

Abstract
Proteasome, consisting of a tube-shaped proteolytic core particle and attached to it regulatory modules, is a multifunctional enzymatic complex essential for the ubiquitin-proteasome metabolic pathway. Due to its immense involvement in regulation of cellular physiology, the proteasome is an acknowledged anti-cancer drug target and potential target to treat inflammatory or degenerative diseases. So far, competitive inhibitors of the core particle gain most consideration as drugs. We postulate that noncompetitively-acting small-molecule compounds would provide excellent means to precisely regulate actions of the proteasome. In this study we evaluated five short peptides based on sequences of two proteins known to interact with the core proteasome: HIV-1 Tat and PA28/REG activator. We performed CD, FT-IR and NMR analysis, supplemented by MD simulations, and tested influence of the peptides on performance of the core particle active sites and functioning of regulatory modules. We found that PP2-containing Tat peptides are noncompetitive inhibitors of the core, interfering with the actions of PA28αβ activator. In addition, at low concentrations the turn-prone Tat2 is able to activate the latent core. The random coil-structured PA28-derived peptides display only weak or nondetectable direct effects on the core activities, exhibiting, however, a positive cooperation with activity-enhancing actions of PA28αβ.
Keywords: proteasome, HIV-1 Tat protein, 11S activator, allosteric, inhibitor
INTRODUCTION
Eukaryotic 26S proteasome is a large (2,500 kDa) proteolytic complex, which plays a major role in ubiquitin-dependent turnover of intracellular proteins. In a process of controlled proteolysis it recognizes and digests short-lived regulatory proteins influencing all major cellular processes such as cell cycle progression, oncogenesis, transcription, tissue differentiation, apoptosis, an influx of substrates through metabolic pathways, and antigen processing. The proteasome is also responsible for the housekeeping chores degrading mutated, misfolded or oxidatively damaged proteins. Since proteasome is involved in many fundamental biological processes, any disturbance in its activity may lead to detrimental physiological consequences usually resulting in cell death [1, 2].
The 26S proteinase is composed of the catalytic core particle (CP) capped at each end by the regulatory ATP-dependent 19S complex (called also PA700, regulatory particle; RP) responsible for recognition, unfolding, deubiquitinylation and translocation of substrates into the lumen of the catalytic particle. The core which is referred to as 20S proteasome is a ~700 kDa hollow cylindrical protein complex consisting of four stacked rings (α-β-β-α) each comprising seven different subunits [3, 4]. The two outer (α) rings are anchor sites for regulatory particles and form gates controlling the access to the axial channel directing substrates to six active sites located at the β-rings and facing the inner chamber of the cylinder [3-6]. The 20S proteasome can degrade only short peptides and poorly structured proteins [2, 5].
The proteasome is classified as an N-terminal nucleophile aminohydrolase since it uses its N-terminal threonine residues from β1, β2 and β5 subunits as catalytic sites. The β1 subunit is responsible for the proteasome postglutamyl (PGPH) or caspase-like activity, β2 for the trypsin-like (T-L), and β5 for the chymotrypsin-like (ChT-L) activity, cleaving peptide bonds on the carboxyl side of acidic, basic and hydrophobic amino acid residues, respectively [2-4]. In human and other higher vertebrates three homologous β subunits: β1i, β2i and β5i induced by interferon-γ (IFN-γ) can replace the respective constitutive catalytic subunits leading to the formation of so called immunoproteasome, as opposed to housekeeping proteasome [7]. The immunoproteasome CP is characterized by an enhanced generation of peptides that constitute ligands for complexes in the major histocompatibility (MHC) class I antigenic presentation [7]. The channel leading to the active sites is closed most of the time in the latent 20S proteasome [3-5]. It is speculated that an attachment of regulators such as the 19S cap to the face of alpha rings opens the gate and activates the core particle [2,5,6]. The crystal structure of 20S and the heptameric activator complex PA26 (proteasome activator; Figure 1) showed that the channel opens upon the attachment. However this assembly, or the assembly of CP with a homolog of PA26 (Figure 1), PA28 is unable to process ubiquitinylated substrates [6,8-10]. Instead, such ‘activated’ 20S proteasome displays up-regulated peptidase activity [6,9], similarly to the CP with the channel permanently opened by molecular deletion of the gate-forming structural elements [2,5]. PA28 (11S) complex naturally occurs as a heteroheptamer of alpha and beta subunits or homoheptamer of gamma subunits [6,8]. The expression of PA28αβ is upregulated by γ-interferon and this activator accelerates production of antigenic peptides by the proteasome [6,7]. The crystal structure of recombinant homoheptameric human PA28α has been solved (1avo) [11]. Assemblies of CP with 19S on one side and PA28 on the opposite have been reported [2].
Figure 1.

Design of PA28-derived peptides was based on the crystal structures of PA28α and PA26-20S complex.
Left: The rectangle represents the complete sequence of PA28 subunits. The designed peptides that include the activation loop (AL; bold) are marked in red whereas a peptide incorporating the C terminal sequence (Ct; bold) is marked in blue. The residue positions of peptides specific for each type of subunit are given next to the peptide sequence. The approximate location of helical structures guided by the crystal structure of human PA28α (1avo [11]) is shown above the sequence rectangle. The arrangement of helices and loops is similar in PA28 and PA26 subunits, however the respective AL fragment in PA26 shown in the right panel, extends from residue 96 to 107 (I96PEHKEEDNLGV107).
Right: the positions of AL (red) and Ct (blue) are shown as ribbons (top right) or spacefilled model (bottom right) in the crystal structure of Trypanosoma brucei PA26 (11S; a homolog of PA28) bound to yeast 20S core particle (1fnt [9]). The interface between the activator and the 20S particle is enlarged. Note seven ALs (the leftmost two ALs closely overlap) and seven C-terminal fragments of PA26 heptamer extending toward the top surface of proteasome α ring. A single AL and Ct is further enlarged below. The zoomed-in fragments are marked by purple frames. The ribbon models correspond to secondary structures detected in the crystal structure.
Proteasome with its rich physiological functions is a target of inhibitors – drugs [12]. Many competitive inhibitors of the proteasome have been already developed. Since blocking of active centers halts cleavage of all substrates, such inhibitors unexpectedly found powerful pharmaceutical applications in the fight against cancer [12]. Namely, it was found that cancer cells are much more susceptible to apoptosis triggered by a partial inhibition of proteasome than normal cells. Although the mechanism of such distinct response of the cells is not completely understood, bortezomib (Velcade®), a competitive inhibitor of proteasome has been approved as a drug to treat multiple myeloma and lymphoma, and is currently tested for other types of cancer [12].
Nevertheless, it would be highly desirable to reach beyond the apoptosis based elimination of cells and gain access to a selective regulation of the proteasome activity, with a precise controlling of gate opening and with enzymatic activity of one type of the active center allosterically influencing performance of other centers [13]. It is conceivable that compounds regulating the proteasome activity in an allosteric manner may offer exactly this type of discriminating capabilities. It is acknowledged that the 19S cap and PA28 activator allosterically regulate the gate status beyond the physical clearing the path for substrates. Moreover, the permanent path opening by simple deletion of the gate elements leads to serious physiological consequences [5,6].
Cathelicidins’ derived proline-arginine-rich (PR) peptides constitute an example of a distinct group of natural compounds that allosterically regulate proteasome activity. It was found that a 39-residue PR39 peptide and its 11-residue N-terminal truncation homologs interact with the α-ring of CP interfering with efficient movements of the gating mechanism. In vitro, PR peptides noncompetitively inhibit the ChT-L and PGPH activities to a different extent and only weakly affect the T-L activity of activated 20S proteasome [14]. Interestingly, in vivo this group of peptides is able to selectively block degradation of NF-κB inhibitor IκBα and HIF-1α without influencing the gross ubiquitin dependent proteolysis normally leading to the accumulation of polyubiquitinated substrates [15].
In our quest to precisely control proteasome activity using the allosteric mechanism, we turned our attention to structural elements of the 11S activator responsible for interactions with CP. We took advantage of the available crystal structures of 11S activator alone and in complex with CP to design short peptides based on sequences of the PA28 activating loop and its C-terminus. These regions are indispensable elements of PA28 to productively interact with proteasome [16]. Additionally, we included in our analysis selected regions of HIV-1-Tat protein since it competes with the 11S activator for binding sites on CP and is known to strongly inhibit proteasome activity [17].
RESULTS AND DISCUSSION
Design of peptides containing the putative CP-binding elements
In our search for allosteric modulators of proteasome enzymatic activity we focused on ligands that modify performance of the catalytic centers by interacting with the alpha ring top surface (alpha face). In particular, we designed and tested several peptides, which sequences were derived from the 11S activator and its in vitro putative competitor, HIV-1 Tat protein (Table 1). We based our design on the available crystal structures of the 11S activator and NMR structures of HIV-1 Tat. They include a heptameric human PA28 activator built solely from the α type subunits (pdb: 1avo) and a Trypanosoma brucei homolog of PA28α, a PA26 heptamer co-crystallized with yeast 20S proteasome (pdb: 1fnt) [9,10,11]. Although naturally found PA28 forms a heptamer of α and β subunits, it is believed that the solved structure of a homoheptamer of PA28α faithfully renders the arrangement of polypeptides in the native complex, and that the PA26 homolog provides a good insight into the activator-proteasome interface. The structures indicate presence of two regions in the PA28α involved in interactions with CP [9,10]) (Figure 1). They include a C-terminal region comprised of 7 residues forming a short α helix (243-249) and a 6 residue loop between helix 3 and 4. It is suggested that the C-terminal tails interact with the N-terminus of helix 1 of selected CP α subunits via their carboxylate. This contact supplies binding affinity that stabilizes the complex and brings the activation loops in close proximity to the binding sites. The helical structure of tails is evident only when the activator is in a complex with CP. Despite their fundamental role in binding to CP, the tails are relatively diverse between different subunits and between species. However, the deletion of C-terminal Tyr or its substitution with a charged residue abolishes the activator binding and activation of CP [8]. An activator prepared exclusively with β subunits does not enhance peptidase activity of CP. On this basis we prepared a peptide, which sequence KKP240RGETKGNleIY249 (abbreviated KKP; the original Met was substituted with isosteric Nle), corresponding to the C terminus of the PA28α subunit, was extended beyond the CP contact region into the conserved Lys239 and Pro240 (Figure 1) The latter is in a direct contact with the activation loop and together they comprise a surface critical for the proteasome activation [16].
Table 1.
Sequences of 12-residue peptides used in the study.
| peptide | sequence | source |
|---|---|---|
| IPK | IPKIEDGNDFGV | activation loop of PA28β |
| IPR | IPRIEDGNNFGV | activation loop of PA28α/γ |
| KKP | KKPRGETKGNleIY | C-terminus of PA28α |
| Tat1 | GRKKRRQRRRPS | fragment of the basic domain of HIV-1 Tat protein |
| Tat2 | RKKRRQRRQDPI | fragment of the basic domain of HIV-1 Tat protein |
We have also synthesized peptides that incorporate in their sequence activation loops (residues Arg141-Gly149, AL) specific for the α and γ subunits - IPR141IEDGNNFG149V (γ: R146 – G154, IPR; loop-αγ) and the β subunit - IPK131IEDGNDFG139V (IPK; loop-β) flanked by the neighboring residues (Figure 1, Table 1). These highly conserved moieties are crucial for the capability of the 11S complex to activate CP. Indeed, it has been shown that although a single substitution of Asp146 by tyrosine in PA28α preserves the binding of the activator at the same time it eliminates the activating effect. Mutants bearing similar substitutions in PA28β subunit (N135Y) and PA28γ (N151Y) also compete with binding of the wild type activators but fail to stimulate CP [17].
Human immunodeficiency virus type 1 (HIV-1) Tat (trans activator) is an 86 residues protein responsible for transactivation of viral and certain host genes (Figure 2). It belongs to a group of early RNA binders. Besides its role in the lentiviral transcription it regulates activity of several genes involved in angiogenesis. Interestingly, it also affects transcription of catalytic immuno subunits of proteasome, inhibits CP activity in vitro, and competes with the PA28 binding markedly influencing MHC class I associated antigen presentation. Tat protein is a relatively poorly structured protein. NMR and MD studies identified two major domains in Tat: a hydrophobic core and glutamine rich domain [18] (Figure 2). Two other regions are more structurally flexible and form a cysteine rich region and a basic domain. The basic region responsible for RNA binding contains an arginine-rich motif (ARM) and shows a weak tendency to form helical structure. This region is also involved in interactions with CP. Additionally, it shares a REG/Tat-proteasome-binding (RTP) site with the 11S activating particle. The site consists of residues Lys51, Arg52 and Asp67 in Tat and Glu235, Lys236 and Lys239 in PA28α (Figures 1, 2) [19]. Mutations of the corresponding residues into alanine abolish the molecular effect on CP but do not disrupt the binding. Therefore, to compare structure function relationship of the 11S activator derived peptides with competing for CP binding HIV-1 Tat protein we prepared two peptides. Both of them are based on a sequence of Tat obtained from HIV-1 Zaire 2 isolate (HV1Z2) structurally characterized with NMR by Bayer et al. (pdb: 1TIV) [18]. The first sequence (Tat1: G48RKKRRQRRRPS59) comprised the basic domain of Tat including Lys51 and Arg52 of RTP. In turn, in the second peptide (Tat2: R49KKRRQRR56Q66DPI69) the RPS59 C-terminal sequence was substituted with the third residue of the RTP motif (Asp67) flanked by three neighboring residues (Figure 2, Table 1).
Figure 2.

Design of HIV-1 Tat derived peptides was based on the sequence of the viral protein and its NMR structure (1TIV [18]).
Left: the rectangle represents the complete sequence of HIV-1 Tat protein. Labels I-V correspond to the mapped regions of the protein. The position of sequences incorporated in proteasome-interacting peptides is marked with color rectangles. The sequence of Tat1 is derived from the RNA-binding arginine-rich motif (ARM; orange) located in the region IV and covering residues 48-59. Tat2 incorporates residues 48-56 of the ARM and a tetrapeptide 66-69 of the glutamine-rich region V (green). Selected residues of both the regions constitute the REG/Tat proteasome-binding site (RTP, underlined). The RTP site is shared by HIV-1 Tat and C-terminal residues of REG/PA28 (Figure 1) [19].
Right: The spatial structure of HIV-1 Tat protein based on the NMR model 1TIV. The identical color coding is used in both the panels. Note the poorly structured nature of the viral protein.
Peptides representing the PA28/REG activation loops and C-terminus productively interact with the CP
We predicted that the individual peptides of sequences derived from the activator loop would not substantially affect any of peptidase activity of the CP since in the native PA28 complex the loops require presence of the C-terminal region, represented here by the KKP peptide, for the CP activation. Indeed, IPR and IPK peptides constituting activation loops alone did not influence performance of 20S proteasome (not shown). Unexpectedly, the unaided KKP peptide inhibited T-L and PGPH peptidases of latent 20S proteasome (Figure 3A, Table 2). In addition, it weakly inhibited ChT-L peptidase of the activated CP (not shown). It appears that these peptides did not constitute substrates for the CP (incubation of the peptides with the activated proteasome for 3 hrs at 37°C, products detection with MALDI-TOF). Since both elements: the activation loop and the C terminus act together to stimulate the CP we attempted to recreate such conditions by the simultaneous exposure of the CP to both types of peptides. We found that when 20S proteasome was incubated with a mixture of KKP and IPK (0.5 μM each), the post-hydrophobic activity was stimulated up to about 50% (Figure 3B). Although this effect was at least an order of magnitude weaker than the activation observed with PA28αβ complex, it demonstrated that the peptides could productively interact with proteasome even without a context of the native activator complex. Surprisingly, a similar small but reproducible stimulating effect was achieved with the mixture of the loop peptides alone (IPR+IPK; Figure 3B). Since no additional activation was noted after an addition of KKP peptide to such a mixture we concluded that the loop peptides albeit poorly could recreate actions of the native PA28αβ complex. It has been shown previously that the heteromeric complex is much better activator of CP than homomeric PA28α ligand. Although the mechanism of activation still remains unclear it could be speculated that the β subunits stabilize the complex and affect a mode of binding of α subunits.
Figure 3.
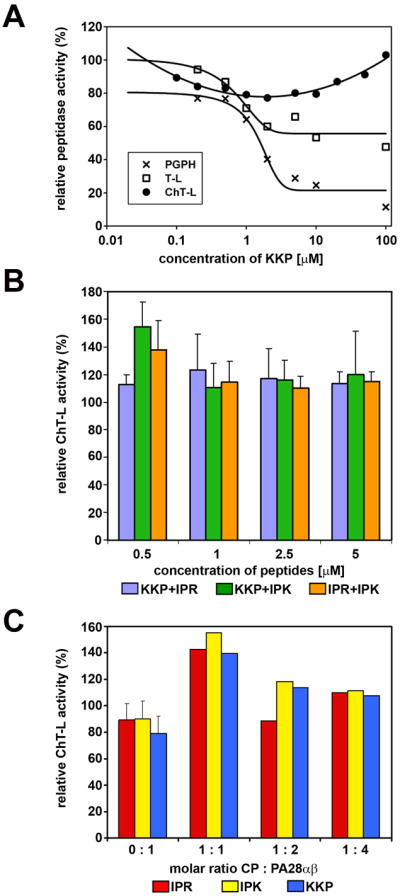
The PA28 derived peptides affected selected peptidase activities of human 20S proteasome and its interactions with native PA28αβ activator. In all graphs 100% of the relative effect (Y axis) refers to activity (nanomoles of released AMC product per mg of CP protein per second) recorded with a solvent (DMSO) without the peptides added to reaction mixture.
(A) KKP peptide inhibited the T-L (I50=0.35 microM) and PGPH (I50=1.42 microM) peptidase activities of latent CP. Ch-L activity remained unaffected both for latent (see graph) and SDS-activated proteasome (for comparison: 107% of activity with 1 mM peptide, 115% with 2 mM, 108% with 5 mM and 106% with 10 mM). The standard deviations (SD bars are omitted from graphs for clarity) for experimental points are in the range of 6% to 13% (n = 3 to 7 experiments). IPR and IPK peptides did not substantially affect any of peptidase activities (not shown).
(B) PA28 peptides combined in pairs showed a weak stimulating effect on the ChT-L activity of CP. (mean ± SD; n = 3 to 5).
(C) Pre-treatment of CP with activator-derived peptides at 1 mM concentration stimulated activation of CP by native PA28αβ complex but only if the proteins were present in a 1:1 (CP : PA28αβ) molar ratio. Under the conditions used, PA28αβ activated CP approximately 2,5x, 5x and 8x (with CP:PA28αβ molar ratio of 1:1, 1:2 and 1:4, respectively). The respective activity of CP+PA28αβ was set as 100%. When SD bars are added, the data represent means from 6 or 7 experiments. When SD bars are omitted, the data are representative of two experiments with activities differing by 6% to 16%.
Table 2.
Summary of effects of the peptides on proteasome peptidase activities. Refer to Figures 3 and 4 for the % of activity changes and the I50 parameters (peptide concentrations required to achieve 50% inhibition of the maximal reaction rate of substrate digest by 20S proteasome).
| KKP | Tat1 | Tat2 | |
|---|---|---|---|
| ChT-L | No effect | Activation | Activation |
| ChT-L+SDS* | No effect | Inhibition | Inhibition |
| T-L | Inhibition | No effect | No effect |
| PGPH | Inhibition | Inhibition | Inhibition |
measured in the presence of 0.005% SDS
Interestingly, when 20S proteasome was individually pretreated with one of these peptides a similar 50% level of further stimulation was observed after an addition of PA28 but only when the complexes were prepared in a 1:1 molar ratio (Figure 3C). The result may support possibility that the peptides bound to one of the α faces of proteasome facilitate binding of PA28 to the same or opposite face likely by changing its conformation. In fact, it has been observed previously that in PA26 complexed with the CP not all seven binding sites are simultaneously occupied by the C-termini [16]. Therefore, it is possible that the binding of PA28 to the CP proceeds through a positive cooperative process and pretreatment with the peptides improves the initial binding affinity. At higher ratios though they interfered with the complex formation likely by simultaneous engagement of too many binding sites on the CP.
Peptides derived from HIV-1 Tat protein are noncompetitive/uncompetitive inhibitors of CP
HIV-1 Tat protein is known as a noncompetitive inhibitor of 20S proteasome. We found that both the Tat peptides affected proteasome activity but to a surprisingly different extent. Analysis of reaction rates of digest of a substrate for ChT-L activity in the presence of the Tat peptides indicates that Tat1 is a mixed type inhibitor of the activated proteasome with noncompetitive and uncompetitive components (Table 3). Likely, this peptide binds to more than one type of receptors on proteasome. In contrast, Tat2 interacts with the CP as a classic noncompetitive inhibitor (Table 3) since only value of Vmax was lowered in the presence of the peptide, whereas the Km value remained unchanged. Interestingly, Tat1 was a much stronger inhibitor of the SDS-activated proteasome than Tat2 (Table 2, Figure 4A).
Table 3.
Kinetic constants of Suc-LLVY-MCA hydrolysis by the activated 20S proteasome in the presence of the Tat peptides.
| Km [μM SucLLVY-MCA] | Vmax [μM MCA/mg protein/hr] | |
|---|---|---|
| control | 76.1 | 5.5 |
| Tat1 | 95.5 | 3.5 |
| Tat2 | 77.2 | 3.7 |
Figure 4.

Tat peptides affected selected peptidase activities of human 20S proteasome and its interactions with native PA28αβ activator. In all graphs 100% of the relative effect (y axis) refers to activity (nanomoles of released AMC product per mg of CP protein per second) recorded with a solvent (DMSO) without the Tat peptides added to reaction mixture.
(A) top left: Tat peptides exhibited a complex influence on ChT-L activity of CP. At lower concentrations Tat2 peptide, and to a much lesser extent Tat1, activated latent CP. Higher concentrations of the peptides inhibited the ChT-L peptidase. In contrast, this activity was inhibited by the whole range of peptides concentrations when CP was activated by 0.005% SDS (top right; I50=0.16 microM with Tat1 and 0.37 microM with Tat2). T-L activity of latent CP was only weakly affected by the peptides (bottom left). PGPH activity of latent CP was similarly inhibited by both the peptides (bottom right; I50=0.063 mM with Tat1 and 0.075 mM with Tat2). The data points are averages from 3 to 7 experiments, with standard deviations in the range of 7% to 15%.
(B) Tat peptides interfered with activation of ChT-L peptidase by native PA28αβ in a dose-dependent manner. Molar ratio of CP to PA28αβ was 1:3. (mean ± SD; n = at least 3)
Both the Tat peptides substantially inhibited PGPH activity of the latent CP but they poorly performed as inhibitors of T-L activity (Figure 4A, Table 1). Surprisingly, Tat2 activated ChT-L activity of the latent CP in relatively narrow range of peptide concentrations reaching the maximum effect at about 1 μM concentration. A similar but much weaker effect at much lower concentrations was also noted for Tat1 (Figure 4A). The mechanism of the activation remains to be established. However, we speculate that the peptides may selectively activate CP either by binding to the α face and opening of the central channel or interacting with internal chambers of the enzyme. Since the Tat peptides are strongly positively charged, likely their interactions are carried out via acidic patches present on the α face of 20S particles.
Similarly to the full-length protein, the Tat peptides competed with PA28 activator for binding sites on the α face diminishing the activation effect of the complex. Tat1 was a better competitor than Tat2 since it eliminated the activation of the CP by about 70% at lower peptide concentrations whereas the maximal effect of Tat2 reached only 50% (Figure 4B). We did not detect further reproducible response of the CP activity beyond purely additive effect when the Tat peptides and any of the PA28 peptides were simultaneously incubated with 20S proteasome (not shown). The result may suggest that these peptides utilize distinct binding domains and/or separate mechanisms to influence proteasome activity. Interestingly, the most recent crystal structure data suggest that distinct structural mechanisms may inflict gate opening and proteasome activation by PA26 and by 19S cap homologue, the PAN complex [20].
CD and FTIR detected a high content of random coil structure with some propensity for PP2 formation in the investigated peptides
To better understand structural properties of the designed peptides that may influence their biological performance we initially used CD and FTIR spectroscopy. Collected CD spectra indicated that IPR and IPK (activation loops) shared a similar conformational composition whereas the remaining peptides constituted a separate group (Figure 5). The CD spectra of IPR and IPK peptides presented a minimum at about 200 nm and a small shoulder at 223 nm implying predominantly a random coil structure [21]. In contrast, Tat1 and Tat2 peptides showed a minimum at 197 nm and a well-developed slightly positive band at about 220 nm. Such spectral properties point to a left-handed 31 helical or polyproline type 2 (PP2)structure [22]. To confirm the presence of a PP2 structure we also recorded CD spectra in a range of temperatures and detected a clear temperature dependence of peak intensities for both Tat peptides. Since the intensity of negative and positive band decreased gradually with the raising temperature forming an isodichroic point at about 206 nm (Figure 5 insert), we concluded unambiguously that Tat peptides were locally ordered with a substantial content of the PP2 conformation [23]. In turn, the main minimum of KKP spectrum was blue shifted to about 190 nm and more shallow than that recorded for the Tat peptides. However, similarly to the Tat spectra it also displayed a small positive ellipticity at 220 nm. The shape of this spectrum was symptomatic for mainly an unordered structure of KKP but with some admixture of the PP2 conformation.
Figure 5.

Far-UV CD spectra of KKP, IPK, IPR, Tat1 and Tat2 peptides shown in molar residue ellipticity (MRE) units. Inset: temperature dependence of Tat1 MRE was indicative for the presence of PP2 conformation. Spectra were recorded in 0.02 M phosphate buffer pH 6.7 or in water (Tat peptides).
To further analyze secondary structure of the peptides we employed FTIR spectroscopy. As a structural probe we used the amide I band that represents primarily the C=O stretching vibration of the amide groups absorbing in the region of 1600-1700 cm-1 [24]. The complex amide band envelope formed by superposition of several peaks was separated into components by calculating second derivative of the spectra [25, 26] presented in Figure 6. One of the striking features of the spectra was the presence of a deep minimum between 1640 and 1650 cm-1 that could represent the superposition of signals from an α-helix and unordered segments [27]. Likely only the latter contributed to this minimum since the CD spectra did not demonstrate any evidence for the presence of helical structure. A second minimum found in all recorded spectra around 1670 cm-1 is usually assigned to turns but the guanidine side chain stretching vibration manifesting at 1673 cm-1 also may contribute to it [28]. That possibility was corroborated by the observation that IPK peptide lacking in its sequence an Arg residue revealed in this region a broad line without a clear minimum. Additionally, spectra of KKP as well as IPK and IPR peptides showed a small dip around 1690 cm-1 usually interpreted as a high-frequency component of a β-sheet. However, taking into account the absence of the β-sheet low-frequency band, typically more intense than the high one, this minimum should be rather ascribed to turns. Tat2 peptide was the only one not displaying connected to random coil minimum at 1650 cm-1, instead of which it reveals two minima at 1665 and 1620 cm-1. The former was assigned to turns, whereas the latter was treated as a spectral feature of the hydrated PP2 structure [29]. These results indicated that the conformation of Tat2 was more ordered than the rest of the peptides and mainly composed of PP2 structure and a kind of a stable turn. A minimum at 1620 cm-1 also appeared in Tat1 and KKP spectra confirming the CD observations that these three peptides were locally organized in the PP2 conformation.
Figure 6.

FTIR second derivative spectra of the investigated peptides prepared as a hydrated film on CaF2 plates.
The cumulative results obtained with CD and FTIR spectroscopy indicate that most of the analyzed peptides exist predominantly in the random coil conformation. The PP2 structure, often associated with protein-ligand interacting sites [30] was detected in Tat1, Tat2, and KKP peptides. We speculate that the flexibility of this type of the structure together with a high content of basic residues in their sequences could be linked to the biological activity of the Tat peptides.
Stable turns are present in activation loops and Tat peptides
The NMR detected proton chemical shifts of the investigated peptides are summarized in Tables 1S-4S (Supplementary Materials). The NMR spectra of KKP displayed cross peaks only for a single conformer with all trans peptide bonds. In turn, the spectra of IPK, IPR, Tat1 and Tat2 demonstrated two distinct sets of proton resonances arising from equilibrium between two conformations. We suggest that their presence may reflect the cis/trans transitions of a peptide bond involving the prone to isomerization Pro residue at position 2 (IPK and IPR) or at position 11 (Tat1 and Tat2). The contributions of more populated conformer were about 75% and 95% for IPK and IPR, respectively, and about 90% each for Tat1 and Tat2. All peptide bonds are trans in the major conformations of IPK and IPR, what was unambiguously verified by Hα(i)-HN(i+1) and Hα(i)-Hδ(i+1) cross peaks of the Pro residues. Comparison of the fingerprint regions of TOCSY spectra of IPR and IPK (Figure 7) showed that the substitution of Arg3 in IPR with Lys3 in IPK and Asp8 in IPR with Asn8 in IPK had a minimal impact on amide proton chemical shifts. The main differences in the amide proton position were detected in the region between residues 3 and 5 pointing at certain variations within a secondary structure of the N-terminal part of the molecules.
Figure 7.
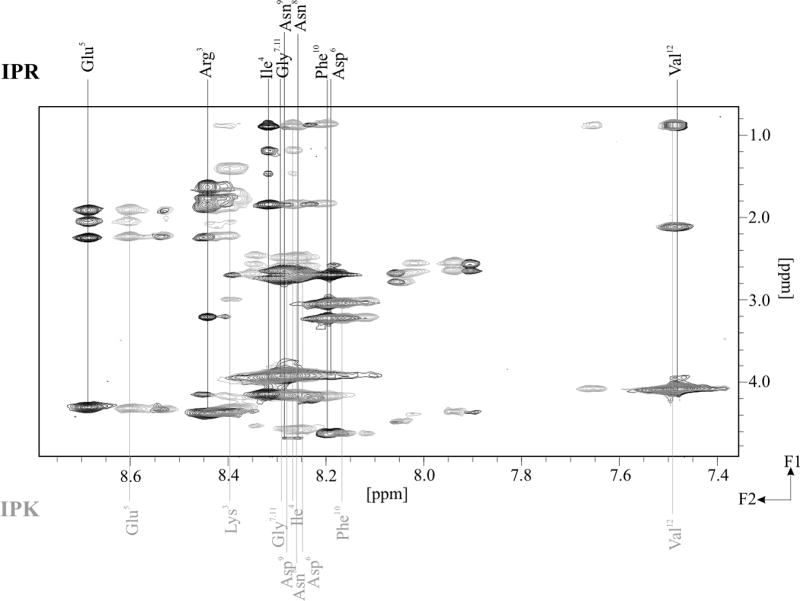
Comparison of fingerprint regions of TOCSY spectra of IPK and IPR measured with mixing time 70 ms.
Since many cross peaks of Arg and Lys residues in Tat1 and Tat2 peptides were superimposed it was difficult to achieve a complete assignment of proton signals with high confidence. The most serious ambiguity concerned Arg residues at positions 5, 6 and 9 of Tat1 and 4, 5 and 8 of Tat2 (Tables 3S and 4S, Supplementary Materials) since the lack of appropriate sequential ROE connectivity made impossible to distinguish these residues. Therefore, we attempted to assign proton chemical shifts based on a spectrum of the Tat protein fragment with a very similar sequence (YGRKKRRQRRRP), already described in the literature [30]. The Hα(10)-Hδ(11) connectivity, confirming the trans Arg10-Pro11 and Asp10-Pro11 peptide bonds for Tat1 and Tat2, respectively, was observed in the ROESY spectra.
PP2 structure is especially plausible in Tat peptides
Positions of chemical shifts depend on backbone dihedral angles and local chemical environment. Therefore, random coil chemical shifts are commonly used to detect secondary structure elements in proteins and peptides in chemical shift index calculations [31]. To decipher the secondary structure of the investigated peptides, the deviations of the chemical shift values ΔδHα and ΔδHN were used (Figure 8). The sequence correction for the statistical-coil chemical shifts of the Hα and HN, proposed by Schwarzinger et al. was implemented [32]. Additionally, corresponding temperature corrections were computed for the HN statistical-coil chemical shifts [30].
Figure 8.
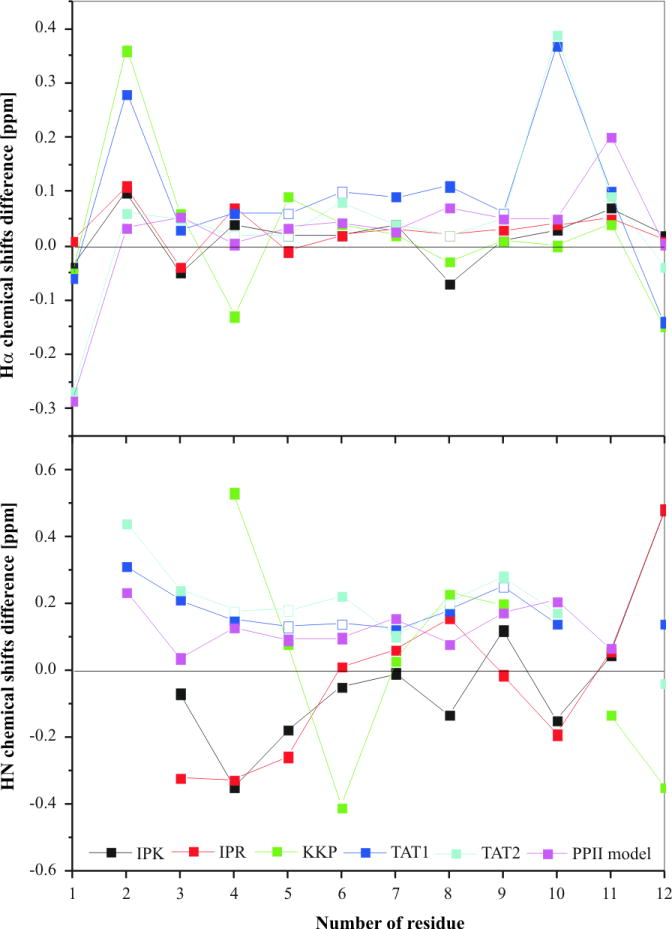
Deviations from random-coil chemical shifts for the Hα and HN resonances of investigated peptides and a model 31-helix (PP2) peptide (YGRKKRRQRRRP). A negative chemical shift difference indicates an upfield chemical shift compared to the random coil values. The deviations were not calculated for Nle10 of KKP. The deviations of Hα and HN chemical shifts of Arg5,6,9 and Arg4,5,8 of Tat1 and Tat2 (Tables 3S and 4S, Supplementary Materials), respectively, which are not unambiguously assigned, exist in random coil range (empty square).
The Hα chemical shifts appeared to match closely their random coil values with the subsequent Hα chemical shift difference (ΔδHα) typically lower than 0.1 ppm (Figure 8). However, ΔδHα values less than 0.05 ppm were also observed for a 12-residue model of 31-helix (PP2) peptide [30]. Therefore, the presence of PP2 structure, especially in Tat1 and Tat2 peptides, which displayed a high sequence similarity to the above model peptide, cannot be excluded. In turn, ΔδHN values of the residues in the middle part of Tat1 and Tat2 were in the range of 0.12-0.21 ppm placing them between the values characteristic for PP2 (0.08 ppm) and β-strand (0.29 ppm). However, the lack of HN(i)-HN(i+1) cross peaks in the ROESY spectra, strongly supported presence of the PP2 structure, devoid of internal hydrogen bonding [33]. The ΔδHN values for the remaining peptides did not corroborate the presence of PP2 structure since they fluctuated within wider limits. KKP is characterized by high values of the vicinal coupling constants, 3JHNHα, which may imply the existence of a β-sheet structure or rather an extended conformation similar to a single strand of a β-sheet.
MD simulations confirm the propensity for PP2 and turn structures in selected peptides
In order to test influence of limited number of NMR restraints on the final conformation (Table 4), the MD simulations without any NMR restraints were carried out. Comparison between the results of the MD simulations without restraints and the ones with TAV showed significant differences in the folding of the peptides. The largest differences were observed in KKP and Tat1. Namely, the analysis of the structures obtained in the last 800 ps of MD simulations without NMR restraints revealed presence of the regular secondary structure in both the peptides (Table 5S, Supplementary Materials). KKP adopted a short antiparallel β-sheet between Arg4 and Gly5 in one and Ile11 and Tyr12 in the other strand. The β-turns, centered in residues 8, 9 and 9, 10 connected the two strands. In turn, an α-helical structure was detected between residues 2 and 7 of Tat1. However, these properties were not preserved after time-averaged NMR distance so dihedral angle restraints were included (Table 4).
Table 4.
Summary of the NMR-derived constraints used for MD with TAV calculations
| Interproton upper distance bounds from ROEs | Peptide |
||||
|---|---|---|---|---|---|
| IPK | IPR | KKP | Tat1 | Tat2 | |
| total number | 51 | 49 | 57 | 24 | 34 |
| intraresidue | 40 | 38 | 40 | 18 | 28 |
| i, i+1 | 11 | 10 | 16 | 6 | 6 |
| i, i+2 | 0 | 1 | 1 | 0 | 0 |
| Total dihedral angle restraints (phi) | 9 | 8 | 8 | 3 | 3 |
The structures of the peptides obtained in the last 800 ps of MD simulations with TAV are shown in Figure 9 and are aligned to their first coordinates using Cα atoms within the entire molecules. Scatter plots of the conformational states of each amino acid of all the conformations of the investigated peptides generated with TAV MD simulations are presented in Figure 10. It can be seen that the conformational states were clustered mainly around two or three vertical lines at about ϕ=-140° and -60° for IPK, IPR and Tat2 and at about ϕ=-140°, -60° and 60° for KKP and Tat1. The first cluster contained mainly extended states, whereas the second cluster included the PP2 and the type I, II and III of β-turn states. Finally, the third cluster comprised the type I’, II’ and III’ of β-turn states.
Figure 9.
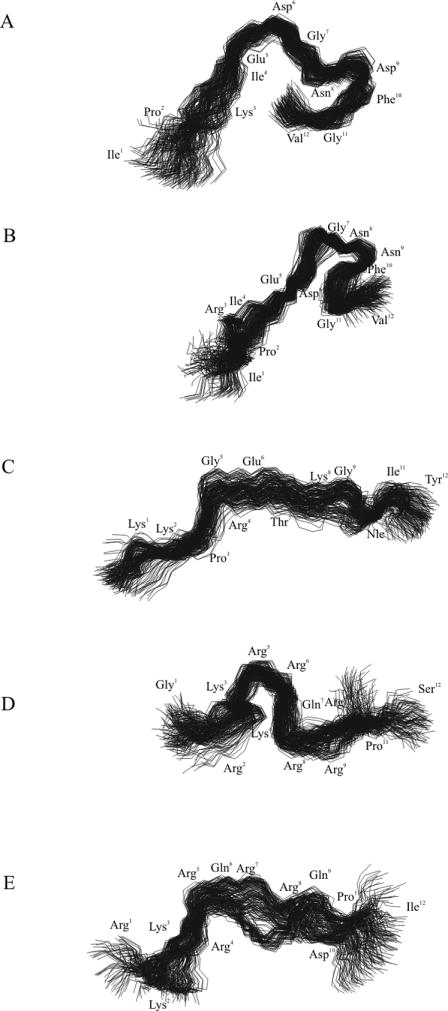
Superposed conformations of the investigated peptides obtained in the last 800 ps of MD simulations with time-averaged restraints (TAV) (A) IPK, (B) IPR, (C) KKP, (D) Tat1 and (E) Tat2. RMSD=1.275, 1.030, 1.476, 1.876 and 1.988 Å for Cα atoms of IPK, IPR, KKP, Tat1 and Tat2, respectively.
Figure 10.
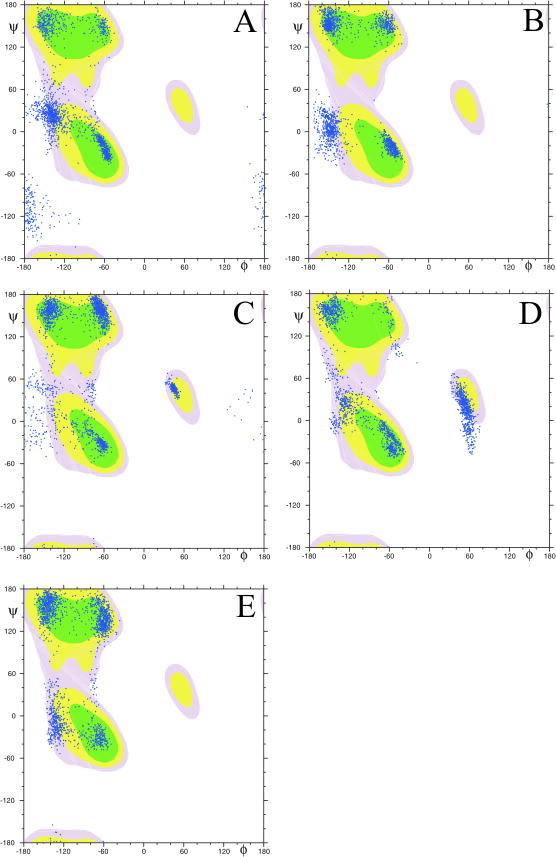
Scatter plots of the dihedral angles (ϕ and ψ of all residues and all conformations of (A) IPK, (B) IPR, (C) KKP, (D) Tat1 and (E) Tat2 calculated by MD simulations with time-averaged restraints (TAV) in the last 800 ps.
In the case of IPK and IPR, the βI- or βIII-turns appeared in a C-terminal part of molecules and they were stabilized by appropriate hydrogen bonds (Table 5). Interestingly, the analysis of three-dimensional structures of IPR revealed the existence of an Asx-turn that was cyclized by a hydrogen bond between the amide proton of Phe10 and an oxygen atom of the Asn8 side chain. The Asx-turn is a local conformation of peptide chains, which is topologically equivalent to a β-turn [34]. A salt bridge between a negatively charged side chain of Glu and a positively charged side chain of Arg residue constituted another characteristic structural element of IPR. Comparison of the averaged structures of IPR and IPK (Fig. 2Sa, Supplementary Materials) showed different location of the corresponding side chains.
Table 5.
Conformational characteristics of the investigated peptides found on the basis of calculated structures obtained in the last 800 ps of MD with TAV.
| Peptide | Structure | Hydrogen Bond | RMSDCα[Å] | Rgheavy[Å] |
|---|---|---|---|---|
| IPK | 5,6 β IV | δHN2 8-CO12 | 1.275 | 7.2 |
| 8,9 β I or III | HN10-CO7 | |||
| 9,10 β I | HN11-CO8 | |||
| IPR | 6,7 β IV | HN6,7-εCO5 | 1.030 | 6.8 |
| 9,10 β I or III | HN10-δCO8 | |||
| 8-10 Asx | HN11,12-CO8 | |||
| Salt bridge Glu5-Arg3 | ||||
| KKP | - | Salt bridge Glu6-Arg4 | 1.476 | 10.2 |
| Tat1 | 2,3 β II or III’ | HN6,8-CO3 | 1.876 | 7.5 |
| 4,5 β I or III | HN7-CO4 | |||
| 5,6 β I | ||||
| 7,8 β III’ | ||||
| Tat2 | - | Salt bridge Asp10-Arg8 | 1.988 | 9.3 |
In contrast with IPK and IPR, Tat1 possesses reverse structures within almost the entire backbone. The β-turns at position 4,5 and 5,6 were mostly of the type I. Both β-turns were hydrogen bonded. In turn, KKP and Tat2 adopted rather extended conformation. However, the middle part of both peptides might assume the PP2 structure. The comparison of average structures of KKP and Tat2 showed a high degree of similarity within residues 4-8 (RMSD4-8Cα = 0.953 Å) (Fig. 2Sb, Supplementary Materials). The radii of gyration (Rg) averaged within the last 800 ps of MD simulations with TAV amount 10.2 Å and 9.3 Å for KKP and Tat2, respectively. Rather surprising is a comparison of the conformations of Tat1 and Tat2. Both peptides possess a high sequence similarity, yet they exhibited quite different conformational preferences (Fig. 2Sc, Supplementary Materials).
The presence of secondary structure motifs correlates with peptides ability to directly affect CP activities
Each of the five analyzed peptides influences in vitro performance of the core proteasome particle. Structure analysis pointed at random coil as their predominant conformation in solution. However, three of the peptides: Tat1, Tat2, and KKP (PA28α C-terminus), can attain stable secondary structure motifs, most notably PP2, often found in ligand interacting interfaces. Interestingly, these are the peptides which directly affect catalytic activities of CP. The effects range from noncompetitive inhibition to activation of selected peptidases of the latent core particle. The presence of stable turns in Tat2 seems to correlate with its ability to activate the ChT-L peptidase of the proteasome. A similar correlation was detected for PR peptides by the authors (unpublished observations). Both Tat and PR peptides display high content of basic residues, another common trait in these proteasome-activating ligands. Interestingly, the same peptide can activate one of the peptidases and inhibit another one, or inhibit two out of three peptidases, as is the case of Tat peptides and KKP, respectively. Such diversification of effects of 20S ligands has been observed before. For example, the PA28γ homoheptamer activates only T-L peptidase and fails to affect or inhibits the other peptidases [6,13]. Moreover, the activation-inhibition pattern displayed by PA28γ complex can be switched to the activation pattern resembling PA28αβ actions by a single amino acid residue mutation in PA28γ subunit [13]. The fact that the mutated residue is not located in AL or C-terminal region underscores the significance of the structural context of proteasome-interacting regions. In the light of these data our result of the inhibitory effects of the activator-derived KKP peptide is less surprising and may be explained by conformational differences between the free peptide fragment and the same fragment within the subunit. There is one more important implication of the observed varied effects of proteasome ligands interacting with α face. The effects strongly suggest that the changes in peptidase activities are allosteric in nature rather than a consequence of physical occlusion of the gate resulting in inhibition, or stabilization of the open gate resulting in unobstructed access of the substrates to the catalytic chamber and activation. Since the peptides affect each peptidase activity in such a distinct manner, they may substantially affect the specificity of proteasome, a valuable notion when application of these or similar small-molecule ligands as regulators of proteasome is considered. Besides direct influence on the CP catalytic properties, the Tat peptides, or homologous peptide-like ligands, may serve as intracellular competitors preventing the 11S activator binding.
The random-coil peptides derived from activation loops can recreate effects of the 11S activator complex
Even if the single activation loop peptides did not significantly and reproducibly affect peptidase activities of CP, their effects are apparent in mixtures with the C-terminal peptide, with each other or with the full PA28αβ complex. Under such conditions the peptides inflict a mild activating effect on the ChT-L peptidase, reminiscent of the much stronger actions of PA28αβ. We speculated above that the positive cooperative effects resulting from occupation of multiple binding sites on α faces contribute to the activation effects. Our observation that random coil is the predominant conformation of activation loop peptides suggests intriguing possibilities. It is plausible that productive interactions of these peptides with the α face are due to a conformational selection of low-incidence peptide conformations. Thus, the primary structure of loop-derived peptides would implicate the interactions with α face under favorable binding conditions. Future studies should determine, whether the incorporation of rigid structural elements into the peptide sequences would increase the peptide - α face affinity and enable stronger modulation of the proteasome activity.
MATERIALS AND METHODS
Peptide synthesis
The following peptides were synthesized (Table 1): IPRIEDGNNFGV (IPR) and IPKIEDGNDFGV (IPK) that correspond to the activation loops of PA28 α/γ and β subunits, respectively; KKPRGETKGNleIY (KKP) constituting C-terminus of PA28 α subunit, GRKKRRQRRRPS (Tat1) and RKKRRQRRQDPI (Tat2) derived from HIV-1 Tat sequence. All the peptides were synthesized on TentaGel R RAM resin (Rapp Polymere) using Fmoc strategy with continuous-flow methodology (9050 Plus PepSynthesizer, Millipore). Crude peptides were purified by reversed-phase high-performance liquid chromatography (RP-HPLC) using a C8 semi-preparative Kromasil (21 × 250mm, 7μm; Eka Chemicals) column. The peptides purity was determined with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) and analytical RP-HPLC using a C18 XTerra column (4.6 × 150mm, 5μm, Waters) and a 30 min. linear gradient of 5-80% acetonitrile in 0.1% aqueous trifluoroacetic acid as a mobile phase.
Analytical data were as follows: IPK: RT = 11.9 min, MS: [M] = 1303.3, calc. 1303.4; IPR: RT = 12.0 min, MS: [M] = 1330.5, calc. 1330.5; KKP: RT = 10.4 min, MS: [M] = 1389.7, calc. 1389.7; Tat1: RT = 5.8 min, MS: [M+H]+ = 1581.8, calc. 1580.9; Tat2: RT = 7.8 min, MS: [M] = 1637.1, calc. 1637.0.
Biochemical assays
Influence of the peptides on the catalytic properties of 20S proteasome was tested with housekeeping CP purified from human erythrocytes (BioMol Inc.) as previously described [14]. Latent CP or CP activated with 0.005% sodium dodecyl sulfate (SDS) at a final concentration of 3 nM was utilized. The following model fluorogenic peptide substrates (Bachem, Calbiochem) were employed to determine the activity of three proteasomal peptidases: succinyl-LeuLeuValTyr-4-methylcoumarin-7-amide (SucLLVY-MCA, for measuring ChT-L activity), butoxycarbonyl-LeuArgArg-MCA (BocLRR-MCA, T-L activity), and carbobenzoxy-LeuLeuGlu-β-naphthylamide (Z-LLE-βNA, PGPH activity). All the assays were performed in the 96 well plate format using a reaction volume of 100 μL. Tests were carried out in a reaction buffer (50 mM Tris/HCl, pH 8.0). The ChT-L activity of the proteasomes tested in our studies was consistently upregulated up to 10–fold by treatment with 0.005% SDS. Stock solutions of substrates (Bachem and Calbiochem) and the designed peptides were dissolved in dimethyl sulfoxide (DMSO). To avoid unspecific effects on the CP the content of DMSO never exceeded 3% of the final reaction volume. After a 10 min. preincubation at room temperature a substrate was added at 50 or 100 μM final concentration (1% of the volume). The release of an aminomethyl coumarin (AMC) group was followed by measuring fluorescence emission at 460 nm in 2 min. intervals for up to 60 min. at 37°C (Fluoroskan Ascent). Then, the peptidolytic activity was calculated as nanomoles of released AMC product per mg of CP protein per second. To test competition between the peptides and activator the purified human PA28αβ activator (BioMol Inc.) was mixed in several molar ratios with the proteasome pretreated with DMSO or the peptides, as indicated. A 5 min. preincubation followed the treatment with the activator and/or designed peptides before the substrate was added. All activity assays were repeated at least three times with different batches of proteasomes, and the variation between results remained in the 5 – 10% range. Representative results are shown in figures and tables.
Circular dichroism (CD)
CD spectra were collected with a JASCO J-815 spectrometer in the range 185-260 nm. The measurements were performed with 0.2 mg/ml peptides dissolved in 0.02 M phosphate buffer pH 6.7. CD spectra of Tat peptides were additionally collected in water. For each peptide the presented spectrum constitutes an average of two independent measurements. Results are expressed as mean residue ellipticity (MRE).
Fourier Transformed Infrared Spectroscopy (FTIR)
Transmission IR spectra were collected in duplicate using a Bruker IFS-66 spectrometer with a DTGS detector continuously purged with dry nitrogen. To obtain the peptides (TFA-free) in a form of hydrated film, water was evaporated from the samples placed on a CaF2 window with a gentle stream of N2. For each sample 240-320 interferograms were averaged at a spectral resolution of 4 cm-1. Data analysis was performed with OPUS (Bruker) and GRAMS 5.22 (Galactic Industries) programs.
Nuclear magnetic resonance (NMR)
All the peptides were at least 95% pure. IPK, IPR and KKP peptides were dissolved in KH2PO4/Na2HPO4 buffer, pH 6.8. Since Tat1 and Tat2 are not fully soluble in this buffer, they were dissolved in water (pH of the solutions 2.9 and 3.0, respectively). The spectra were recorded with a 500MHz Varian spectrometer equipped with a Performa II gradient generator unit (WFG, Ultrashims), high stability temperature unit and a 5 mm 1H{13C/15N} PFG triple resonance inverse probe head.
The 2D NMR spectra were collected at 30°C. The temperature coefficients of the amide proton chemical shifts were measured from 1D NMR spectra for the following temperatures: 22, 30, 35, 40 and 45°C. Proton resonance assignments were achieved by use of the proton-proton total chemical shift correlation spectroscopy (TOCSY) [35], the rotating-frame Overhauser enhancement spectroscopy (ROESY) [36,37] as well as the gradient heteronuclear single quantum coherence (1H-13C gHSQC) [38,39]. For each sample, the mixing time of 70 ms for the TOCSY measurements was applied. The mixing time of the ROESY experiments was set to 175 and 300 ms for IPK and IPR, respectively, and the volumes of cross peaks were picked up for the ROESY spectra with a mixing time of 300 ms.
Vicinal coupling constants, 3JHNHα, were assigned using DQF-COSY [40]. The DQF-COSY spectra were processed to enhance resolution to 1.2 Hz per point in F2. For Gly9, the two 3JHNHα coupling constants with Hα protons are equal within the error limits. In order to ensure that the measurements were not a subject to line-width errors, 3JHNHα were also measured using certain well-resolved signals in the 1D 1H spectra. The 3JHNHα coupling constant values obtained from both DQF-COSY and 1D 1H spectra were averaged.
The proton chemical shifts were referenced to the H2O frequency measured with respect to 2,2-dimethyl-2-silapentanesulfonic acid (DSS). The 13C chemical shifts were referenced to DSS according to the following relationship: 13C/1H = 0.251449530 [41]. Spectral processing was carried out using VNMR (Varian Inc., Palo Alto, CA) and analyzed with XEASY [42].
MD Simulations in aqueous solution
Molecular dynamics (MD) simulations were carried out using the AMBER force field [43]. MD calculations were initiated from random conformations, which were put into a water solution. The initial solvent configuration around each peptide was obtained by filling a cubic box with water molecules. The overall box size was enlarged by about 10 Å in each direction. Sodium (IPK and IPR) or chloride ions (KKP, Tat1 and Tat2) were used to neutralize the system. To equilibrate the solution density, the initial simulations were carried out at 303 K, in a periodic box, until a density was close to 1.0 g/ml.
Afterwards, we used two molecular dynamics procedures. One included time-averaged (TAV) distance and dihedral angle restraints derived from the NMR spectroscopy. The other was divested an experimental information. The calculations were performed only for major conformations with all peptide bonds in trans configuration. The geometry of the peptide groups was kept fixed with the force constants f = 50 kcal/(mol × rad2). In the case of MD with TAV, the inter-proton distances were restrained with f = 20 kcal/(mol × Å2), and the dihedral angles with f = 2 kcal/(mol × rad2). During MD simulation with TAV, a 9 Å cutoff radius was chosen. The MD simulations were carried out at 303 K in a periodic box of constant volume, with the Particle-mesh Ewald (PME) procedure. The time step was 2 fs. The total duration of the run was 4 ns. The coordinates were collected every 2000th step. The conformations obtained during the last 800 ps of simulation were used in a subsequent analysis. As a result, 200 conformations for each peptide were collected.
The inter-proton distances used in TAV were calculated by the CALIBA algorithm of the DYANA program [44]. The backbone 3JHNHα coupling constants were converted to backbone torsion angle ϕ constraints according to the following rules: 3JHNHα < 6 Hz constrained the ϕ angle to the range of -90°÷-30°, 6 Hz < 3JHNHα < 8 Hz constrained to the range of -120°÷-60°, and 3JHNHα > 8 Hz constrained to the range of -140°÷-100° [45]. The accumulated results were analyzed using the Carnal and Ptraj programs constituting a part of the AMBER 8.0 package [43]. Molecular structures were drawn and analyzed with the graphic programs RASMOL [46] and MOLMOL [47].
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01 GM069819 and by William and Ella Owens Medical Foundation award (M.G.) as well as grant BW-8000-5-0449-8 (E.J.).
References
- 1.Varshavsky A. Trends Bioch Sci. 2005;30:283–286. doi: 10.1016/j.tibs.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Groll M, Bochtler M, Brandstetter H, Clausen T, Huber R. ChemBioChem. 2005;6:222–256. doi: 10.1002/cbic.200400313. [DOI] [PubMed] [Google Scholar]
- 3.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 4.Unno M, Mizushima T, Morimoto Y, Tomisugi Y, Tanaka K, Yasuoka N, Tsukihara T. Structure. 2002;10:609–618. doi: 10.1016/s0969-2126(02)00748-7. [DOI] [PubMed] [Google Scholar]
- 5.Bajorek M, Glickman MH. Cell Mol Life Sci. 2004;61:1579–1588. doi: 10.1007/s00018-004-4131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rechsteiner M, Hill CP. Trends Cell Biol. 2005;15:27–33. doi: 10.1016/j.tcb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Rock KL, York IA, Saric T, Goldberg AL. Adv Immunol. 2002;80:1–70. doi: 10.1016/s0065-2776(02)80012-8. [DOI] [PubMed] [Google Scholar]
- 8.Song X, von Kampen J, Slaughter CA, DeMartino GN. J Biol Chem. 1997;272:27994–28000. doi: 10.1074/jbc.272.44.27994. [DOI] [PubMed] [Google Scholar]
- 9.Whitby FG, Masters EI, Kramer L, Knowlton JR, Yao Y, Wang CC, Hill CP. Nature. 2000;408:115–20. doi: 10.1038/35040607. [DOI] [PubMed] [Google Scholar]
- 10.Forster A, Masters EI, Whitby FG, Robinson H, Hill CP. Mol Cell. 2005;18:589–599. doi: 10.1016/j.molcel.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 11.Knowlton JR, Johnston SC, Whitby FG, Realini C, Zhang ZG, Rechsteiner M, Hill CP. Nature. 1997;390:639–643. doi: 10.1038/37670. [DOI] [PubMed] [Google Scholar]
- 12.Borissenko L, Groll M. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 13.Tan X, Osmulski PA, Gaczynska M. Curr Med Chem. 2006;13:155–165. doi: 10.2174/092986706775197926. [DOI] [PubMed] [Google Scholar]
- 14.Gaczynska M, Osmulski PA, Gao Y, Post MJ, Simons M. Biochemistry. 2003;42:8663–8670. doi: 10.1021/bi034784f. [DOI] [PubMed] [Google Scholar]
- 15.Gao Y, Lecker S, Post MJ, Hietaranta AJ, Li J, Volk R, Li M, Sato K, Saluja AK, Steer ML, Goldberg AL, Simons M. J Clin Invest. 2000;106:439–448. doi: 10.1172/JCI9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Clawson A, Realini C, Jansen CC, Knowlton JR, Hill CP, Rechsteiner M. Proc Nat Acad Sci. 1998;95:2807–2811. doi: 10.1073/pnas.95.6.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seeger M, Ferrel K, Frank R, Dubiel W. J Biol Chem. 1997;272:8145–8148. doi: 10.1074/jbc.272.13.8145. [DOI] [PubMed] [Google Scholar]
- 18.Bayer P, Kraft M, Ejchart A, Westendorp M, Frank R, Rösch P. J Mol Biol. 1995;247:529–535. doi: 10.1006/jmbi.1995.0158. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Seifert U, Salzmann U, Henklein P, Preisner R, Henke W, Sijts AJ, Kloetzel P-M, Dubiel W. J Mol Biol. 2002;323:771–782. doi: 10.1016/s0022-2836(02)00998-1. [DOI] [PubMed] [Google Scholar]
- 20.Stadtmueller BM, Ferrell K, Whitby FG, Heroux A, Robinson H, Myszka DG, Hill CP. J Biol Chem. 2009 doi: 10.1074/jbc.C109.070425. papers in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed J, Reed TA. Anal Biochem. 1997;254:36–40. doi: 10.1006/abio.1997.2355. [DOI] [PubMed] [Google Scholar]
- 22.Sreerama N, Woody RW. Biochemistry. 1994;33:10022–10025. doi: 10.1021/bi00199a028. [DOI] [PubMed] [Google Scholar]
- 23.Chen K, Liu Z, Kallenbach NR. Proc Nat Acad Sci. 2004;101:15352–15357. doi: 10.1073/pnas.0406657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Twardowski J, Anzenbacher P. Raman and IR Spectroscopy in Biology and Biochemistry. chapter 3.1 Ellis Horwood / Polish Scientific Publishers PWN; New York – London – Toronto – Sydney – Tokyo –Singapore – Warsaw: 1994. [Google Scholar]
- 25.Krimm S, Bandekar J. Adv Protein Chem. 1986;38:181–364. doi: 10.1016/s0065-3233(08)60528-8. [DOI] [PubMed] [Google Scholar]
- 26.Surewicz WK, Mantsch HH, Chapman D. Biochemistry. 1993;32:389–394. doi: 10.1021/bi00053a001. [DOI] [PubMed] [Google Scholar]
- 27.Casal HL, Kőhler U, Mantsch HH. Biochim Biophys Acta. 1988;957:11–20. doi: 10.1016/0167-4838(88)90152-5. [DOI] [PubMed] [Google Scholar]
- 28.Venyaminov SY, Kalnin NN. Biopolymers. 1990;30:1243–1257. doi: 10.1002/bip.360301309. [DOI] [PubMed] [Google Scholar]
- 29.Elangovan S, Margolis HC, Oppenheim FG, Beniash E. Langmuir. 2007;23:11200–11205. doi: 10.1021/la7013978. [DOI] [PubMed] [Google Scholar]
- 30.Lam SL, Hsu VL. Biopolymers. 2003;69:270–281. doi: 10.1002/bip.10354. [DOI] [PubMed] [Google Scholar]
- 31.Wishart DS, Sykes BD, Richards FM. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 32.Schwarzinger S, Kroon GJA, Foss TR, Wright PE, Dyson HJ. J Biomol NMR. 2000;18:43–48. doi: 10.1023/a:1008386816521. [DOI] [PubMed] [Google Scholar]
- 33.Dyson HJ, Rance M, Hoighten RA, Lerner RA, Wright PE. J Mol Biol. 1988;201:161–200. doi: 10.1016/0022-2836(88)90446-9. [DOI] [PubMed] [Google Scholar]
- 34.Abbadi A, Mcharfi M, Aubry A, Prémilat S, Boussard G, Marraud M. J Am Chem Soc. 1991;113:2729–2735. [Google Scholar]
- 35.Bax A, Freeman R. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 36.Bothner-By AA, Stephens RL, Lee JM, Warren CD, Jeanloz RW. J Am Chem Soc. 1980;106:811–813. [Google Scholar]
- 37.Bax A, Davis DG. J Magn Reson. 1985;63:207–213. [Google Scholar]
- 38.Kay LE, Keifer P, Saarinen T. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 39.Kontaxis G, Stonehouse J, Laue ED, Keeler J. J Magn Reson Ser A. 1994;111:70–76. [Google Scholar]
- 40.Rance M, Sorenson OW, Bodenhausen G, Wagner G, Ernst RR, Wüthrich K. Biochem Biophys Res Commun. 1983;117:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 41.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. J Biomol NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 42.Bartles C, Xia T, Billeter M, Günter P, Wütrich K. J Biomol NMR. 1995;5:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 43.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER 8. University of California; San Francisco: 2004. [Google Scholar]
- 44.Güntert P, Mumenthaler C, Wüthrich K. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 45.Pardi A, Billeter M, Wüthrich K. J Mol Biol. 1984;180:741–751. doi: 10.1016/0022-2836(84)90035-4. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez-Ferrer A, Nunez-Delicado E, Bru R. Trends Biochem Sci. 1995;20:286–288. doi: 10.1016/s0968-0004(00)89046-5. [DOI] [PubMed] [Google Scholar]
- 47.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:52–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.