoptosis in GL261 cells, as evidenced by increased caspase 3/7 activity (Fig. 1f). Notably, apoptosis could be rescued by ectopic expression of ATF5. Similarly, the RAF inhibitor sorafenib (Fig. 1g) and inhibitors of FGFR, EGFR, RAS or PI3K (Supplementary Fig. 1i) induced apoptosis in GL261 cells but not in cells ectopically expressing ATF5. By contrast, JNK inhibitor I and SB203580, which did not affect Atf5 expression (see Fig. 1c), failed to induce apoptosis in GL261 cells (Fig. 1g).
Lastly, we investigated whether the ATF5-mediated survival pathway was required for malignant glioma cell proliferation in a mouse syngenic model. We injected GL261 cells stably expressing ATF5 or empty vector into the flanks of mice, and subsequently administered vehicle or sorafenib by gavage. As expected, control mice injected with GL261 cells developed tumors, whereas tumors were not detected in GL261-injected mice treated with sorafenib (Fig. 1h). Notably, ectopic expression of ATF5 in GL261 cells substantially reduced sensitivity to sorafenib. Taken together, these data indicate that RAS/MAPK- or PI3K-mediated activation of ATF5 expression is essential for survival of malignant glioma cells both in culture and in mouse xenografts.
ATF5 promotes survival through up-regulation of MCL1
Previous studies have shown that sorafenib can induce apoptosis by decreasing the level of myeloid cell leukemia sequence 1 (MCL1), an anti-apoptotic B-cell leukemia/lymphoma 2 (BCL2) family member24, which we confirmed also occurred in GL261 cells (Fig. 2a). This observation raised the possibility that ATF5 may prevent apoptosis by up-regulating MCL1 expression; we therefore performed a series of experiments to investigate this possibility. Knockdown of ATF5 decreased levels of MCL1, but had no effect on expression of several other anti-apoptotic BCL2 proteins including BCL2, BCL2-like 1 (BCL2L1, also called BCL-XL), and baculoviral IAP repeat-containing 3 (BIRC3, also called cIAP2) (Fig. 2b).
Figure 2.

ATF5 promotes survival through up-regulation of MCL1. (a) qRT-PCR analysis of Mcl1 expression in GL261 cells treated with DMSO, sorafenib (10 µM), U0126, FR180204, JNK inhibitor I or SB203580. (b) Immunoblot analysis monitoring ATF5, MCL1, BCL2, BCL-XL or cIAP2 levels in GL261 cells transiently transfected with a Luc or ATF5 siRNA. β-actin (ACTB) was monitored as a loading control. (c) qRT-PCR analysis of Mcl1 expression in GL261 cells transiently transfected with a Luc or ATF5 siRNA. (d) ChIP analysis monitoring binding of ATF5 to three regions of the Mcl1 promoter, as indicated, in the presence or absence of sorafenib (10 µM). (e) Immunoblot analysis monitoring cleaved caspase 3 (c-CASP3) levels in GL261 cells stably expressing either Flag or Flag-MCL1, and transiently transfected with a Luc or ATF5 siRNA. (f) A model depicting the ATF5-mediated survival pathway in solid tumors. (g) Immunoblot analysis monitoring ATF5, MCL1 and c-CASP3 levels in U87MG, DU145, UACC62, A549 and OVCAR3 cells treated with a Luc or ATF5 siRNA. All error bars shown, ±s.d.; *P < 0.05; **P > 0.05.
We next sought to determine whether ATF5 regulates Mcl1 transcription. Knockdown of ATF5 decreased Mcl1 mRNA levels (Fig. 2c) and, conversely, over-expression of ATF5 increased Mcl1 mRNA levels (Supplementary Fig. 2). To determine whether ATF5 directly bound to the Mcl1 promoter, we performed ChIP experiments using a series of primer-pairs that spanned the Mcl1 promoter. ATF5 binding was enriched at two regions of the Mcl1 promoter corresponding to −1531/−1358 base pairs (bp) and −853/−718 bp upstream of the transcription start-site (Fig. 2d). Importantly, binding of ATF5 to both Mcl1 promoter regions was inhibited by sorafenib (Fig. 2d).
We next asked whether MCL1 is required for ATF5-mediated survival by monitoringGL261 cells stably expressing MCL1 for apoptosis following siRNA-mediated knockdown of ATF5. ATF5 knockdown induced apoptosis in control GL261 cells, as evidenced by the production of cleaved caspase 3 (Fig. 2e). Notably, apoptosis was substantially inhibited by ectopic expression of MCL1. Collectively, these results indicate that MCL1 is a downstream effector of an ATF5-mediated survival pathway (Fig. 2f).
We next asked whether the ATF5-mediated survival pathway is generally active in human cancer cell lines. A search of the Oncomine cancer profiling database25,26 revealed that ATF5 expression is elevated in a wide range of tumors relative to normal tissue (Supplementary Table 2). Moreover, siRNA-mediated knockdown of ATF5 in several diverse human cancer cell lines, U87MG (glioblastoma), DU145 (prostate), UACC62 (melanoma), A549 (lung) and OVCAR3 (ovary), resulted in inhibition of MCL1 expression and induction of apoptosis (Fig. 2g). These results suggest that the ATF5 pathway mediates cell survival in malignant glioma and diverse human cancer cell lines.
ATF5 expression correlates with poor prognosis
Several lines of evidence indicate that the components of the ATF5-mediated survival pathway are highly expressed in malignant glioma. First, as mentioned above, ATF5 is highly expressed in human glioblastoma cells but not normal brain tissues5. Second, the FRS2 gene is amplified in a subset of malignant gliomas27. Finally, previous studies have shown that the activities of PAK1 and ERK correlate with prognosis in glioblastoma28,29. A search of the Oncomine database revealed that, like ATF5, FRS2, CREB3L2 and MCL1 are up-regulated in human glioblastoma compared to normal brain samples (Supplementary Fig. 3a). Moreover, genome-wide analysis of focal copy number in human glioblastoma30 also indicates that components of the ATF5-mediated survival pathway are amplified in the disease (Supplementary Fig. 3b).
To confirm and extend these results, we performed immunohistochemistry on a panel of four normal human brain samples and 38 glioblastomas; representative examples are shown in Fig. 3a, and the results summarized in Supplementary Fig. 3c. Positive staining for phosphorylated ERK (pERK), ATF5, CREB3L2 or MCL1 was found in 50–74% of malignant gliomas and, notably, 42% of malignant gliomas stained positively for all four proteins. By contrast, none of the four proteins were detected by immunohistochemical analysis in normal brain. Statistical analysis revealed a significant correlation in malignant gliomas between pERK and either CREB3L2 or ATF5, and between CREB3L2 and either ATF5 or MCL1 (Supplementary Fig. 3d). By contrast, a significant correlation was not observed between MCL1 and either pERK or ATF5, suggesting that MCL1 expression may be regulated by alternative pathway(s) in malignant glioma. This conclusion is consistent with previous studies demonstrating that MCL1 expression can be regulated by both transcriptional and post-transcriptional mechanisms31,32.
Figure 3.

ATF5 expression correlates with poor prognosis in human malignant glioma. (a) Immunohistochemical analysis of phosphorylated ERK (pERK), CREB3L2, ATF5, and MCL1 in brain tissue sections from a normal individual and glioblastoma subject. Samples were also stained with hematoxylin and eosin (H&E) (left). (b) Kaplan-Meier analysis assessing the correlation between survival of 23 individuals with malignant glioma and the expression of ATF5 (left) or pERK (right). The P-value of both ATF5 and pERK expression in subject survival was 0.03. “Month” refers to the time of death after the malignant glioma was first diagnosed.
To determine whether there was a correlation between ATF5 expression and prognosis of individuals with glioblastoma, we analyzed the clinical history of 23 individuals with malignant glioma and performed Kaplan-Meier analysis. This analysis revealed that individuals with ATF5-positive glioblastomas had substantially shorter survival times than those with ATF5-negative glioblastomas (Fig. 3b). As expected from previous studies29, pERK showed a similar correlation with clinical outcome. Thus, ATF5 expression is a marker of clinical outcome in individuals with malignant glioma.
Role of the ATF5 pathway in human malignant glioma cells
Malignant glioma is thought to arise from self-renewing progenitors termed glioma stem cells (GSCs)33, which are highly resistant to chemo- and radio-therapies34. To test whether components of the ATF5-mediated survival pathway are over-expressed in GSCs, we isolated GSCs from either a human malignant glioma primary cell line (GS9-6) or human glioblastoma cell line (D456MG) maintained as a xenograft, and analyzed expression of ATF5-mediated survival pathway components by immunoblot and qRT-PCR analysis. As a control we also monitored the expression of ATF5 pathway components in GS9-6 and D456MG GSCs following induction of differentiation by serum treatment. As expected, serum treatment induced differentiation of GS9-6 GSCs, as evidenced by up-regulation of glial fibrillary acidic protein (GFAP), an astrocytic marker, and down-regulation of nestin, a stem cell marker (Fig. 4a). Upon differentiation, CREB3L2, ATF5 and MCL1 levels decreased, indicating that these proteins are enriched in undifferentiated GS9-6 GSCs. Furthermore, qRT-PCR analysis verified that expression of CREB3L2, ATF5 and MCL1 decreased following differentiation of GS9-6 and D456MG GSCs (Fig. 4b).
Figure 4.

The ATF5-mediated survival pathway is essential for viability of human malignant glioma cells. (a) Immunoblot analysis of pERK, ERK, CREB3L2, ATF5, MCL1, GFAP and nestin (NES) in GS9-6 GSCs treated in the presence or absence of serum. (b) qRT-PCR analysis monitoring CREB3L2, ATF5, MCL1, CD133, NES and GFAP in D456MG and GS9-6 GSCs treated in the presence relative to absence of serum. (c) Immunoblot analysis of CREB3L2, ATF5, MCL1 and c-CASP3 in GS9-6 GSCs treated in the absence or presence of 40 µM sorafenib. (d) qRT-PCR analysis of CREB3L2, ATF5 and MCL1 in GS9-6 GSCs treated in the absence or presence of 40 µM sorafenib. (e) Cell viability, monitored by MTT assay, of GS9-6 GSCs incubated with or without serum, and then treated with sorafenib for 96 h. Cell viability was significantly affected by sorafenib treatment (P = 3.53e–07). All error bars shown, ±s.d.; *P < 0.05; **P > 0.05.
To determine whether, as in malignant glioma cells, the ATF5-mediated survival pathway was regulated by MAPK signaling, we treated GS9-6 GSCs with sorafenib, and assessed expression of CREB3L2, ATF5 and MCL1 by immunoblot (Fig. 4c) and qRT-PCR analysis (Fig. 4d). The results show that sorafenib reduced expression of CREB3L2, ATF5 and MCL1 at both the protein and mRNA levels. Furthermore, sorafenib treatment induced apoptosis in GS9-6 GSCs, as evidenced by elevated levels of cleaved caspase 3 (Fig. 4c). Consistent with this result, sorafenib treatment resulted in decreased cell viability of both undifferentiated and differentiated GS9-6 GSCs (Fig. 4e). Thus, the ATF5-mediated survival pathway we identified in mouse malignant glioma is also essential for survival of human malignant glioma cells.
Sorafenib suppresses growth of malignant glioma in mice
To test whether sorafenib could suppress development of brain tumors, we used an established mouse xenograft model of human malignant glioma35. In brief, GSCs were injected intracranially into mice, animals were treated in the presence or absence of sorafenib, and tumor development was monitored by magnetic resonance imaging (MRI). Representative examples are shown in Fig. 5a, and the results quantified in Fig. 5b. All of the mice treated with vehicle harbored a tumor >5 mm3 and 50% had a tumor >25 mm3. By contrast, only 60% of the mice treated with sorafenib had a tumor >5 mm3 and none had a tumor >25 mm3. Immunohistochemical staining of tumor sections for cleaved caspase 3 confirmed that sorafenib treatment reduced tumor growth by inducing apoptosis (Fig. 5c,d). Taken together, these data demonstrate that inhibition of MAPK signaling suppresses development of malignant glioma in a mouse model.
Figure 5.

Inhibition of MAPK signaling suppresses development of malignant glioma in mouse xenografts. (a) Mouse xenograft experiments. Tumor formation was monitored by MRI (left); the position of the tumor is indicated by the arrow. Brain sections were stained with H&E (middle) or an antibody against GFAP (right) to verify the glial origin of the tumor. Expression of CREB3L2, ATF5 and MCL1 in these sections is shown in Supplementary Fig. 5. (b) Quantification of tumor size based on MRI images. Sorafenib significantly inhibited tumor formation (P = 0.0344). (c) c-CASP3 staining in brain sections from vehicle- or sorafenib-treated mice; arrows indicate c-CASP3-positive (apoptotic) cells. (d) c-CASP-3-positive cells were counted in three different fields under the microscope, and the average value is shown. All error bars shown, ±s.d.; *P < 0.05; **P > 0.05.
Finally, we investigated whether sorafenib could synergize with temozolomide (TMZ), a chemotherapeutic agent used to treat glioblastomas36, to inhibit tumor cell growth. We found that sorafenib and TMZ synergistically decreased viability of U87MG human glioblastoma cells (Fig. 6a), which express high levels of ATF5 and CREB3L2 (Supplementary Fig. 4a), but not SF295 human glioblastoma cells (Supplementary Fig. 4b), which express low levels of ATF5 and CREB3L2 (Supplementary Fig. 4a). We also observed a synergistic effect on tumor growth in U87MG mouse xenografts (Fig. 6b). Taken together, these results indicate that sorafenib can sensitize glioblastoma cells to TMZ, and further suggest that such sensitization selectively occurs in cells expressing high levels of ATF5.
Figure 6.

Sorafenib synergizes with temozolomide to inhibit tumor cell growth. (a) U87MG cells were treated with combinations of sorafenib and TMZ, and cell viability was measured using an MTT assay. The combination of sorafenib and TMZ significantly decreased cell viability (P = 1.468e–05). (b) Mouse xenograft experiments. The combination of sorafenib and TMZ treatment significantly decreased tumor volume compared to sorafenib (P = 4.4e–05) or TMZ (P = 5.6e–06) alone. All error bars shown, ±s.d.
DISCUSSION
ATF5 is expressed at high levels in malignant glioma but not in mature neurons or astrocytes6. Moreover, interference with ATF5 function results in glioma cell death in primary tumors without affecting normal cells surrounding the tumor6. These findings suggest that ATF5 is an attractive target for the development of therapeutics for malignant glioma4. However, ATF5 is a transcription factor, a class of molecules for which the development of small molecule drugs has proven challenging37. An alternative strategy to targeting ATF5 directly is to interfere with upstream regulators of ATF5 expression or downstream effectors through which ATF5 promotes survival.
In this report, we have described a novel negative-selection strategy using genome-wide RNAi screening to elucidate RAS/MAPK- and PI3K-directed signaling pathways by which ATF5 is expressed and promotes survival of malignant glioma cells. The genome-wide RNAi screening strategy we have developed can be generally applied to identify other survival pathways in cancer cells. Our results enable us to propose a model for an essential survival pathway in malignant glioma and other solid tumors (Fig. 2f). An FRS2/PAK1-activated RAS/MAPK signaling cascade up-regulates CREB3L2, which directly binds to the ATF5 promoter resulting in ATF5 transcription. FRS2 may also activate ATF5 transcription through PI3K signaling. ATF5 then stimulates MCL1 transcription by binding to its promoter, resulting in increased levels of the anti-apoptotic protein MCL1, which promotes cell survival. Our results do not rule out the possibility that other ATF5 targets, which remain to be identified, may also contribute to the inhibition of apoptosis.
We have shown that pharmacological inhibition of this pathway substantially decreases ATF5 levels and blocks proliferation of malignant glioma cells both in cell culture and in mouse tumor models. Our experiments were carried out using the well-characterized RAF inhibitor sorafenib at concentrations in cultured cells and at doses in mice38–40 that approximate the levels of sorafenib achieved in subjects treated with the drug41–44. Moreover, we have found that in the presence of TMZ, the drug most commonly used to treat malignant glioma, the concentration of sorafenib required for growth inhibition is dramatically reduced. Thus, when combined with TMZ, the sorafenib concentration obtainable in subjects may be well above that required for an effective response. Finally, although our sorafenib results clearly demonstrate that the pro-survival pathway we have identified can be pharmacologically inhibited, it is likely that more recently developed RAF inhibitors45, and inhibitors of other components of this ATF5-mediated survival pathway, such as MEK, may be even more efficacious.
METHODS
RNAi
For pATF5-DTR, we amplified 4117 bp of the mouse Atf5 promoter from a BAC clone and cloned it into pEF4/Myc-His B (Invitrogen), then amplified the human DTR gene from mCD11C-DTR-EGFP46 and cloned it downstream of the Atf5 promoter. For pCMV-ATF5, we amplified ATF5 cDNA from pCMV-SPORT6-ATF5 (Open Biosystems) and cloned it into p3XFlag-myc-CMV™-26 (Sigma). We transduced GL261 cells stably expressing pATF5-DTR and pCMV-ATF5 with retroviral pools of a mouse shRNAmir library (Open Biosystems) as described47. We incubated cells with 100 pM DT for 7 d, and cultured surviving cells in media without DT for 2 weeks. To validate candidates, we transiently transfected GL261 cells with an individual shRNA, and assessed expression of endogenous Atf5 by RT-PCR (using primers listed in Supplementary Table 3) quantified using Image J software. For RNAi, we transiently transfected GL261 cells (5×105) with an siRNA or shRNA (Supplementary Table 3) using Effectene (QIAGEN).
Quantitative RT-PCR
Quantitative RT-PCR was performed as previously described47 using primers listed in Supplementary Table 3.
ChIP
ChIP assays were performed as previously described48 using antibodies listed in Supplementary Table 3. Immunoprecipitated DNA was amplified by real-time PCR (primers listed in Supplementary Table 3).
Luciferase reporter assay
We cloned the mouse Atf5 promoter into pGL4.14 (luc2/Hygro; Promega), and mutated the CRE element into a Not I site. We co-transfected this plasmid and pGL4.73 (hRluc/SV40; Promega) into GL261 cells and . assessed luciferase activity 48 h later using the Dual-Luciferase Reporter Assay System (Promega). Relative luciferase activity was calculated and normalized to empty vector.
Apoptosis assays
We measured apoptosis using a Caspase-Glo® 3/7 Assay (Promega). To eliminate background caspase activity, caspase 3/7 activity was normalized to cell number (obtained from an MTT assay), and then to the activity obtained with the control shRNA.
Preparation of GSCs
For GS9-6, a freshly-resected tumor, never treated with radio- or chemotherapy, was dispersed with trypsin and placed into neurosphere culture in DMEM/F12 medium supplemented with B27 (Invitrogen), 20 ng ml−1 fibroblast growth factor (PeproTech) and 20 ng ml−1 epidermal growth factor (PeproTech). For passaging, we dispersed neurospheres using a basic solution49 and maintained them in the media described above. D456MG GSCs were prepared as previously described34.
Mouse tumorigenesis assays
For the syngenic model, we injected 1×106 GL261 cells subcutaneously into the flank of C57/BL6 mice (Taconic), aged 8 weeks. Tumor volume was calculated [(length × width2) × (π/6)]. We fed mice daily with sorafenib (60 mg kg−1) or vehicle (cremphor:ethanol:water 12.5%:12.5%:75%) through gavage starting at 4 d. Mice were sacrificed 31 d after injection. For the xenograft model, we implanted 5×105 GS9-6 cells stereotaxically in the right frontal lobe [from bregma, anteroposterior , 2 mm; mediolateral, 2 mm; ventral (from dura), 2 mm] of NIH-bg-nu-xidBR mice (Charles River Laboratories) as described35. We administered sorafenib (100 mg kg−1) or vehicle daily through gavage starting at 7 d. Tumor growth was monitored at the UMMS Image Core of Advanced MRI using Philips DICOM Viewer R2.4, and tumor volume was calculated (length × width × depth). After 81 d mice were imaged and sacrificed, and their brains sectioned and fixed in formalin. We performed all animal experiments in accordance with the Institutional Animal Care and Use Committee guidelines.
Temozolomide experiments
We treated U87MG or SF295 cells with sorafenib, TMZ (Sigma) or both for 4 d, and monitored cell viability by MTT assay. For the xenografts, we injected 5×106 U87MG cells subcutaneously into the flank of nude mice (Taconic), aged 6–8 weeks. Starting at 15 d, we fed mice daily with sorafenib (20 mg kg−1), TMZ (7.5 mg kg−1) or a combination of both through gavage, then treated with sorafenib for another 10 d. Mice were sacrificed 30 d after injection. Tumor dimensions were measured every 3 d and tumor volume was calculated.
Statistical analyses
We performed all analyses in R 2.9.050. We performed Welch two sample t-test51 on log-transformed qPCR data (Figs. 1b,c,e; 2a,c; 4d) and log-transformed relative caspase activity data (Fig 1f,g). We performed repeated measure analysis of variance on log-transformed tumor volume (Figs. 1h, 6b). In addition, we performed a list of pre-determined contrasts to test whether sorafenib treatment decreases tumor volume in the absence or presence of ATF5, and whether combined sorafenib and TMZ treatment decreases tumor volume significantly compared with sorafenib or TMZ treatment alone. We applied ANOVA to the rank-transformed cell viability data to test the effect of serum, sorafenib and the interaction of serum and sorafenib (Fig. 4e) and of TMZ, sorafenib and the interaction of TMZ and sorafenib (Fig. 6a). A linear mixed-effects model was fit to the log-transformed tumor volumes (Fig. 5b) or number of apoptotic cells (Fig. 5d) with treatment as a fixed effect and experiment day as random effect to determine whether the tumor size differs between treatments using R package nlme_3.1–90.
Supplementary Material
Acknowledgements
We thank Y. Gillespie (University of Alabama at Birmingham), D. Bigner (Duke University) and R. Lang (University of Cincinnati) for reagents; the University of Massachusetts Medical School RNAi Core Facility for providing shRNAs; the UMMS Department of Pathology for providing human tissue sections, and the UMMS DERC Morphology core for performing the immunohistochemistry; K. Rock for suggesting the diphtheria toxin strategy; Y. Sun for critical advice on MRI; C. Gilbert for maintaining GS9-6 cells; S. Griggs for technical support; and S. Evans for editorial assistance. This work is supported by US National Institutes of Health grant RO1CA115817 to M.R.G. M.R.G is an investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions
Z.S. and M.R.G designed all experiments. Z.S performed all experiments. Z.S. and M.R.G. prepared the manuscript. L.L. and A.H.R. assisted with intracranial injections. T.W.S, A.D. and R.P.M. helped analyze human malignant gliomas. L.J.Z. performed all statistical analyses.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Persengiev SP, Green MR. The role of ATF/CREB family members in cell growth, survival and apoptosis. Apoptosis. 2003;8:225–228. doi: 10.1023/a:1023633704132. [DOI] [PubMed] [Google Scholar]
- 4.Greene LA, Lee HY, Angelastro JM. The transcription factor ATF5: role in neurodevelopment and neural tumors. J. Neurochem. 2009;108:11–22. doi: 10.1111/j.1471-4159.2008.05749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monaco SE, Angelastro JM, Szabolcs M, Greene LA. The transcription factor ATF5 is widely expressed in carcinomas, and interference with its function selectively kills neoplastic, but not nontransformed, breast cell lines. Int. J. Cancer. 2007;120:1883–1890. doi: 10.1002/ijc.22469. [DOI] [PubMed] [Google Scholar]
- 6.Angelastro JM, et al. Selective destruction of glioblastoma cells by interference with the activity or expression of ATF5. Oncogene. 2006;25:907–916. doi: 10.1038/sj.onc.1209116. [DOI] [PubMed] [Google Scholar]
- 7.Persengiev SP, Devireddy LR, Green MR. Inhibition of apoptosis by ATFx: a novel role for a member of the ATF/CREB family of mammalian bZIP transcription factors. Genes Dev. 2002;16:1806–1814. doi: 10.1101/gad.992202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collier RJ. Diphtheria toxin: mode of action and structure. Bacteriol. Rev. 1975;39:54–85. doi: 10.1128/br.39.1.54-85.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva JM, et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- 10.Sato T, Gotoh N. The FRS2 family of docking/scaffolding adaptor proteins as therapeutic targets of cancer treatment. Expert. Opin. The.r Targets. 2009;13:689–700. doi: 10.1517/14728220902942330. [DOI] [PubMed] [Google Scholar]
- 11.da Fonseca CO, et al. Recent advances in the molecular genetics of malignant gliomas disclose targets for antitumor agent perillyl alcohol. Surg. Neurol. 2006;65(Suppl 1):S1:2–S1:8. doi: 10.1016/j.surneu.2005.06.030. discussion S1:8-1:9. [DOI] [PubMed] [Google Scholar]
- 12.Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat. Rev. Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 13.Kondo S, et al. BBF2H7, a novel transmembrane bZIP transcription factor, is a new type of endoplasmic reticulum stress transducer. Mol. Cell. Biol. 2007;27:1716–1729. doi: 10.1128/MCB.01552-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 15.Mohammadi M, et al. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 1998;17:5896–5904. doi: 10.1093/emboj/17.20.5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Discafani CM, et al. Irreversible inhibition of epidermal growth factor receptor tyrosine kinase with in vivo activity by N-[4-[(3-bromophenyl)amino]-6-quinazolinyl]-2-butynamide (CL-387,785) Biochem. Pharmacol. 1999;57:917–925. doi: 10.1016/s0006-2952(98)00356-6. [DOI] [PubMed] [Google Scholar]
- 17.Hara M, et al. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc. Natl. Acad. Sci. U. S. A. 1993;90:2281–2285. doi: 10.1073/pnas.90.6.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilhelm S, Chien DS. BAY 43-9006: preclinical data. Curr. Pharm. Des. 2002;8:2255–2257. doi: 10.2174/1381612023393026. [DOI] [PubMed] [Google Scholar]
- 19.Favata MF, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 20.Ohori M, et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- 21.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J. Biol. Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 22.Cuenda A, et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 23.Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 24.Yu C, et al. The role of Mcl-1 downregulation in the proapoptotic activity of the multikinase inhibitor BAY 43-9006. Oncogene. 2005;24:6861–6869. doi: 10.1038/sj.onc.1208841. [DOI] [PubMed] [Google Scholar]
- 25.Rhodes DR, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun L, et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9:287–300. doi: 10.1016/j.ccr.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Fischer U, et al. A different view on DNA amplifications indicates frequent, highly complex, and stable amplicons on 12q13-21 in glioma. Mol Cancer Res. 2008;6:576–584. doi: 10.1158/1541-7786.MCR-07-0283. [DOI] [PubMed] [Google Scholar]
- 28.Aoki H, et al. Phosphorylated Pak1 level in the cytoplasm correlates with shorter survival time in patients with glioblastoma. Clin. Cancer Res. 2007;13:6603–6609. doi: 10.1158/1078-0432.CCR-07-0145. [DOI] [PubMed] [Google Scholar]
- 29.Pelloski CE, et al. Prognostic associations of activated mitogen-activated protein kinase and Akt pathways in glioblastoma. Clin. Cancer Res. 2006;12:3935–3941. doi: 10.1158/1078-0432.CCR-05-2202. [DOI] [PubMed] [Google Scholar]
- 30.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahmani M, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol. Cell. Biol. 2007;27:5499–5513. doi: 10.1128/MCB.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J. Biol. Chem. 2005;280:35217–35227. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- 33.Alcantara Llaguno S, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 35.Li L, et al. EGFRvIII expression and PTEN loss synergistically induce chromosomal instability and glial tumors. Neuro. Oncol. 2009;11:9–21. doi: 10.1215/15228517-2008-081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villano JL, Seery TE, Bressler LR. Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother. Pharmacol. 2009;64:647–655. doi: 10.1007/s00280-009-1050-5. [DOI] [PubMed] [Google Scholar]
- 37.Weiss WA, Taylor SS, Shokat KM. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat. Chem. Biol. 2007;3:739–744. doi: 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sparidans RW, et al. Liquid chromatography-tandem mass spectrometric assay for sorafenib and sorafenib-glucuronide in mouse plasma and liver homogenate and identification of the glucuronide metabolite. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009;877:269–276. doi: 10.1016/j.jchromb.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 39.Lagas JS, et al. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol. Cancer Ther. 2010;9:319–326. doi: 10.1158/1535-7163.MCT-09-0663. [DOI] [PubMed] [Google Scholar]
- 40.Hu S, et al. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin. Cancer Res. 2009;15:6062–6069. doi: 10.1158/1078-0432.CCR-09-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Awada A, et al. Phase I safety and pharmacokinetics of BAY 43-9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumours. Br. J. Cancer. 2005;92:1855–1861. doi: 10.1038/sj.bjc.6602584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strumberg D, et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J. Clin. Oncol. 2005;23:965–972. doi: 10.1200/JCO.2005.06.124. [DOI] [PubMed] [Google Scholar]
- 43.Clark JW, Eder JP, Ryan D, Lathia C, Lenz HJ. Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43-9006, in patients with advanced, refractory solid tumors. Clin. Cancer Res. 2005;11:5472–5480. doi: 10.1158/1078-0432.CCR-04-2658. [DOI] [PubMed] [Google Scholar]
- 44.Richly H, et al. Results of a Phase I trial of sorafenib (BAY 43-9006) in combination with doxorubicin in patients with refractory solid tumors. Ann. Oncol. 2006;17:866–873. doi: 10.1093/annonc/mdl017. [DOI] [PubMed] [Google Scholar]
- 45.McCubrey JA, et al. Emerging Raf inhibitors. Expert. Opin. Emerg. Drugs. 2009;14:633–648. doi: 10.1517/14728210903232633. [DOI] [PubMed] [Google Scholar]
- 46.Jung S, et al. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449:1073–1077. doi: 10.1038/nature06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheng Z, Wang SZ, Green MR. Transcription and signalling pathways involved in BCR-ABL-mediated misregulation of 24p3 and 24p3R. EMBO J. 2009;28:866–876. doi: 10.1038/emboj.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sen A, Kallos MS, Behie LA. New tissue dissociation protocol for scaled-up production of neural stem cells in suspension bioreactors. Tissue Eng. 2004;10:904–913. doi: 10.1089/1076327041348554. [DOI] [PubMed] [Google Scholar]
- 50.Ihaka R, Gentleman R. A language for data analysis and graphics. J. Comput. Graph. Stat. 1996;5:299–314. [Google Scholar]
- 51.Ruxton GD. The unequal variance t-test is an underused alternative to Student's t-test and the Mann-Whitney U test. Behavioral Ecology. 2006;17:688–690. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.