

Abstract
In response to growth factors, class IA phosphoinositide 3-kinases (PI3Ks) phosphorylate PtdIns(4,5)P2, converting it to PtdIns(3,4,5)P3 to activate protein kinase B/Akt. This is widely reported to promote tumorigenesis via increased cell survival, proliferation, migration and invasion and many tumor types, including colorectal cancer, exhibit increased PI3K signaling. In order to investigate the effect of inhibiting PI3K and as an alternative to the use of small molecular inhibitors of PI3K with varying degrees of selectivity, HT29 and HCT116 colorectal cancer cells bearing mutant PIK3CA were generated that could be induced with doxycycline to express synchronously a dominant negative subunit of PI3K, Δp85α. Upon induction, decreased levels of phosphorylated PKB were detected, confirming PI3K signaling impairment. Induction of Δp85α in vitro reduced cell number via accumulation in G0/G1 phase of the cell-cycle in the absence of increased apoptosis. These effects were recapitulated in vivo; HT29 cells expressing Δp85α and grown as tumor xenografts had a significantly slower growth rate upon administration of doxycycline with reduced Ki67 staining without increased levels of apoptotic tissue biomarkers. Furthermore, in vitro Δp85α expression did not sensitize HT29 cells to oxaliplatin- or etoposide-induced apoptosis, irrespective of drug treatment schedule. Further analysis comparing isogenic HCT116 cells with and without mutation in PIK3CA showed no impact of the mutation in either proliferative or apoptotic response to PI-3K inhibition. These data demonstrate in colorectal cancer cells that PI3K inhibition does not provoke apoptosis per se nor enhance oxaliplatin- or etoposide-induced cell death.
Keywords: PI3-kinase, Colorectal Cancer, Cell-Cycle Arrest, Apoptosis, Oxaliplatin
Introduction
The phosphoinositide 3-kinases (PI3K) family of lipid kinases phosphorylate inositol rings on the D3 position (1). The most thoroughly studied class of PI3K are class IA PI3Ks which are constitutive heterodimers composed of a catalytic subunit, p110, bound to a regulatory subunit, p85 (2). Upon receptor tyrosine kinase (RTK) activation class IA PI3Ks are activated primarily by the recruitment of the p110 subunit to the RTK or RTK associated adaptor molecules, by the p85 subunit via recognition of specific phospho-tyrosine residues. This causes the re-localization of the heterodimer to the plasma membrane (2), in close proximity to its substrate, whilst potentially also removing allosteric inhibition of p110 by p85 (3, 4). Once activated, the class IA PI3Ks substrate phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) is converted into the secondary messenger PtdIns(3,4,5)P3 to activate many downstream kinases, most notably protein kinase B (PKB, also know as Akt) (5). The PI3K/PKB signaling network has been reported to promote cell survival, proliferation and tumor growth, as well as enhancing angiogenesis and migration, all important factors in tumorigenesis, suggesting a role for PI3K signaling in cancer (2). Indeed, numerous elements of the PI3K/PKB network are mutated, up-regulated or down-regulated in several tumor types, all leading to increased PI3K signaling. The most well studied alteration is the down-regulation or mutation of PTEN, the antagonistic phosphatase to class I PI3K, which leads to an increase in levels of PtdIns(3,4,5)P3 and consequently activation of PKB (6, 7). More recently activating mutations have been discovered in PIK3CA, the gene that encodes the class IA catalytic subunit p110α, in numerous cancers including lung, breast and colorectal cancer (CRC), (8). Furthermore, amplifications in the genes encoding p110α, p85α and PKB have been described and there is recent evidence of an activating mutation in PKBα in breast, colorectal and ovarian cancers (9). In addition to this direct evidence of deregulation of the PI3K/PKB network, it is well documented that RTKs, such as EGFR, can be aberrantly activated in various cancers, leading to activation of the PI3K/PKB network (10). Indeed, many of the most novel mechanism based cancer therapeutics, such as Gefitinib, Cetuximab and Trastuzumab, act through the inhibition of aberrantly activated RTKs, one effect of which will be to reduce signaling through the PI3K/PKB network.
The overwhelming evidence of PI3K/PKB involvement in tumorigenesis means that inhibition of this signaling network is an attractive and tractable avenue of investigation for pharmacological intervention and there are several small molecule inhibitors (SMIs) of PI3Ks that have been developed and historically used as pharmacological tools with which to study the effect of PI3K inhibition. The most widely used PI3K inhibitor is LY294002, however it inhibits other members of the PI3K family (11), as well as non-PI3K family members (12). Wortmannin, another widely used PI3K inhibitor has a narrower selectivity profile than LY294002, although it also inhibits PLK1 (13). Wortmannin is also unstable in aqueous solutions (14) precluding it from chronic use. More ‘drug-like’ PI3K inhibitors are approaching or entering early clinical trials, although their specificity is only now starting to be reported (12). Therefore, to determine the effect of PI3K inhibition in CRC cells, without the confounding factor of ‘off-target’ effects associated with small molecule inhibitors, two CRC cell lines with mutant PIK3CA, HT29 and HCT116, were engineered to contain an inducible Myc-tagged dominant negative form of the regulatory PI3K subunit p85α, MycΔp85α, which lacks the domain required to interact with catalytic PI3K subunits. These models of inducible and synchronous inhibition of PI3K activity were used to examine the impact of PI3K inhibition on CRC cell survival and proliferation in vitro and in vivo. The effects of SMI of PI-3K were also compared in isogenic HCT116 CRC cells that had either mutant or wild type PIK3CA and in SW620 with wild type PIK3CA. Since PI3K signaling SMIs will most likely be used in combination drug regimes in the clinic, the effect of inhibiting PI3K was also investigated in HT29 cells in combination with oxaliplatin (a DNA platinating agent that promotes cytostasis in HT29 cells and is routinely used to treat CRC) and with etoposide (a topoisomerase II inhibitor that induces concentration and time dependent apoptosis in this cell line).
Results
Generation of HT29 inducible MycΔp85α cell lines
In order to study the effect of PI3K inhibition in CRC cell lines, a cDNA encoding MycΔp85α, a Myc-tagged version of the bovine regulatory PI3K subunit p85α lacking the internal SH2 domain required for binding to the catalytic p110 subunit, was cloned into the pTRE vector. pTRE contains a disabled CMV promoter that is only active in the presence of a tetracycline transactivator protein and tetracycline or its analogues e.g. doxycycline (dox). pTREMycΔp85α was fused with the previously described pN1pβ-actin-rtTA2S-M2-IRES-EGFP vector, which confers neomycin resistance (15) to generate a single vector, pSMVMycΔp85α. In addition to MycΔp85α, pSMVMycΔp85α encoded the improved tetracycline transactivator protein rtTA2S-M2 linked via an IRES sequence to EGFP, allowing fluorescent activated cell sorting of stably transfected clones. pSMVMycΔp85α was electroporated into HT29 cells which have been characterized extensively with regards to their PI3K signaling pathway (16, 17), are mutant for PIK3CA (P449T (18) - confirmed by sequencing (data not shown)) and have been previously used to successfully generate inducible cell lines (15). Following neomycin selection and cell sorting based on EGFP signal, 48 single HT29 cell clones were generated and screened for the inducible expression of a Myc-tagged protein by western blotting. Approximately 25% of clones showed robust induction of a Myc-tagged protein at the appropriate size of ~85 kDa; three clones were selected for further evaluation. To confirm that this Myc-tagged protein was indeed MycΔp85α parental HT29 cells and HT29 pSMVMycΔp85α clones 12, 15 and 17 (referred to as Δ12, Δ15 and Δ17) were grown in the absence or presence of dox for 24 h and the resultant cell lysates were assayed by western blotting for the level of Myc-tagged protein and p85 (Figure 1A). A Myc-tagged protein was detected only in the presence of dox in all three clones and not detected in the parental cells. Consistent with this Myc-tagged protein being functional MycΔp85α there was also an increase in the level of p85 in all three clones 24 h after dox treatment, while the level of actin remained constant across all eight samples, indicative of even protein loading. This demonstrated that the addition of dox to the three HT29 pSMVMycΔp85α clones induced the expression of MycΔp85α, however it was noticeable that the level of expression varied between the three clones, with clone Δ15 expressing the greatest amount of MycΔp85α. Therefore, to determine whether this was due to heterogeneity within cell populations, where some of the cells do not express MycΔp85α or whether there were different expression levels between the clones, IHC was carried out with an anti-Myc-tag antibody (Figure 1B). This demonstrated that in each clone all the cells expressed a Myc-tagged protein, while clone Δ15 exhibited the strongest staining intensity. Thus the differences in MycΔp85α levels observed in cell lysates were due to different expression levels between the three clones.
Figure 1. Inducible expression of MycΔp85α inhibits PI3K activity.
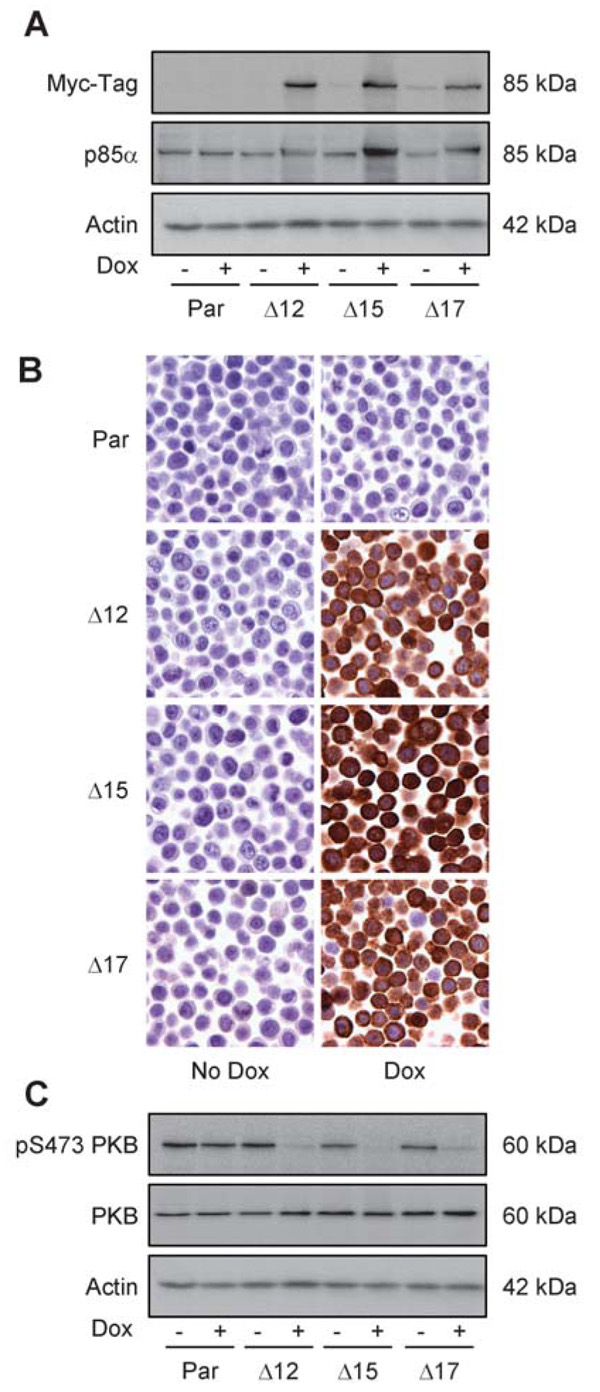
Parental HT29 cells (Par) or clones 12, 15 and 17 containing pSMVMycΔp85α (Δ12, Δ15 and Δ17) were grown in the absence (− or no dox) or presence (+ or dox) of 0.5 mg.ml−1 doxycycline (dox) for 24 h and either lysed for western blot analysis (A and C) or fixed with 10% formalin (B). A Cell lysates were assayed for the expression of Myc-tagged proteins, p85 and actin (loading control) by western blotting. B Formalin fixed cells were analyzed for the expression of Myc-tagged protein by IHC analysis. C Cell lysates were assayed for the level of PKB phosphorylated on serine residue 473 (pS473 PKB), total-PKB and actin (loading control). Data are representative images from at least three independent experiments.
Expression of MycΔp85α inhibited PI3K signaling
To determine whether expression of MycΔp85α led to the inhibition of PI3K signaling cells were treated with dox for 24 h and phosphorylation status of downstream targets of the PI3K pathway were assessed by western blotting. While the addition of dox to parental cells had no effect on phosphorylation of serine 473 of PKB (Figure 1C), the expression of MycΔp85α promoted down-regulation of PKB phosphorylation in all three clones. However, it seemed there was greater down-regulation of PKB-phosphorylation in clone Δ15 than in the other clones, consistent with the observation of increased induction of MycΔp85α in this clone. Taken together, these data demonstrated that expression of MycΔp85α leads to inhibition of PI3K signaling in HT29 cells.
Expression of MycΔp85α reduced proliferation but did not provoke apoptosis in vitro
The effect of MycΔp85α expression on cell population growth kinetics was investigated in order to explore whether MycΔp85α expression modulated cell-cycle progression and/or cell death. Cells were left untreated or treated with dox on day 0 and the increase in cell number (as measured by protein stain) with time was calculated relative to day 0. All of the clonal cell populations increased at the same rate as the parental cell line in the absence of dox and dox did not affect the population growth rate of the parental cell line (Figure 2A). In the presence of dox, cell numbers of all clonal populations increased more slowly than seen in the absence of dox, with a significant reduction in cell population observed as early as day 2. To determine the underlying mechanism of the MycΔp85α mediated reduction in cell population, the cell-cycle profile was analyzed for untreated cells and cells that had been treated with dox for 24 h. The DNA profiles of the three clones in the absence of dox were the same as parental cells and dox had no effect on the DNA profile of parental cells. The effect of MycΔp85α expression was to significantly increase the proportion of cells in the G0/G1 phase of the cell-cycle (Figure 2B – upper panel), with a corresponding decrease of cells in S phase and G2/M phase (data not shown). This suggested that the reduction in cell number observed in cells with impaired PI3K signaling was due, at least in part, to reduced proliferation associated with a delay in cell-cycle progression.
Figure 2. MycΔp85α expression prevents cell proliferation by causing a G0/G1 arrest without inducing apoptosis in vitro.

A Cells were seeded into 96 well plates and after 24 h treated with 0.5 mg.ml−1 dox or left untreated. A plate was harvested every 24 h for four days and the amount of protein in each well relative to day 0 was determined by SRB staining. B Cells were grown in the absence or presence of dox for 24 h and harvested by trypsinisation and fixed in 70% ethanol. Cells were then stained with PI and the amount of PI incorporated into cells determined by flow cytometry. % of cells in the G0/G1 stage of the cell-cycle was then calculated using Modfit LT 3.2 software. C Cells grown in the absence or presence of Dox for 24 h were harvested by trypsinisation and stained with anti-annexin V and 7-AAD. Samples were analyzed by flow cytometry and the percentage of annexin V positive, 7-AAD negative cells were calculated. D Cells were grown in the presence or absence of dox for 24 h and lysed. Lysate containing 20 μg of total protein was analyzed in duplicate on Mesoscale Discovery cleaved- and total-caspase 3 duplex plates and the percentage of cleaved caspase 3 determined. All graphs represent the mean from three independent experiments +/− standard error of the mean (S.E.M.). * p<0.05, ** p<0.01, *** p<0.001 according to two-tailed unpaired t-test compared to corresponding no dox treatment. Blots are representative examples from three independent experiments.
One mechanism whereby PI3K inhibition could lead to a G1 arrest would be by the enhanced expression of the cyclin dependent kinase (CDK) inhibitor p27, which is translated in a FKHR-dependent fashion inhibitable by PI3K activity (19). p27 expression was assessed in lysates from parental cells and clones treated with dox for 24 h. While the addition of dox to parental cells had no effect on p27 expression level, the induction of MycΔp85α led to a marked increase in p27 level (Figure 2B – lower panel). Therefore, PI3K inhibition in HT29 cells leads to a G1-delay consistent with increased expression of p27. However, while cell-cycle delay contributed to the reduction in cell number it remained possible that inhibition of PI3K might also have promoted cell death. Moreover, as PI3K activity has been widely reported to have pro-survival affects (2), the impact of MycΔp85α expression on apoptosis was investigated.
Cell-cycle profiles had not indicated any increase in cells with an apoptotic sub-G1 DNA content regardless of dox addition, suggesting that MycΔp85α expression did not cause apoptosis (data not shown). To more thoroughly test this observation, MycΔp85α expressing cells were analyzed using two different methods to detect apoptosis. Untreated and 24 h dox-treated cells were analyzed by flow cytometry for phosphatidylserine exposure and 7-AAD exclusion using 100 μM etoposide-treated parental HT29 cells as a positive control. In parental cells, the level of non-permeable cells with exposed phosphatidylserine (annexin V positive, 7-AAD negative – indicative of apoptosis – Figure 2C) and permeable cells (annexin V positive, 7-AAD positive – indicative of primary or secondary necrosis – data not shown) was negligible, regardless of dox-treatment, while etoposide-treatment caused a significant increase in both sub-populations. MycΔp85α expression had no significant effect on the amount of non-permeable-annexin V positive or permeable cells although there was a trend to suggest that MycΔp85α expression might have increased the number of non-permeable-annexin V positive cells from 0.8% to 1.6%, especially in clone Δ15. This result was verified by examining the level of cleaved-caspase 3 in cell lysates after 24 h dox treatment using a duplex ELISA based assay that detects both cleaved- and total-caspase 3. Caspase 3 cleavage was not induced in parental cells by dox treatment, while etoposide treatment significantly increased the level of caspase 3 cleavage (Figure 2D). MycΔp85α expression did not cause a significant level of caspase 3 cleavage in any of the clones, although again there is a slight (1%) increase in clone Δ15. Taken together these data suggest that PI3K inhibition for 24 h did not induce a significant level of apoptosis per se in HT29 cells.
MycΔp85α induction in HCT116 CRC cells also caused cell-cycle arrest
In order to determine whether the effect of MycΔp85α induction in HT29 cells also occurred in another CRC cell line, clones containing pSMVMycΔp85α were generated in HCT116. Like HT29 cells, HCT116 contain a mutant PIK3CA (H1047R (20, 21)). Data presented here is for HCT116 MycΔp85α clone 23 (Δ23), but similar data has also been obtained from another two clones (data not shown). Initially, the dox inducible expression of MycΔp85α was tested by western blotting for levels of Myc-tagged protein and p85 in lysates from parental and Δ23 cells grown in the presence or absence of dox for 24 h (Figure 3A – top two panels). HCT116 lysates contained an unrelated 85 kDa protein which was detected by the Myc-tag antibody observed in both parental and Δ23 cell lysates, however, there is a clear increase in the intensity of a band at 85 kDa upon the addition of dox to Δ23 cells. Furthermore, there is increased expression of protein detected by p85 antibodies which migrates slightly more slowly than endogenous p85α in Δ23 dox treated lysates. This demonstrated that MycΔp85α is induced upon addition of Dox to Δ23 cells. To determine whether MycΔp85α expression also impaired PI3K signaling, the level of phospho-PKB in the same lysates was investigated (Figure 3A – lower two panels). The addition of dox to parental HCT116 cells had no effect on the level of PKB-phosphorylation, while the addition of dox to Δ23 cells caused a clear decrease in phospho-PKB, consistent with MycΔp85α expression inhibiting PI3K activity. The effect of MycΔp85α expression on HCT116 cell population growth kinetics was assessed, using the SRB assay, and were significantly reduced in Δ23 cells in the presence of dox compared to all other groups (Figure 3B). This reduction in population growth kinetics correlated with a cell-cycle delay, as demonstrated by an increase in Δ23 cells in the G0/G1 stage of the cell-cycle after MycΔp85α induction (Figure 3C). Furthermore, MycΔp85α expression did not cause apoptosis, as assessed by annexin V / 7-AAD assay and the level of cleaved caspase 3 (Figure 3D). This suggested that in HCT116 cells, inhibition of PI3K activity lead to a reduction in cell proliferation that was caused by cell-cycle delay and not apoptosis, as seen in HT29 cells. These results were phenocopied with the relatively specific PI3K SMI PI-103 (Figure 4 described below), consistent with PI3K inhibition, and not simply an artifact of protein over-expression.
Figure 3. MycΔp85α expression inhibits PI3K signaling and causes a cell-cycle arrest in HCT116 cells.

A Parental HCT116 and clone Δ23 cells were grown in the presence or absence of 0.5 mg.ml−1 Dox for 24 h and lysed. Lysates were assayed for the level of Myc-tagged protein, p85, phospho-PKB and total-PKB by western blotting. B Cells were seeded into 96 well plates and after 24 h treated with 0.5 mg.ml−1 dox or left untreated. A plate was harvested every 24 h for five days and the amount of protein in each well relative to day 0 was determined by SRB staining. *** p<0.001 according to two-tailed unpaired t-test compared to corresponding no dox treatment. C Cells were grown in the absence or presence of dox for 24 h and harvested by trypsinisation and fixed in 70% ethanol. The cell-cycle profile was determined and % of cells in the G0/G1 stage of the cell-cycle calculated as in the previous figure. ** p<0.01 according to two-tailed unpaired t-test compared to all other groups. D Cells were grown in the absence or presence of dox for 24 h. The % annexin V +ve / 7-AAD −ve cells and the % cleaved caspase 3 was determined as in the previous figure. All graphs represent the mean from three independent experiments +/− S.E.M. Blots are representative examples from three independent experiments.
Figure 4. PI3K inhibition causes cell-cycle arrest but not apoptosis in PIK3CA wild-type cells.

HCT116 and SW620 cells (left panel) or HCT116 PIK3CA mutant or wild-type cells (right panel) were grown in the absence or presence of 1 μM PI-103 for 24 h. Cells were lysed or fixed in 70% ethanol. A Lysates were assayed for the level of phopsho-PKB and total-PKB. B The cell-cycle profile was determined and % of cells in the G0/G1 stage of the cell-cycle calculated as in previous figures. C Fixed cells were stained with DAPI and the percentage of nuclei with an apoptotic morphology were counted. D The % of cleaved caspase 3 in lysates was determined as in previous figures. Blots are representative examples of two independent experiments. Graphs represent the mean from three independent experiments +/− S.E.M. * p<0.05, ** p<0.01, *** p<0.001 according to two-tailed unpaired t-test.
PI3K inhibition mediated cytostasis was not dependent on PIK3CA mutation
The data described above pertained to cell lines that are mutant for PIK3CA. Therefore, to determine whether the cytostatic effect of PI3K inhibition occurring without apoptosis was dependent on PIK3CA mutation, the effect of inhibiting PI3K activity was compared between SW620 cells, which are wild-type for PIK3CA (18), and HCT116 cells. Moreover, as a more stringent test, the response to PI-3K inhibition of isogenic HCT116 cells expressing only wild-type or mutant PIK3CA was compared; this was achieved through targeted insertion of a disruptive DNA sequence at the start of either the wild-type or mutant allele (21).
HCT116 and SW620 cells treated with 1 μM PI-103 for 24 h and the level of pPKB and total PKB, the cell-cycle profile and level of apoptosis were analyzed. In both cell lines PI-103 treatment caused the reduction of levels of pPKB, without effecting levels of PKB (Figure 4A – left panels). It is, however, interesting to note that a higher level of pPKB was observed in HCT116 cells (mutant PIK3CA) than in SW620 cells (wild type PIK3CA). Cell-cycle analysis revealed that inhibition of PI3K signaling was associated with a significant increase in the number of cells with a 2n DNA content in both cell lines (Figure 4B), consistent with a G0/G1 cell-cycle arrest. The percentage of cells with a 2n DNA content in the absence of PI-103 was significantly lower in HCT116 cells than SW620 cells, suggesting that HCT116 spend less time in the G1 stage of the cell-cycle than SW620 cells, consistent their increased level of pPKB. PI-103 treatment had no effect on apoptosis in either HCT116 cells or SW620 cells, as assessed by apoptotic morphology (Figure 4C) and cleaved caspase 3 (Figure 4D).
While SW620 cells are PIK3CA wild-type, they carry a KRAS mutation (18), which may activate PI3K signaling. As previously described (21), Figure 4A (right hand panels) shows HCT116 cells expressing only mutant PIK3CA (Mut) have higher levels of phosphorylated PKB that those expressing only wild-type PIK3CA (WT), but in both cases 1 μM PI-103 reduced the level of phospho-PKB. PI-103 treatment caused G0/G1 arrest in both Mut and WT PIK3CA HCT116 cells (Figure 4B). Furthermore, PI-103 treatment did not cause apoptosis in either cell line (Figures 4C and D). Taken together these data suggest that the effect of PI3K inhibition (by SMI or induction of dominant negative p85α) seen in HT29 and HCT116 cells is not specific to CRC cells that contain a mutation in PIK3CA.
MycΔp85α expression was induced in vivo leading to reduced tumor growth without increased apoptosis
To determine whether the inhibition of cell proliferation and the lack of apoptosis caused by the induction of MycΔp85α were recapitulated under more physiological conditions clone Δ15 and parental HT29 cells were grown as s.c. tumor xenografts in nude mice. Once the tumors reached 300 mm3, mice were fed with either dox-impregnated-feed or control-feed. To confirm that dox feed induced the expression of MycΔp85α in clone Δ15 xenografts, mice were sacrificed three days after being switched onto dox or control feed and the presence of Myc-tagged protein expression in the xenografts was determined by IHC and western blotting (Figure 5A). Exogenous Myc-tagged protein was detected in Δ15 xenografts when the tumor-bearing animal had been fed with dox feed and not in any other xenografts. The cells in the xenografts that did not stain with the anti-Myc-tag antibody had a fibroblast-like morphology and are therefore likely to be murine tumor stroma that should not express MycΔp85α, as opposed to HT29 cells that had not induced MycΔp85α. The effect of MycΔp85α expression in vivo on tumor growth was investigated by measuring the volume of the tumors every 2-3 days (Figure 5B). The tumor xenografts derived from parental cells grew at 37 mm3.day−1 and 33 mm3.day−1 on animals fed with control or dox-laced feed respectively (no significant difference). The growth rate of Δ15 xenografts grown on animals fed with control-feed was 17mm3.day−1 (not significantly different from parental cells). However, the growth rate of Δ15 Dox-fed xenografts was reduced to 2 mm3.day−1, (p<0.05 vs. all other conditions) demonstrating that expression of MycΔp85α reduced the growth rate of HT29 xenografts. As was the case in vitro, the reduced HT29 tumor growth rate could be due to reduced cell proliferation and/or increased apoptosis. Therefore, xenografts harvested after 3 days of control- or dox-feed were stained for the proliferation marker Ki67 (Figure 5C) or cleaved PARP, which is indicative of apoptosis (Figure 5D). The number of Ki67 positive cells was not significantly different between parental xenografts from animals fed with control- or dox-feed or Δ15 xenografts fed with control feed (38%, 34% and 31% respectively). However, the number of Ki67 positive cells was significantly less in Δ15 xenografts of mice on dox-feed (23%; p<0.01 vs. all other conditions) suggesting that the reduction in tumor growth was, at least in part, due to a reduced cell proliferation. There were no significant differences between the four groups of mice with respect to the level of cleaved PARP (Figure 5D); while there is a suggestion of more cleaved PARP positive cells in the Δ15 Dox treated xenografts than the untreated xenografts, this was within the normal variation for this assay (as demonstrated by the difference between the untreated and Dox treated parental xenografts). Taken overall, the data demonstrate that induced MycΔp85α expression did not provoke apoptosis in HT29 xenografts, consistent with the lack of Δ15 xenograft regression after animals were switched onto dox-feed and with the in vitro findings.
Figure 5. MycΔp85α expression inhibits xenografts tumor growth rate and Ki67 staining but has no effect on the level of PARP cleavage.

Parental HT29 cells and clone Δ15 cells were grown as tumor xenografts on Balb/c-nude mice and tumors were measured three times a week by callipers. Once tumors reached 300 mm3 mice were switched onto feed containing dox or control-feed. Four animals were sacrificed three days after being switched onto dox/control-feed and their tumors were bisected and either snap frozen or fixed in 10% formalin. A further six mice from each group were used for tumor growth analysis. A Top Panel – Tumors were assayed for the presence of Myc-tagged proteins by IHC: Bottom Panel – Tumor lysates were assayed for the presence of MycΔp85α by western blotting. +ve = positive control of a dox induced clone Δ15 cell lysate B The mean (+/− S.E.M) of the tumor growth rate from each group of mice after the tumor reached 300 mm3. C and D Tumors were stained for Ki67 (C) and cleaved PARP (D) by IHC and the percentage of positive cells counted. Data represents the mean +/− S.E.M. * p<0.05, ** p<0.01 compared to all other groups according to two-tailed unpaired t-test.
PI3K inhibition did not enhance oxaliplatin-induced apoptosis
If PI3K inhibitors were to be approved for use in the clinic for the treatment of CRC current practice would suggest that they would be used in combination with the best available treatment. One of the drugs most commonly used to treat CRC is oxaliplatin, whose main mechanism of action is thought to be DNA damage brought about by the formation of DNA-platinum adducts (22). While expression of MycΔp85α did not enhance apoptosis per se in HT29 cells grown in vitro or in vivo it is possible that it primes for drug induced apoptosis by reducing the threshold at which apoptosis can be engaged. Therefore, cells were treated with a concentration of oxaliplatin that caused between 5 and 10% apoptosis after a 48h constant challenge (10 μM for parental, Δ12 and Δ17 cells, 3 μM for Δ15 cells), or an equivalent vehicle control, and MycΔp85α expression was either not induced (no dox), or induced 24 h before the addition of oxaliplatin (dox 1st), at the same time as oxaliplatin (tog) or 24 h after oxaliplatin (ox 1st). The apoptotic index was determined by staining nuclei with DAPI and counting the % nuclei with a classical apoptotic morphology. For all four cell lines, parental, Δ12, Δ15 and Δ17, oxaliplatin treatment caused a significant increase in the number of apoptotic cells in the absence of dox (Figure 6A). As expected, in the parental cells the addition of dox did not affect the levels of apoptosis induced by oxaliplatin treatment. Furthermore, with the exception of clone Δ15, the expression of MycΔp85α did not affect the level of apoptosis, irrespective of when it was induced relative to the oxaliplatin treatment. For clone Δ15, induction of MycΔp85α before oxaliplatin treatment reduced oxaliplatin-induced apoptosis, while the addition of dox at the same time as or after oxaliplatin had no effect on the levels of apoptosis. This could be due to the higher level of MycΔp85α expression in clone Δ15 (relative to the other clones) and that PI3K inhibition is more pronounced in this clone, or it could be due to other clonal variations. In either case, inhibition of class IA PI3K signaling in HT29 cells did not enhance oxaliplatin-induced apoptosis. To confirm this observation the level of caspase 3 cleavage in cells treated in the same way was determined using the MSD ELISA based system (Figure 6B). In the absence of dox, oxaliplatin-treatment caused a significant increase in the level of cleaved-caspase 3 (relative to total-caspase 3). The addition of dox to parental cells had no effect on the levels of apoptosis, and the induction of MycΔp85α did not change the levels of oxaliplatin-induced caspase 3 cleavage relative to no dox in any clones for any schedule. These data demonstrate that PI3K inhibition did not enhance oxaliplatin-induced apoptosis in HT29 cells.
Figure 6. MycΔp85α expression does not enhance oxaliplatin-induced or etoposide-induced apoptosis.
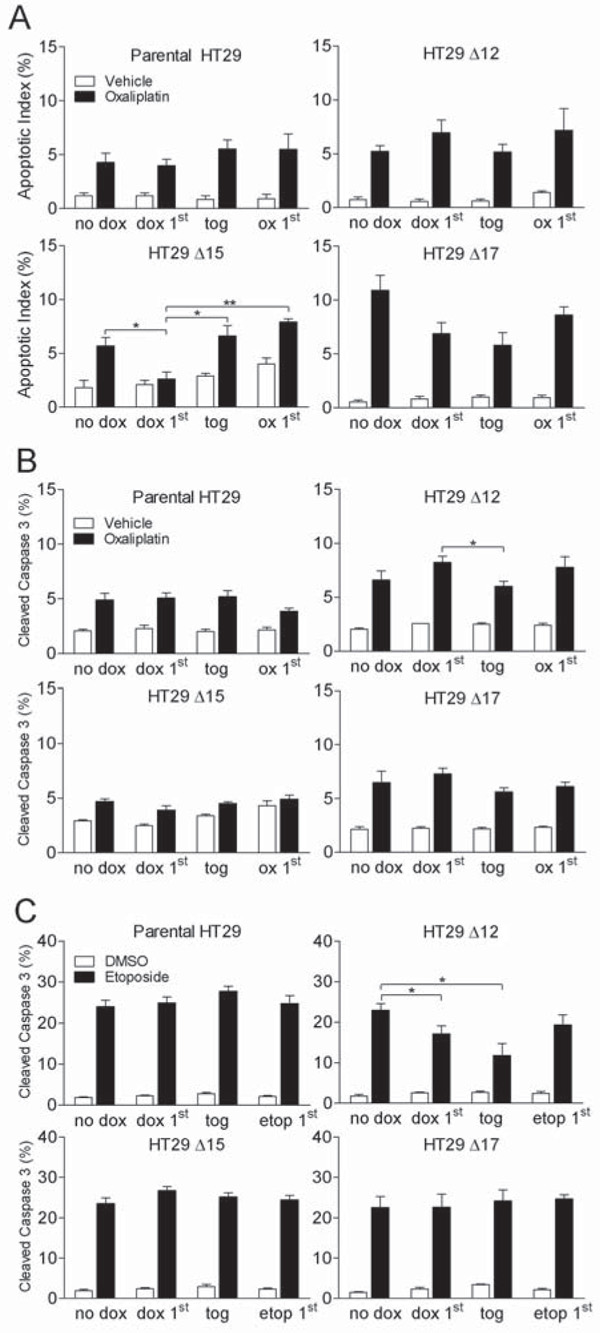
A and B Cells were treated for 48 h with a concentration of oxaliplatin which gave 5-10% apoptosis or vehicle control (PBS). This was either the only treatment the cells received (no dox) or the cells were also treated with dox 24 h prior to oxaliplatin (dox 1st), at the same time as oxaliplatin (tog) or 24 h after oxaliplatin (ox 1st). 48 h after the start of oxaliplatin treatment cells were harvested by trypsinisation and either fixed in 70% ethanol or lysed. A Fixed cells were stained with DAPI and the percentage of cells with an apoptotic nuclei were counted. B The percentage of cleaved-caspase 3 within lysates was determined as described in previous figures. C Cells were treated for 48 hours with 100 μM etoposide or DMSO equivalent. This was either the only treatment the cells received (no dox) or the cells were also treated with dox 24 h prior to etoposide (dox 1st), at the same time as etoposide (tog) or 24 h after etoposide (etop 1st). 48 h after the start of etoposide treatment cells were harvested by trypsinisation and lysed. The percentage of cleaved-caspase 3 within lysates was determined using Mesoscale Discovery cleaved- and total-caspase 3 duplex plates. All graphs represent the mean from three independent experiments +/− S.E.M. * p<0.05, ** p<0.01 according to two-tailed unpaired t-test.
PI3K inhibition did not enhance etoposide-induced apoptosis
While oxaliplatin can induce apoptosis when used at high concentrations it is primarily thought to act as a cytostatic agent at concentrations achieved in the clinic (23). Therefore, the effect of MycΔp85α induction on the apoptotic-response of a cytotoxic drug, etoposide, which is associated with apoptotic responses in vitro and in vivo was explored using the caspase 3 and cleaved-caspase 3 ELISA. Cells were treated for 48 h with etoposide and MycΔp85α expression was either not induced (no dox) or induced 24 h before (dox 1st), at the same time as (tog) or 24 h after (etop 1st) the start of etoposide treatment. Etoposide treatment (100 μM) in parental cells, clone Δ15 and clone Δ17 lead to ~20% cleaved-caspase 3 and this level was not affected by the addition of dox, irrespective of the schedule (Figure 6C). In clone Δ12, etoposide-treatment in the absence of dox also lead to ~20% cleaved-caspase 3, however MycΔp85α expression induced before or at the same time as etoposide-treatment led to a significant decrease in the level of cleaved-caspase 3. It seems unlikely that this represents a bona fide inhibition of etoposide-induced apoptosis by PI3K inhibition as it only occurs in one clone and is therefore more likely due to clonal variation. However, what this data does demonstrate is that PI3K inhibition did not enhance etoposide-induced apoptosis in HT29 colorectal cancer cells.
Discussion
This study has demonstrated that inhibition of PI3K signaling in HT29 and HCT116 colorectal cancer cell lines by inducible over-expression of a dominant negative isoform of the regulatory PI3K subunit p85α leads to cell-cycle delay in the G0/G1 phase of the cell-cycle without apoptosis in vitro. Furthermore, inhibition of PI3K with the SMI PI-103 gave a similar effect in CRC cell lines with mutant or wild-type PIK3CA. Consistent with these data, induced expression of MycΔp85α in HT29 xenografts led to a reduction in tumor growth rate, a reduction in Ki67 positive cells and no change tumor apoptosis. In addition, MycΔp85α expression in HT29 cells did not enhance oxaliplatin- or etoposide-induced apoptosis regardless of whether MycΔp85α was induced before, at the same time, or after the start of drug treatment.
The rationale for studying PI3K inhibition in colorectal cancer cells using models which allowed the inducible expression of a dominant negative PI3K isoform were three fold. Firstly, using a genetic system to inhibit PI3K rather than SMIs allowed for a greater degree of confidence that any results were due to inhibition of PI3K activity, as opposed to ‘off-target’ effects of any SMI. Secondly, the inducible expression of MycΔp85α allowed the study of cells immediately after inhibition of PI3K activity, whereas stable constitutive over-expression would only allow the study of PI3K inhibition weeks to months after PI3K inhibition has been initiated after the appropriate clones had been selected and screened. Finally, all the commercially available PI3K inhibitors are problematic for in vivo studies due to poor pharmacokinetic (PK) profiles whereas the PK of dox was not a hindrance to robust induction of MycΔp85α in vivo.
The main effect of PI3K inhibition in both HT29 and HCT116 cells was the accumulation of cells with 2n DNA content and accumulation of p27, suggesting G0/G1 arrest. This was somewhat to be expected as treatment of a variety of cell lines with various different PI3K SMIs has been reported to induce a G0/G1 arrest (24-26). What is perhaps more surprising is that inhibition of PI3K signaling, which is commonly associated with cell survival signaling, did not increase apoptosis, although a similar lack of apoptosis but reduction of cell proliferation has been reported with PI3K signaling inhibition in hematopoietic cells (27). However, the majority of research implicating PI3K signaling with apoptosis has been carried out with the SMI LY294002 which, as well as potently inhibiting PI3K activity, also inhibits many other members of the PI3K superfamily as well as caesin kinase 2 (28). Furthermore, LY294002 can lead to an increase in intracellular H2O2 in prostate, leukemic and bladder cancer cell lines via a PI3K independent mechanism, enhancing drug-induced apoptosis (29). Therefore, the expectation that PI3K inhibition should drive apoptosis per se may be an over-simplification. Indeed, recent work with a more selective PI3K inhibitor, PI-103, in a range of cell lines failed to detect apoptosis at PI-103 concentrations that clearly inhibited PI3K activity (25) consistent with the findings reported here.
While it could be argued that PI3K inhibition does not cause apoptosis in vitro because the cells are grown under non-physiological conditions, in the presence of 10% FBS and normal oxygen levels, this is not the case in vivo, where there are reduced levels of growth/survival factors and oxygen tension due to irregular tumor xenograft vasculature. Indeed, even under these more stringent conditions no evidence could be found that PI3K inhibition causes apoptosis, while a reduction in tumor growth and proliferation was readily detected.
The hypothesis that PI3K inhibition could reduce the threshold for apoptosis was tested using with two different drugs, oxaliplatin and etoposide and in neither case did PI3K inhibition lead to enhanced levels of apoptosis. As MycΔp85α expression caused a cell-cycle delay and drug-induced apoptosis is often cell-cycle dependent different schedules of PI3K inhibition and drug treatment were evaluated but no schedules lead to enhanced apoptosis. Of interest and possible clinical benefit, PI3K inhibition prior to oxaliplatin treatment did not reduce the levels of oxaliplatin- or etoposide-induced apoptosis as might have been predicted for an agents reported to require proliferation for cytotoxicity.
This study does not rule out the possibility that PI3K inhibition could enhance apoptosis in other cell contexts or that induced by other chemotherapeutics or novel kinase inhibitors that are now entering the clinic.
While PI3K inhibition did not appear to affect apoptosis in HT29 or HCT116 cells, the reduction in proliferation that was observed is encouraging. Many current therapeutics have cytostatic properties and the almost total inhibition of cell proliferation and tumor growth seen both in vitro and in vivo suggests that PI3K inhibition may be beneficial in the clinic.
PI3K inhibition also caused cell-cycle arrest in CRC cells (SW620 and HCT116) that did not contain a PIK3CA mutation (Fig 4) but which are mutant for KRAS,(18), one effect of which is likely to be activation of the PI3K pathway (30). The loss of PTEN is a further mechanism observed in CRC cells to activate PI-3K signaling (31). This may lead one to speculate that PI3K inhibition might be a valuable cytostatic therapy in the majority of CRC cancers, and perhaps in other cancer types, such as glioblastoma (32), where aberrations in the PI3K signaling pathway are common.
Finally, these inducible cell lines are being exploited as useful tools to develop imaging biomarkers of tumor cell proliferation, where decreased biomarker detection after synchronous induction of MycΔp85α in HT29 tumor xenografts acts as a first hurdle for further biomarker optimisation and development.
Materials and Methods
Molecular Biology
The cDNA encoding MycΔp85α (27) was inserted into pTRE (Clontech) to generate pTREMycΔp85α. This was digested with AseI (NEB) and fused with AseI linerised pN1pβactin-rtTA2S-M2-IRES-EGFP (15) to generate pSMVMycΔp85α.
Cell Culture
HT29 cells (ATCC) were cultured in RPMI1640 (Gibco), SW620 cells (ATCC) in DMEM (Gibco) and HCT116 cells (ATCC) and HCT116 mutant PIK3CA and wild-type PIK3CA (a kind gift from B. Vogelstein) were cultured in McCoys 5A (Gibco), all supplemented with 10% foetal bovine serum (Biowest) in a humidified atmosphere at 37°C and 5% CO2. Cells were routinely monitored for mycoplasma and were free from infection. To generate MycΔp85α clones cells were electroporated with 15 μg of pSMVMycΔp85α and cultured in the presence of 800 μg.ml−1 G418 (Gibco) for approximately two weeks. The top 10% of GFP expressing cells were then selected by FACS (BD FACS Vantage SE), 200 cells were seeded into a 10 cm dish and discrete colonies picked when visible (33). MycΔp85α expression was induced by treating cells with 0.5 μg.ml−1 dox (Clontech) and clones that expressed the lowest level of MycΔp85α while reducing levels of phosphorylated PKB were selected for further study, to minimise non-specific protein over-expression effects. A stock solution of 10 mM oxaliplatin (Alexis Biochemicals) was dissolved in PBS, a stock solution of 100 mM etoposide (Sigma) was dissolved in DMSO and a stock solution of 10 mM PI-103 (Calbiochem) was dissolved in DMSO. All were diluted to the appropriate concentration in media.
Western Blotting
Cells grown in vitro were washed either with ice cold PBS and lysed directly into cell lysis buffer (Cell Signaling) supplemented with Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail I and II (Sigma) or detached and adherent cells were pooled after trypsinisation and lysed in the same lysis buffer for analysis of apoptosis. Tumor xenografts were ground to a fine powder in a pestle and mortar under liquid nitrogen and lysed with the same lysis buffer. Protein concentration was determined by BCA assay (Thermo Scientific) following manufacturers instructions and 20 μg of total protein was used per assay as previously described (15). The following primary antibodies were used; p85 (#06-195, Upstate), Myc-tag (#05-419, Upstate), pSer473-PKB (#4058, Cell Signaling), total-PKB (#9272, Cell Signaling) p27 (#sc-528, Santa Cruz Biotechnology) and Actin (#A4700, Sigma).
Immunohistochemistry
Staining was performed as previously described (34). The following primary antibodies were used: Myc-tag (#05-419, Upstate), Ki67 (#M7420, Dako) and cleaved-PARP (#552597, BD Pharmingen). Slides were scanned using a Zeiss Mirax Scan fitted with a 20x objective and four images from each slide were selected at random with Mirax Viewer software. The total number of cells per slide was determined using ‘Nucleus Counter’ plug-in for ImageJ software and the number of positive cells in each image was counted by two independent counters.
Electrochemiluminescent Immunoassay
Detached and adherent cells were pooled after trypsinisation and the resulting cell pellets lysed as for western blotting. 25 μl of 0.8 μg.μl−1 lysate were analyzed in duplicate on a cleaved-caspase 3 and total-caspase 3 duplex electrochemiluminescent plate (Meso Scale Discovery, MSD) and analyzed on a Sector Imager 6000 (MSD). As cleaved-caspase 3 and total-caspase 3 levels were measured in the same well % cleaved-caspase 3 was determined using the following formula; (2 × cleaved-signal)/(cleaved-signal + total-signal) × 100.
Growth Assay
Cells were seeded in 96 well plates and 24 h later (day 0) one plate was fixed with ice cold 10% trichloroacetic acid 60 min and left to dry. The rest of the plates were treated with dox or dox free media and one plate was fixed every 24 h. Once fixed, cells were stained with 0.4% Sulforodamine B (SRB) for 15 min, washed with 1% acetic acid, the dye released with 1.5 M Tris.HCl (pH 8.8) and the absorbance at 540 nm read on a 96 well plate spectrophotometer.
Cell-cycle Analysis
Detached and adherent cells were harvested by trypsinisation, re-suspended in PBS and fixed by adding ice cold ethanol drop-wise to a final concentration of 70% v/v. Fixed cells were incubated with 40 μg.ml−1 propidium iodide and 50 μg.ml−1 RNAse A in PBS for 30min at 37°C in the dark. Propidium bound DNA was then detected by flow cytometry (BDFacsCalibur) and the % cells in each stage of the cell-cycle determined using ModFit LT 3.2 modeling software (Verity Software).
Measurement of Apoptosis
Apoptotic index was calculated by counting the % cells with apoptotic nuclear morphology. Briefly, cells were fixed as for cell-cycle analysis and re-suspended in Prolong Gold antifade reagent with DAPI (Invitrogen), spotted onto a microscope slide and a minimum of 300 nuclei were analyzed on an Olympus BX51 microscope with a 100x objective lens. In addition, cells harvested by trypsinisation were re-suspended in annexin Binding Buffer (BD Biosciences) and incubated with annexin V-APC and 7-AAD (BD Bioscience) for 15 min at room temp in the dark and analysed on a BD FACSArray Bioanalyzer (BD Bioscience).
Tumor Xenografts
HT29 xenografts were grown by subcutaneous injection of 5×106 cells in 0.1 ml of serum free medium into the mid-dorsal region of the back of 6 to 8-wk-old female Balb/c-NUDE mice (Paterson Institute for Cancer Research, Manchester, UK). Mice were housed in individually vented caging systems on a 12 h light/12 h dark environment and maintained at uniform temperature and humidity. Tumor size was measured three times a week with callipers and the volume calculated as tumor length x tumor width2/2. When tumors reached ~300 mm3 animals were switched onto feed containing 625 mg.kg−1 of dox or control feed (Harlan-Teklad). Upon sacrifice, tumors were excised and bisected. One half was snap frozen in liquid nitrogen and used to prepare lysates, the other half was fixed in 10% formalin for immunohistochemistry. All experiments were conducted according to Home Office Regulations (UK) under Project Licence (40-2746) held by Prof Dive.
Acknowledgements
The authors would like Burt Vogelstein for providing HCT116 PIK3CA mutant and wild-type cells, Karen Morris for technical assistance with the annexin V / 7-AAD analysis and Alison Hogg for technical assistance with the immunohistochemistry.
References
- 1.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 2.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 3.Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 4.Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 7.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 8.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 9.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 10.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin Cancer Res. 1997;3:1149–1156. [PubMed] [Google Scholar]
- 12.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Shreder KR, Gai W, Corral S, Ferris DK, Rosenblum JS. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem Biol. 2005;12:99–107. doi: 10.1016/j.chembiol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Holleran JL, Egorin MJ, Zuhowski EG, Parise RA, Musser SM, Pan SS. Use of high-performance liquid chromatography to characterize the rapid decomposition of wortmannin in tissue culture media. Anal Biochem. 2003;323:19–25. doi: 10.1016/j.ab.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 15.Welman A, Cawthorne C, Barraclough J, Smith N, Griffiths GJ, Cowen RL, Williams JC, Stratford IJ, Dive C. Construction and characterization of multiple human colon cancer cell lines for inducibly regulated gene expression. J Cell Biochem. 2005;94:1148–1162. doi: 10.1002/jcb.20342. [DOI] [PubMed] [Google Scholar]
- 16.Wang Q, Kim S, Wang X, Evers BM. Activation of NF-kappaB binding in HT-29 colon cancer cells by inhibition of phosphatidylinositol 3-kinase. Biochem Biophys Res Commun. 2000;273:853–858. doi: 10.1006/bbrc.2000.3034. [DOI] [PubMed] [Google Scholar]
- 17.Yeates LC, Gallegos A, Kozikowski AP, Powis G. Down regulation of the expression of the p110, p85 and p55 subunits of phosphatidylinositol 3-kinase during colon cancer cell anchorage-independent growth. Anticancer Res. 1999;19:4171–4176. [PubMed] [Google Scholar]
- 18.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, Santarius T, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Hunter C, Jenkinson A, Jones D, Kosmidou V, Lugg R, Menzies A, Mironenko T, Parker A, Perry J, Raine K, Richardson D, Shepherd R, Small A, Smith R, Solomon H, Stephens P, Teague J, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Reinhold W, Weinstein JN, Stratton MR, Futreal PA, Wooster R. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–2612. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrow CJ, Gray A, Dive C. Comparison of phosphatidylinositol-3-kinase signaling within a panel of human colorectal cancer cell lines with mutant or wild-type PIK3CA. FEBS Lett. 2005;579:5123–5128. doi: 10.1016/j.febslet.2005.07.096. [DOI] [PubMed] [Google Scholar]
- 21.Samuels Y, Diaz LA, Jr., Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 22.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.William-Faltaos S, Rouillard D, Lechat P, Bastian G. Cell-cycle arrest by oxaliplatin on cancer cells. Fundam Clin Pharmacol. 2007;21:165–172. doi: 10.1111/j.1472-8206.2007.00462.x. [DOI] [PubMed] [Google Scholar]
- 24.Casagrande F, Bacqueville D, Pillaire MJ, Malecaze F, Manenti S, Breton-Douillon M, Darbon JM. G1 phase arrest by the phosphatidylinositol 3-kinase inhibitor LY 294002 is correlated to up-regulation of p27Kip1 and inhibition of G1 CDKs in choroidal melanoma cells. FEBS Lett. 1998;422:385–390. doi: 10.1016/s0014-5793(98)00043-x. [DOI] [PubMed] [Google Scholar]
- 25.Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, Henley A, Di-Stefano F, Ahmad Z, Guillard S, Bjerke LM, Kelland L, Valenti M, Patterson L, Gowan S, de Haven Brandon A, Hayakawa M, Kaizawa H, Koizumi T, Ohishi T, Patel S, Saghir N, Parker P, Waterfield M, Workman P. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 26.Weng LP, Smith WM, Dahia PL, Ziebold U, Gil E, Lees JA, Eng C. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999;59:5808–5814. [PubMed] [Google Scholar]
- 27.Craddock BL, Orchiston EA, Hinton HJ, Welham MJ. Dissociation of apoptosis from proliferation, protein kinase B activation, and BAD phosphorylation in interleukin-3-mediated phosphoinositide 3-kinase signaling. J Biol Chem. 1999;274:10633–10640. doi: 10.1074/jbc.274.15.10633. [DOI] [PubMed] [Google Scholar]
- 28.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poh TW, Pervaiz S. LY294002 and LY303511 sensitize tumor cells to drug-induced apoptosis via intracellular hydrogen peroxide production independent of the phosphoinositide 3-kinase-Akt pathway. Cancer Res. 2005;65:6264–6274. doi: 10.1158/0008-5472.CAN-05-0152. [DOI] [PubMed] [Google Scholar]
- 30.Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 31.Chang JG, Chen YJ, Perng LI, Wang NM, Kao MC, Yang TY, Chang CP, Tsai CH. Mutation analysis of the PTEN/MMAC1 gene in cancers of the digestive tract. Eur J Cancer. 1999;35:647–651. doi: 10.1016/s0959-8049(98)00411-0. [DOI] [PubMed] [Google Scholar]
- 32.The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welman A, Barraclough J, Dive C. Generation of cells expressing improved doxycycline-regulated reverse transcriptional transactivator rtTA2S-M2. Nat Protoc. 2006;1:803–811. doi: 10.1038/nprot.2006.117. [DOI] [PubMed] [Google Scholar]
- 34.Barraclough J, Hodgkinson C, Hogg A, Dive C, Welman A. Increases in c-Yes expression level and activity promote motility but not proliferation of human colorectal carcinoma cells. Neoplasia. 2007;9:745–754. doi: 10.1593/neo.07442. [DOI] [PMC free article] [PubMed] [Google Scholar]