

Abstract
Thalassaemia and sickle cell disease have been recognized by the World Health Organization as important inherited disorders principally impacting on the populations of low income countries. To create a national and regional profile of β-thalassaemia mutations in the population of India, a meta-analysis was conducted on 17 selected studies comprising 8,505 alleles and offering near-national coverage for the disease. At the national level 52 mutations accounted for 97.5% of all β-thalassaemia alleles, with IVSI-5(G>C) the most common disease allele (54.7%). Population stratification was apparent in the mutation profiles at regional level with, for example, the prevalence of IVSI-5(G>C) varying from 44.8% in the North to 71.4% in the East. A number of major mutations, such as Poly A(T>C), were apparently restricted to a particular region of the country, although these findings may in part reflect the variant test protocols adopted by different centres. Given the size and genetic complexity of the Indian population, and with specific mutations for β-thalassaemia known to be strongly associated with individual communities, comprehensive disease registries need to be compiled at state, district and community levels to ensure the efficacy of genetic education, screening and counselling programmes. At the same, time appropriately designed community-based studies are required as a health priority to correct earlier sampling inequities which resulted in the under-representation of many communities, in particular rural and socioeconomically under-privileged groups.
Electronic supplementary material
The online version of this article (doi:10.1007/s11568-010-9132-3) contains supplementary material, which is available to authorized users.
Keywords: β thalassaemia, Haemoglobinopathies, Mutation screening, Regional profiling, Genetic counselling, Genetic education, Population genetics, Population stratification, Community genetics, Bioinformatics
Introduction
With an estimated 1,171 million inhabitants, India is second only to China in population numbers and currently accounts for over 17% of the global population (PRB 2009). Unlike China where some 90% of the population are of Han origin (Black et al. 2007), India has multiple geographical, ethnic, religious and language divisions (Bittles 2002). As the peoples of India have traditionally married and reproduced within these sub-divisions, major problems are encountered in estimating the impact of genetic disease at national, regional, state or even local levels. Data of this nature are, however, essential as despite the current national infant mortality rate of 55/1000 (PRB 2009), there is an increasingly rapid transition in the burden of disease across all age groups from a primarily communicable to a non-communicable pattern, with non-communicable diseases already estimated to account for 42% of deaths (Census of India 2001–2003).
The haemoglobinopathies typify these issues. It has been estimated that the prevalence of pathological haemoglobinopathies in India is 1.2/1,000 live births (Christianson et al. 2006), and with approximately 27 million births per year (PRB 2009) this would suggest the annual birth of 32,400 babies with a serious haemoglobin disorder. Within this overall disease classification a 1989 WHO Working Group on guidelines for the control of haemoglobin disorders estimated a 3.9% carrier frequency for β-thalassaemia in India, encompassing all types of β-thalassaemia trait (WHO 1989). This estimate was mainly derived from data collected prior to 1984 and relied on basic haematological methods of analysis supplemented by information sourced from Livingstone (1985). However, in the absence of more comprehensive, quantitative epidemiological information it continues to be widely cited as the baseline national prevalence for β-thalassaemia in India.
A WHO update on β-thalassaemia in India indicated a similar overall carrier frequency of 3–4%, which given the current national population would translate to between 35.1 and 46.8 million carriers of the disorder nationwide (WHO 2008; PRB 2009). At the same time, a screening project based on 56,814 college students and pregnant women recruited in the states of Maharashtra, Gujarat, Punjab, Karnataka, West Bengal and Assam indicated a carrier rate of 2.78% (Mohanty et al. 2008). These different carrier frequency estimates have been used to approximate the numbers of new affected births per year, which have been calculated to range from 10,000 to 15,000 cases (Edison et al. 2008; Sheth et al. 2008; Tamhankar et al. 2009), of which 8,000–10,000 would present with a severe form of the disease (Colah et al. 2009). If accurate, the figures would indicate a cumulative total of 100,000 children with thalassaemia major in India (WHO 2008).
Unfortunately, there are no adequately representative data sets to confirm or deny these approximations, and with 50,000–60,000 strictly endogamous communities in India (Gadgil et al. 1998), it is dubious whether any average disease prevalence estimate could realistically be applied to each and every community and sub-population. This contention is supported by estimates that the carrier frequency for β-thalassaemia ranges from 0.3 to 17% in different local communities (Agarwal and Mehta 1982; Weatherall and Clegg 2001; WHO 2008).
The initial studies on β-thalassaemia in Indian populations were undertaken among overseas migrant communities and so primarily established the presence of thalassaemia mutations in individuals from the states of Gujarat and Punjab, and in the Sindhi community, many of whom originated in Pakistan (Kazazian et al. 1984; Thein et al. 1988). Five mutations, IVSI-5(G>C), IVSI-1(G>T), 619-bp del, Codon 41/42(−TCTT) and Codon 8/9(+G) accounted for 90% of all mutations (Kazazian et al. 1984; Thein et al. 1988). The results were replicated in follow-up collaborative studies undertaken in Indian and Western centres, mainly focused on the populations of Gujarat, Punjab and Maharashtra (Varawalla et al. 1991a, 1992; Garewal et al. 1994). On the basis of these findings it therefore was assumed that in India the prevalence of β-thalassaemia was highest in the Sindhi and Punjabi communities, and it was only towards the end of the twentieth century that reports from other Indian states demonstrated the wide distribution and extensive heterogeneity of β-thalassaemia mutations in different Indian sub-populations.
Given the partial nature of the available information, the establishment of effective national and regional treatment and prevention programmes for a disorder such as β-thalassaemia is extremely difficult, especially with 229 mutations so far described for the disorder in the locus-specific HbVar database (Giardine et al. 2007), 184 of which are β+ or β0 mutations (http://globin.bx.psu.edu/hbvar). The primary aim of the present study was to systematically collate and critically assess the data so far published on β-thalassaemia in India and within the Indian diaspora, and from the results of this meta-analysis to identify the predominant causative mutations at national, regional and state levels. In acknowledgement of the size of the Indian population and the genetic complexity which follows from the numerous sub-divisions (Bittles 2002; Reich et al. 2009), attention also was directed to mutations that to date have been reported as being largely community-specific in their distribution.
Subjects and methods
The geographical locations of the states and regions of India are shown in Fig. 1. To minimize undue bias towards sample collection from individuals of specific geographical or ethnic origin, and to encourage future more representative sampling across states and regions, only studies reporting allelic frequencies for at least 10 β-globin gene mutations and with a minimum of 50 subjects specifically identified by their state of origin were selected for inclusion. Seventeen published studies met these criteria and were accepted for inclusion, with rigorous cross-checking of data to avoid duplicate entries (Table 1). The information on β-globin chain mutations was initially entered by state origin (n = 28), with subsequent collation into six geographical regions as defined in Fig. 1.
Fig. 1.

Map of India by state and region
Table 1.
Profile of studies included in the meta-analysis of β-thalassaemia mutations in India
| Region | State(s) | Study period | Study population | No of subjects (alleles) | Reference |
|---|---|---|---|---|---|
| All India | 12 of 28 states | 1992–1998 | Referral | 1,228 (1,228) | Vaz et al. (2000) |
| 12 of 28 states | 1997–2006 | Referral | 1,029 (1,544) | Edison et al. (2008) | |
| 12 of 28 states | 1995–2007 | Referral and screening programmes | 2,089 (2,089) | Colah et al. (2009)a | |
| West | Gujarat, Maharashtra, other non-West | Not stated | Referral | 269 (269) | Varawalla et al. (1991a)b |
| Gujarat | Not stated | Referral | 248 (248) | Sheth et al. (2008) | |
| North | Punjab, other non-North | 1991–1993 | Referral | 124 (195) | Garewal et al. (1994) |
| Punjab, Haryana, Uttar Pradesh, Other North, Other non-North | Not stated | Referral | 474 (474) | Verma et al. (1997)c | |
| Uttar Pradesh | 1988–1998 | Referral | 376 (376) | Agarwal et al. (2000b) | |
| Punjab | 1998–2002 | Referrals | 176 (352) | Garewal and Das (2003) | |
| Uttar Pradesh, Punjab, Other non-North | 1998–2002 | Referral | 328 (328) | Gupta et al. (2003) | |
| Punjab | 1998–2004 | Referral | 35 (88) | Garewal et al. (2005) | |
| Uttar Pradesh | 2003–2007 | Referral and field study | 578 (626) | Tamhankar et al. (2009) | |
| East | West Bengal | 1995–1998 | Referral and field study | 291 (221) | Das et al. (2000)d |
| West Bengal | Not stated | Referral | 60 (80) | Kukreti et al. (2002) | |
| West Bengal, Jharkand, Orissa, Other Northeast | 2000–2003 | Referral | 63 (110) | Bandyopadhyay et al. (2004)d,e | |
| South | Andhra Pradesh, Karnataka, Other non-South | 2001–2003 | Referral | 77 (77) | Bashyam et al. (2004)d |
| Andhra Pradesh | 2005–2007 | Referral | 190 (200) | Munshi et al. (2009)d |
Data deemed ineligible for the study are separately presented in Supplementary Information (S1). They comprise
a291 subjects listed as ‘immigrants’
b167 subjects listed as North West Pakistan and 142 as ‘Punjab’
c53 subjects listed as Pakistan (Sindh)
dHomozygous and heterozygous HbE subjects, and HbE alleles from HbE thalassaemia cases
e1 subject and 650 chromosomes of ambiguous geographical origins
Data were excluded from the analysis where information on the regional, state or community origins of subjects was unclear, including 1,150 alleles omitted from persons identified only as being of Sindhi or Punjabi origin but lacking any other identifying details (Supplementary Information, S1). The mean IVSI-5(G>C) allele frequency among these excluded individuals was just 12.7%, compared with the national average figure of 54.7%, raising major doubts as to their provenance. Results from the seven Union Territories, the Andaman and Nicobar Islands, Chandigarh, Dadra and Nagar Haveli, Daman and Diu, Delhi, Lakshadweep, and Pondicherry also were excluded because of the mixed and highly mobile populations in Delhi, the national capital, and Chandigarh the joint capital of the states of Punjab and Haryana, and the local and numerically small populations of the other five Territories.
Only 46 alleles were reported for the populations of the Northeast region, which comprises eight individual states with a combined population of 39.0 million (Census of India 2001a), and is home to many tribal communities of Tibeto-Burmese origin. Of these Northeast samples 34 (73.9%) of alleles were IVSI-5(G>C) while the remaining 12 alleles consisted of five rare mutations and two uncharacterized alleles. Given the small and unrepresentative number of alleles tested, the Northeast data were not separately presented by region in Table 1 and Fig. 2, but the results were incorporated into the All-India data analysis.
Fig. 2.
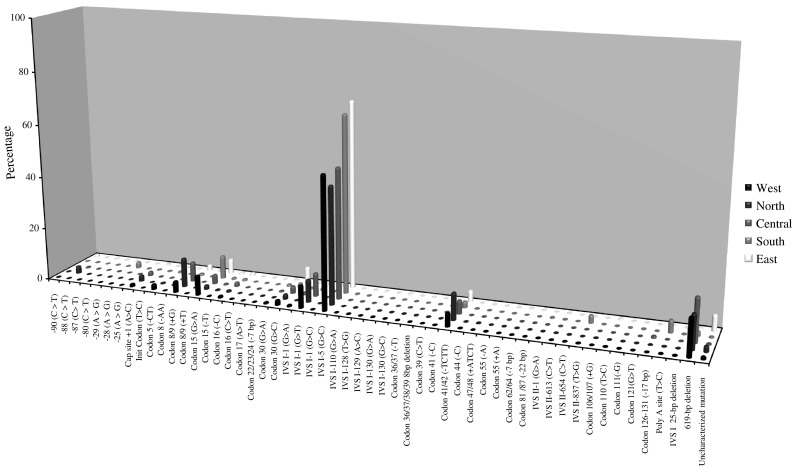
Regional distributions of the most common β-thalassaemia alleles in India (n = 52)
As might have been expected in studies conducted over an extended time period, the methods of genomic analysis employed in the 17 studies varied quite widely and included gap-polymerase chain reaction (PCR), denaturing gradient gel electrophoresis (DGGE), temporal temperature gel electrophoresis (TTGE), amplification refractory mutation system (ARMS), reverse dot blot hybridization (RDB), and direct DNA sequencing (Varawalla et al. 1991a; Verma et al. 1997; Vaz et al. 2000; Old et al. 2001; Agarwal et al. 2003; Bashyam et al. 2004; Sheth et al. 2008; Edison et al. 2008; Colah et al. 2009). For this reason, some variability may inadvertently have resulted in the mutation profiles reported by individual study centres.
Results
National profile of β-thalassaemia mutations
Information on 8,505 alleles was collated, with 64 β-globin gene mutations causing β-thalassemia identified in the Indian population. The profile of the 52 most prevalent and widespread disease alleles, representing 97.5% of the total β-thalassaemia alleles reported at national level, is portrayed by region from the 3′ to 5′ end of the β-globin gene (Fig. 2). Equivalent information on the β-globin mutations identified at individual state level is reproduced in Supplementary Information (S2).
The ten most common β-thalassaemia mutations reported for All-India and by region are listed in Table 2. Nationally, IVSI-5(G>C) was the single most common mutant allele and represented 54.7% of all β-thalassaemia mutations reported. IVSI-5(G>C), 619-bp del, IVSI-1(G>T), Codon 41/42(−TCCT) and Codon 8/9(+G) comprised the five most common disease mutations at the national level and totalled 82.5% of all mutations, with Codon 15(G>A), Codon 30(G>C), Cap site +1(A>C), Codon 5(−CT) and Codon 16(−C) accounting for an additional 11.0% of all mutant alleles (Table 2).
Table 2.
National and regional frequencies (%) of the most common β-thalassaemia mutations in India

It is important to note that 47.0% of the alleles analysed nationally were from subjects who originated either in the western states of Maharashtra and Gujarat or the northern state of Punjab. Furthermore, 15.8% of the national β-thalassaemia allele profile describes persons specifically identified as belonging to Sindhi or Punjabi ethnic groups, which collectively account for just 3.1% of the total population of India (Census of India 2001b). Therefore, as discussed below in terms of regional mutation profiles, over-sampling of these groups significantly influenced the national β-thalassaemia mutation profile reported in previous studies. To complete the national profile of the β-thalassaemia mutations so far described in India the remaining 12 alleles, a number of which have been reported in one or several subjects only, are listed in Table 3.
Table 3.
Less common β-thalassaemia mutations reported in the population of India
| Mutation | State or community of origin | Reference |
|---|---|---|
| -87(C>G) | Uttar Pradesh | Agarwal et al. (2006) |
| IVSII-591(T>C) | Uttar Pradesh | Agarwal et al. (2000a) |
| Codon 8(A>G) | Uttar Pradesh | Agarwal et al. (1999) |
| Codon 13(C>T) | Uttar Pradesh | Agarwal et al. (2000a) |
| Codon 27/28(−C) | Uttar Pradesh | Agarwal et al. (2000a) |
| Codon 5(C>T) | Uttar Pradesh | Agarwal et al. (2000a, 2000b) |
| Codon 4(T>A) | Uttar Pradesh | Agarwal et al. (2000a, 2000b) |
| Codon 57/58(+C) | Sikh | el-Kalla and Matthews (1995) |
| Codon 26(G>T) | Maharashtra, Karnataka | Edison et al. (2008); Colah et al. (2009) |
| IVSII-745(C>G) | Tamil Nadu | Colah et al. (2009) |
| IVSI-5(G>T) | Sindhi | Sheth et al. (2008) |
| Cd88(+T) | Not specified | Varawalla et al. (1991b) |
Regional profile of β-thalassaemia mutations in India
The percentage distribution of five representative β-thalassaemia mutations is illustrated in Fig. 3 according to state of origin. IVSI-5 (G>C) accounts for 54.7% of all β-thalassaemia alleles nationally, and the majority of subjects with this mutation originate from or are resident in the major states of Maharashtra and Gujarat (West region), Uttar Pradesh (North region) and West Bengal (East region). Codon 15 (G>A) also has widespread national distribution but with 35.3% of all subjects resident in Maharashtra. The high percentage of −88(C>T) alleles in cases from Punjab (74.3%) can be ascribed to the frequency of this mutation in the Jat-Sikh community (Garewal et al. 2005). Likewise, the high prevalence of Codon 5(−CT) in Gujarat (79.7%) is associated with the Lohana and Prajapti communities in that state (Sheth et al. 2008). Although the Poly A(T>C) allele has been reported in the populations of nine states, 65.6% of cases were subjects who originated in the adjacent southern states of Tamil Nadu and Karnataka (Edison et al. 2008; Colah et al. 2009).
Fig. 3.
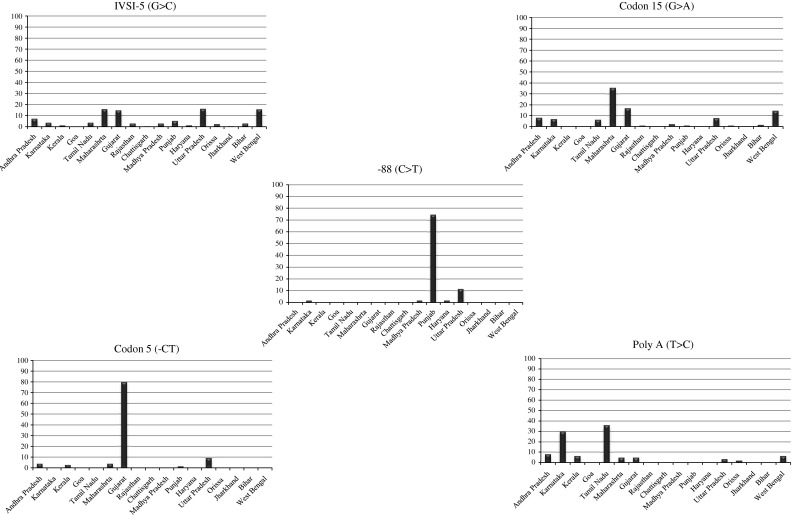
Percentage distributions of five illustrative β-thalassaemia mutations at state level
The West region, comprising the major states of Maharashtra, Gujarat and Rajasthan and the small state of Goa, had a combined population in 2001 of 205.4 million (Census of India 2001a). The West is the most widely represented region in terms of sampling with 3,238 alleles analysed (38.1% of the total sample), and IVSI-5(G>C) accounts for 50.7% of all β-thalassaemia mutations. However, the West region deviates from the national pattern of five common mutations in the somewhat higher prevalence of the 619-bp deletion (14.2%) and IVSI-1(G>T) (8.7%), and with Codon 15(G>A) as the fourth commonest regional mutation with a frequency of 7.6%.
The North region is genetically heterogeneous and ranges from Uttar Pradesh on the Gangetic Plain in the east to Punjab, the westernmost state which adjoins the Pakistani province of Punjab. Haryana with a large agricultural community of Jats, and the Himalayan states of Himachal Pradesh, Uttarakhand and Jammu and Kashmir are the remaining four states in the region. No data are available from Jammu and Kashmir because of ongoing civil unrest. Sampling across the region was non-uniform. Of the 2,484 alleles reported (29.2% of the total sample), 997 were obtained from residents of the state of the Punjab which has a population of 24.3 million, as opposed to the 1,368 alleles representing the 166.1 million strong population of Uttar Pradesh. Although IVSI-5(G>C) accounts for just 44.8% of β-thalassaemia alleles the five most common mutations reported closely match the national pattern, probably due to the high representation of samples from Punjab, but with Codon 16(−C) and −88(C>T) in the list of ten common mutations along with Codon 15(G>A), Codon 30(G>C) and Cap site +1(A>C).
The Central region consisting of the quite sparsely populated states of Madhya Pradesh and Chhattisgarh is grossly under-represented with only 259 reported alleles. Importantly, the Central region is home to many indigenous Scheduled Tribes which in the 2001 Census of India constituted 26.0% of the total regional population of 81.2 million, and with another 13.4% of the population belonging to Scheduled Castes. There is no evidence that either of these predominantly rural and impoverished communities is represented in the regional data set analysed.
Four states Andhra Pradesh, Karnataka, Tamil Nadu and Kerala make up the South region which has a predominantly Dravidian population, ethnically and culturally quite distinct from the largely Indo-European populations of northern, central and western India that represent later population flows into the Indian sub-Continent (Reich et al. 2009). In the 2001 Census of India 20.8% of people countrywide indicated a Dravidian mother tongue, which closely parallels the 21.7% of the national population resident in the South region. IVSI-5(G>C) has a prevalence of 67.9% in the South, suggesting that it may have been the ancestral mutation in the Dravidian founder population of the sub-Continent. The other five and ten most common disease alleles in the South region differ significantly from the overall national pattern; the 619-bp deletion is present in only 1.8% of cases, whereas Codon 15(G>A) is the second most common southern disease allele (8.8%), Poly A site (T>C) is the third most common allele (4.7%) and in 6.3% of cases the disease mutation is rare or unknown.
The East region exhibited by far the highest prevalence of IVS I-5(G>C) at 71.4%, with Codon 30(G>C) and Codon 15(G>A) the second and third most common alleles, accounting for 5.8% and 5.4% of the total respectively, followed by Codon 41/42−TCCT) with a prevalence of 4.3%. The data for the East region are mainly drawn from West Bengal, with the other three constituent states, Bihar, Jharkhand and Orissa, contributing just 311 alleles to the regional total of 1,410 disease alleles. As in the South region there are a large number of alleles (8.0%) which are rare or unknown nationally, probably indicative of the substantial Scheduled Tribal populations in Jharkhand (26.3%) and Orissa (22.1%), the Scheduled Caste communities in West Bengal (23.1%) (Census of India 2001c), and very substantial population movement into the region from Bangladesh (formerly East Pakistan) to the east, during and following the Independence of India in 1947 and the establishment of Bangladesh in 1971.
Discussion
Although it is estimated that more than 300,000 babies are born each year with a major inherited haemoglobin disorder (Christianson et al. 2006) and the lives of many millions of children, adolescents and adults are adversely affected, until quite recently these diseases were rarely included in the health priorities of national governments or international health agencies (Weatherall and Clegg 2001). This situation changed in 2006 with recognition by the Executive Board of the World Health Organization that thalassaemia and sickle cell anaemia were major global health problems which needed to be urgently addressed (WHO 2006), a move reinforced by their inclusion in the current Global Burden of Disease Study (http://globalburden.org).
Given the demonstrated high frequency of β-thalassaemia alleles in India and the immense size of the national population, the present study is necessarily preliminary and any conclusions drawn need to be assessed in that light. As previously noted, with a total population of 1,171 million and a rate of natural population increase of 1.6% (PRB 2009), collecting accurate and representative health information in India is a major problem. The highly endogamous nature of Indian society, traditionally based on castes which claim long and unbroken genealogical histories, means that each community effectively functions as a separate breeding pool, with the consequent probability that recent mutations may be unique to single communities (Bittles 2008, 2009; Bittles and Black 2010). Representative sampling can therefore become extremely difficult, given the population stratification that results from the multiple ethnic, social and religious subdivisions which are a central facet of everyday existence.
When dealing with an autosomal recessive disorder such as β-thalassaemia, an additional important factor that has to be considered is the widespread preference for intra-familial unions in the southern Dravidian states of Andhra Pradesh, Karnataka and Tamil Nadu, where 30+% of marriages are consanguineous, mainly uncle-niece (F = 0.125) or first cousin (F = 0.0625), and with substantial levels of consanguineous unions in neighbouring Kerala and southern Maharashtra (Bittles et al. 1991; Bittles 2002; www.consang.net). In these states it would be expected that a high proportion of β-thalassaemia cases would be homozygous for the causative mutation, and indeed 98% of affected subjects investigated in Andhra Pradesh were homozygotes for a specific mutant allele (Bashyam et al. 2004).
At first sight the situation might be considered different in the other regions of India where exogamy in the Hindu population is practised at gotra level, i.e. involving extended male lineages, but with marital endogamy at caste level. However, since an overwhelming percentage of marriages continue to be contracted on an intra-caste and intra-community basis, even though spouses may not be known to be biologically related, there is a very strong chance that they have a large proportion of their genes in common (Bittles 2008). Thus, even in West, North, and East India, a higher than expected proportion of patients with β-thalassaemia probably are homozygous for a single mutant allele rather than being compound heterozygotes. This probability is increased by the high frequency of the IVSI-5(G>C) mutation in each region (Table 2), and by the 15.8% to 33.0% prevalence of consanguineous marriage at state level in the large Muslim minority population (Bittles and Hussain 2000).
The prevailing wisdom has been that β-thalassaemia in India principally affects the Sindhi, Gujarati, Bengali, Punjabi and Muslim communities (Agarwal 2005), although this supposition has been strongly influenced by the more extensive testing undertaken in these sub-populations. As a large majority of communities have yet to be sampled, especially among the Scheduled Castes and Scheduled Tribes and the group of lower caste communities collectively defined by the Government of India as Other Backward Classes, this opinion may well require significant future revision, and it seems highly probable that previously uncharacterized mutations remain to be identified. In the interim, it is important that public education programmes, in combination with opportunities for premarital and prenatal screening, should be made available to as wide a range of couples, families and communities as possible.
Table 2 showed that while IVSI-5 (G>C) was the predominant mutation throughout India, the prevalence varied from 44.8% in the North to 71.4% in the East. It also was apparent from Table 2 and Fig. 3 that the profile of other mutations showed significant inter-regional variation, to the extent that this variation merited serious consideration in the design and implementation of future screening programmes. The higher the mutation detection rate with as small a number of markers employed, the more efficient the testing protocol will be in terms of staff time expended and the costs involved.
As summarized in Table 4, this type of approach already appears feasible at regional level. Importantly, testing for the five most common mutations at national level would detect 82.5% of cases, and for the ten most common mutations 93.5% of cases would be identified. But by changing the testing protocols to incorporate the most appropriate mutation profiles identified at regional level, the potential levels of detection could be increased to 87.7% (North) for the five most common mutations, and 97.6% (Central) for the ten most common β-thalassaemia mutations. Given the size of the potential β-thalassaemia case-load in India, due accommodation for these differences in the potential efficiency of screening programmes could produce substantial savings in both time and costs.
Table 4.
National and regional frequencies (%) of the five and ten most common β-thalassaemia mutations in India
| All India | West | North | Central | South | East | |
|---|---|---|---|---|---|---|
| Five most common mutations | 82.5 | 86.4 | 87.7 | 87.6 | 86.7 | 89.0 |
| Ten most common mutations | 93.5 | 96.7 | 96.5 | 97.6 | 93.7 | 92.0 |
Could this level of performance be further improved if community-based rather state or regional mutation data were available? To answer this question, previously unreported data on 1,031 β-thalassaemia alleles in the large northern state of Uttar Pradesh (S Agarwal, unpublished) were examined in a separate analysis. As indicated in Table 5 the results have been subdivided into seven categories, corresponding to the main religious, caste and socioeconomic subdivisions within the population of the state.
Table 5.
Community-specific profiles of β-thalassaemia mutations in Uttar Pradesh, India
| Mutation frequencies | Hindus | Muslims | |||||
|---|---|---|---|---|---|---|---|
| Brahmins | Kshatriyas | Vaishyas | Kayasthas | Other Backward Classesa | Scheduled castes | ||
| >50% | Codon 6 | Codon 30 | Other mutations | ||||
| 10–50% | Cap Site +1 (A>C) IVS1-5 (G>C) Codon 41/42 (−TCTT) Codon 8/9 (+G) Codon 30 (G>C) Other mutations |
IVS1-5 (G>C) Codon 15 (G>A) Cap Site +1 (A>C) Codon 41/42 (−TCTT) Codon 16 (−C) Uncharacterized |
IVS1-5 (G>C) Codon 8/9 (+G) Codon 16 (−C) |
IVS1-5 (G>C) | Codon 16 (−C) IVS1-5 (G>C) Codon 15 (G>A) |
Codon 30 (G>C) Cap Site +1 (A>C) Codon 41/42 (−TCTT) Codon 16 (−C) IVS1-1 (G>T) Codon 15 (G>A) |
Codon 15 (G>A) Codon 41/42 (−TCTT) Codon 8/9 (+G) Uncharacterized Cap Site +1 (A>C) |
| <10% | IVS1-1 (G>T) Codon 16 (−C) Codon 15 (G>A) Uncharacterized |
Codon 8/9 (+G) IVS1-1 (G>T) 619-bp del |
Codon 41/42 (−TCTT) Codon 8/9 (+G) 619-bp del |
Codon 41/42 (−TCTT) IVS1-1 (G>T) Codon 30 (G>C) Codon 8/9 (+G) Cap site + 1(A>C) Uncharacterized |
IVS1-5 (G>C) Codon 8/9 (+G) 619-bp del |
IVS1-5 (G>C) Codon 16 (−C) Codon 30 (G>C) IVS1-1 (G>T) 619-bp del |
|
| <1% | Codon 30(G>C) | Codon 41/42 (−TCTT) IVS1-1 (G>T) 619-bp del Codon 30 (G>C) Cap Site +1 (A>C) Codon 15 (G>A) |
IVS1-1 (G>T) Codon 16 (−C) Cap Site +1 (A>C) Codon 15 (G>A) Other mutations Uncharacterized |
619-bp del | Other mutations | Other mutations | |
aOther Backward Classes are defined by the Government of India as economically disadvantaged communities, mainly ‘Backward castes’
The ten most common β-thalassaemia mutations identified are as listed for the North region in Table 2. Although the numbers within each sub-division are small and significant mutation overlap exists between a number of the communities, such as the Hindu upper caste Brahmins and Kshatriyas, there also are major differences in community mutation profiles, e.g., comparing the Brahmin community in which Cap Site +1(A>C), IVSI-5 (G>C) and Codon 41/42(−TCTT) are the three most common diseases alleles, with the communities classified as Other Backward Classes where ‘Other mutations’, Codon 16(−C), IVSI-5(G>C), and Codon 15 (G>A) alleles predominate.
There also is clear evidence of over-sampling of economically more privileged groups. Thus while the four Hindu upper and middle castes, the Brahmins, Kshatriyas, Vaishyas and Kayasthas comprise ~19% of the population of Uttar Pradesh they account for 56.8% of the β-thalassaemia alleles tested, whereas the Hindu Other Backward Classes who form ~31% of the total state population (NSSO 2005) comprised just 11.4% of alleles.
From a genetic screening and genetic counselling perspective the data do indicate that community-specific mutation profiles could be highly effective in helping to screen for and prevent β-thalassaemia. At the same time it has to be acknowledged that to establish similar community-specific mutation profiles throughout India would be an extremely difficult logistic task within the near future. But the potential benefits are very high in health, social and economic terms, and the creation of more detailed databases of β-thalassaemia alleles will facilitate better focused, more efficient, and cost-effective testing and treatment protocols that can concentrate on individual communities and sub-populations.
Conclusions
The outcomes derived from the basic data collated in the present study should provide a sound platform on which future health care planning for the prevention and treatment of β-thalassaemia in India can be undertaken. The need for a paradigm shift in β-thalassaemia-related research is, however, indicated. While determination of the broad-based geographical distribution of causative mutations has been an important initial step, there is a clear need for structured sampling programmes to be planned and instituted to provide representative information on regions, such as Central India and the Northeast, for which data are currently inadequate. Additionally, in a country with a population as large and ethnically and socially diverse as India, the further extension of sampling to facilitate state, district and village registers of persons with β-thalassaemia and carriers of the disorder is warranted (WHO 2008). Indeed, given the continuing marked hereditary sub-divisions within Indian society that result from intra-caste and intra-community marriage, community-specific mutation testing would provide the basis for the optimum delivery of genetic education, screening and prevention programmes.
Electronic supplementary material
Acknowledgments
The authors acknowledge the generous financial contribution provided by the Western Australian State Government in the establishment of the WA Centre of Excellence for Comparative Genomics and support of this project. Technical advice and assistance was kindly provided by Paula Moolhuijzen. The Thalassemia Working Group, Varanasi comprises: S. Sinha, Group Coordinator, R. Raman, Research Coordinator, V. P. Singh, A. Kumar, M. Jain, K. Singh, R. Nagar, Banaras Hindu University, and S. Kumar, P. Rai. B. L. Gupta, Thalassemia/Haemoglobinopathies Programme, Mata Anandmayee Hospital, Varanasi. During the course of this study SS was a Visiting Senior Research Fellow in the Centre for Comparative Genomics, Murdoch University. AHB was supported by National Science Foundation Grant 0527751.
References
- Agarwal MB. The burden of haemoglobinopathies in India—time to wake up? J Assoc Physician India. 2005;53:1017–1018. [PubMed] [Google Scholar]
- Agarwal MB, Mehta BC. Genotypic analysis of symptomatic thalassaemia syndromes (A study of 292 unrelated cases from Bombay) J Postgrad Med. 1982;28:1–3. [PubMed] [Google Scholar]
- Agarwal S, Hattori Y, Gupta UR, Agarwal SS. A novel Indian β-thalassemia mutation: Hb Lucknow [PS(AS)Lys + Arg] Hemoglobin. 1999;23:263–265. doi: 10.3109/03630269909005707. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Hattori Y, Agarwal SS. Rare β-thalassemia mutations in Asian Indians. Amer J Hematol. 2000;65:322–323. doi: 10.1002/1096-8652(200012)65:4<322::AID-AJH14>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Pradhan M, Gupta UR, Sarwai S, Agarwal SS. Geographic and ethnic distribution of β-thalassemia mutations in Uttar Pradesh, India. Hemoglobin. 2000;24:89–97. doi: 10.3109/03630260009003427. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Gupta A, Gupta UR, Sarwai S, Phadke S, Agarwal SS. Prenatal diagnosis in beta-thalassaemia: an Indian experience. Fetal Diagn Therapy. 2003;18:328–332. doi: 10.1159/000071975. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Arya V, Stolle CA, Pradhan M. A novel Indian β-thalassemia mutation in the CACCC box of the promoter region. Eur J Haematol. 2006;77:530–532. doi: 10.1111/j.0902-4441.2006.t01-1-EJH2923.x. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A, et al. Profile of β-thalassemia in eastern India and its prenatal diagnosis. Pren Diagn. 2004;24:992–996. doi: 10.1002/pd.1049. [DOI] [PubMed] [Google Scholar]
- Bashyam MD, Bashyam L, Gorinabele R, Sangal MGV, Rama Devi AR. Molecular genetic analyses of β-thalassemia in South India reveals rare mutations in the β-globin gene. J Hum Genet. 2004;49:408–413. doi: 10.1007/s10038-004-0169-9. [DOI] [PubMed] [Google Scholar]
- Bittles AH. Endogamy, consanguinity and community genetics. J Genet. 2002;81:91–98. doi: 10.1007/BF02715905. [DOI] [PubMed] [Google Scholar]
- Bittles AH. A community genetics perspective on consanguineous marriage. Commun Genet. 2008;11:324–330. doi: 10.1159/000133304. [DOI] [PubMed] [Google Scholar]
- Bittles AH. Consanguinity, genetic drift, and genetic diseases in populations with reduced numbers of founders. In: Vogel F, Motulsky AG, Antonarakis SE, Speicher M, editors. Human genetics—principles and approaches. 4. Heidelberg: Springer; 2009. pp. 507–528. [Google Scholar]
- Bittles AH, Black ML. Consanguinity, human evolution and complex diseases. Proc Natl Acad Sci USA. 2010;107:1779–1786. doi: 10.1073/pnas.0906079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittles AH, Hussain R. An analysis of consanguineous marriage in the Muslim population of India at regional and state levels. Ann Hum Biol. 2000;27:163–171. doi: 10.1080/030144600282271. [DOI] [PubMed] [Google Scholar]
- Bittles AH, Mason WH, Greene J, Appaji Rao N. Reproductive behavior and health in consanguineous marriages. Science. 1991;252:789–794. doi: 10.1126/science.2028254. [DOI] [PubMed] [Google Scholar]
- Black ML, Wang W, Bittles AH. Unity and diversity: genetic studies on the population of China. In: Santos C, Lima M, editors. Recent advances in molecular biology and evolution: applications to biological anthropology. Trivandrum: Research Signpost; 2007. pp. 347–371. [Google Scholar]
- Census of India (2001–2003) Report on Causes of Death, Office of Registrar General, India. http://www.censusindia.gov.in/Vital_Statistics/Summary_Report_Death_01_03.pdf, p 2
- Census of India (2001a) Office of the Registrar General and & Census Commissioner, India. http://www.censusindia.gov.in/population_finder/State_Master.aspx
- Census of India (2001b) Office of Registrar General & Census Commissioner, India. http://www.censusindia.gov.in/Census_Data_2001/Census_Data_Online/Language/parta.htm
- Census of India (2001c) Office of Registrar General & Census Commissioner, Government of India, http://www.censusindia.gov.in/Census_Data_2001/Census_data_finder/A_Series/SC_ST.htm
- Christianson A, Howson CP, Modell B. March of Dimes global report on birth defects. White Plains: March of Dimes Birth Defects Foundation; 2006. [Google Scholar]
- Colah R, Gorakshakar A, Nadkarni A, Phanasgaonkar S, Surve R, Sawant P, Mohanty D, Ghosh K. Regional heterogeneity of β-thalassemia mutations in the multi ethnic Indian population. Blood Cells Mol Dis. 2009;42:241–246. doi: 10.1016/j.bcmd.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Das SK, Madhusnata DE, Bhattacharya DK, Sengupta B, Das N, Talukder G. Interaction of different hemoglobinopathies in Eastern India with a view to establish genotype-phenotype correlation. Am J Hum Biol. 2000;12:452–459. doi: 10.1002/1520-6300(200007/08)12:4<454::AID-AJHB4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Edison ES, Shaji RV, Devi SG, Moses A, Viswabandhya A, Matthews V, George B, Srivastava A, Chandy M. Analysis of β globin mutations in the Indian population: presence of rare and novel mutations and region-wise heterogeneity. Clin Genet. 2008;73:331–337. doi: 10.1111/j.1399-0004.2008.00973.x. [DOI] [PubMed] [Google Scholar]
- el-Kalla S, Matthews AR. A novel frameshift mutation causing β-thalassemia in a Sikh. Hemoglobin. 1995;19:183–189. doi: 10.3109/03630269509036938. [DOI] [PubMed] [Google Scholar]
- Gadgil M, Joshi NV, Manoharan S, Patil S, Prasad UVS. Peopling of India. In: Balasubramanian D, Appaji Rao N, editors. The Indian human heritage. Hyderabad: Universities Press; 1998. pp. 100–129. [Google Scholar]
- Garewal G, Das R. Spectrum of β-thalassemia mutations in Punjabis. Int J Hum Genet. 2003;3:217–219. [Google Scholar]
- Garewal G, Fearon CW, Warren TC, Marwaha N, Marwaha RK, Mahadik C, Kazazian HH., Jr The molecular basis of β thalassaemia in Punjabi and Maharashtran Indians includes a multilocus aetiology involving triplicated α-globin loci. Brit J Haematol. 1994;86:372–376. doi: 10.1111/j.1365-2141.1994.tb04742.x. [DOI] [PubMed] [Google Scholar]
- Garewal G, Das R, Ahluwalia J, Marwaha RK, Varma S. Nucleotide -88 (C-T) promoter mutation is a common β-thalassemia mutation in the Jat Sikhs of Punjab, India. Am J Hematol. 2005;79:252–256. doi: 10.1002/ajh.20445. [DOI] [PubMed] [Google Scholar]
- Giardine B, Van Baal S, Kaimakis P, Riemer C, Miller W, Samara M, Kollia P, Anagnou NP, Chui DH, Wajcman H, Hardison RC, Patrinos GP. HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum Mut. 2007;28:206. doi: 10.1002/humu.9479. [DOI] [PubMed] [Google Scholar]
- Gupta A, Hattori Y, Gupta UR, Sarwai S, Nigam N, Singhal P, Agarwal S. Molecular genetic testing of β-thalassemia patients of Indian origin and a novel 8-bp deletion mutation at codons 36/37/38/39. Genet Test. 2003;7:163–168. doi: 10.1089/109065703322146894. [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Orkin SH, Antonarakis SE, Sexton JP, Boehm CD, Goff SC, Waber PG. Molecular characterization of seven β-thalassemia mutations in Asian Indians. EMBO J. 1984;3:593–596. doi: 10.1002/j.1460-2075.1984.tb01853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukreti R, Dash D, Vineetha KE, Chakravarty S, Das SK, De M, Talukder G. Spectrum of β-thalassemia mutations and their association with allelic sequence polymorphisms at the β-globin gene cluster in an Eastern Indian population. Am J Hematol. 2002;70:269–277. doi: 10.1002/ajh.10117. [DOI] [PubMed] [Google Scholar]
- Livingstone FB. Frequencies of hemoglobin variants: thalassemia, the glucose-6-phosphate dehydrogenase deficiency, G6PD variants, and ovalocytosis in human populations. New York: Oxford University Press; 1985. [Google Scholar]
- Mohanty D, Colah R, Gorakshakar A (eds) (2008) Jai Vigyan S & T mission project on community control of thalassaemia syndromes—awareness, screening, genetic counselling and prevention. A national multicentric task force study of ICMR (2000–2005), Indian Council of Medical Research, New Delhi
- Munshi A, Anandraj MPJS, Joseph J, Shafi G, Anila AN, Jyothy A. Inherited hemoglobin disorders in Andhra Pradesh, India: a population study. Clin Chim Acta. 2009;400:117–119. doi: 10.1016/j.cca.2008.10.025. [DOI] [PubMed] [Google Scholar]
- NSSO . Press Note, July 2004–2005 Report, National Sample Survey Organization, Ministry of Statistics and Program Implementation. New Delhi: Press Information Bureau, Government of India; 2005. [Google Scholar]
- Old JM, Khan SN, Verma I, Fucharoen S, Kleanthous M, Ioannou P, Kotea N, Fisher C, Riazuddin S, Saxena R, Winichagoon P, Kyriancou K, Al-Quobaili F, Khan B. A multi-center study in order to further define the molecular basis of β-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymerase chain reaction. Hemoglobin. 2001;25:397–407. doi: 10.1081/HEM-100107877. [DOI] [PubMed] [Google Scholar]
- PRB . World population data sheet. Washington DC: Population Reference Bureau; 2009. [Google Scholar]
- Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461:489–495. doi: 10.1038/nature08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth JJ, Sheth FJ, Pandya P, Priya R, Davla S, Thakur C, Vaz F. β-thalassemia mutations in Western India. Ind J Pediatr. 2008;75:567–570. doi: 10.1007/s12098-008-0109-3. [DOI] [PubMed] [Google Scholar]
- Tamhankar PM, Agarwal S, Arya V, Kumar R, Gupta UR, Agarwal SS. Prevention of homozygous beta thalassemia by premarital screening and prenatal diagnosis in India. Prenat Diagn. 2009;29:637–638. doi: 10.1002/pd.2176. [DOI] [PubMed] [Google Scholar]
- Thein SL, Hesketh C, Wallace RB, Weatherall DJ. The molecular basis of thalassaemia major and thalassaemia intermedia in Asian Indians: application to prenatal diagnosis. Brit J Haematol. 1988;70:225–231. doi: 10.1111/j.1365-2141.1988.tb02468.x. [DOI] [PubMed] [Google Scholar]
- Varawalla NY, Old JM, Sarkar R, Venkatesan R, Weatherall DJ. The spectrum of β-thalassaemia mutations on the Indian subcontinent: the basis for prenatal diagnosis. Brit J Haemat. 1991;78:242–247. doi: 10.1111/j.1365-2141.1991.tb04423.x. [DOI] [PubMed] [Google Scholar]
- Varawalla NY, Old JM, Weatherall DJ. Rare beta-thalassaemia mutations in Asian Indians. Brit J Haemat. 1991;79:640–644. doi: 10.1111/j.1365-2141.1991.tb08094.x. [DOI] [PubMed] [Google Scholar]
- Varawalla NY, Fitches AC, Old JM. Analysis of β-globin gene haplotypes in Asian-Indians: origin and spread of β-thalassaemia on the Indian subcontinent. Hum Genet. 1992;90:443–449. doi: 10.1007/BF00220475. [DOI] [PubMed] [Google Scholar]
- Vaz FEE, Thakur (Mahadik) CB, Banerjee MK, Gangal SC. Distribution of β-thalassemia mutations in the Indian population referred to a diagnostic center. Hemoglobin. 2000;24:181–194. doi: 10.3109/03630260008997526. [DOI] [PubMed] [Google Scholar]
- Verma IC, Saxena R, Thomas E, Jain PK. Regional distribution of β-thalassemia mutations in India. Hum Genet. 1997;100:109–113. doi: 10.1007/s004390050475. [DOI] [PubMed] [Google Scholar]
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull WHO. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- WHO (1989) Guidelines for the control of haemoglobin disorders: report of the VIth Annual Meeting of the WHO Working Group on Haemoglobinopathies, Cagliari, Sardinia, 8–9 April, 1989. Geneva, World Health Organization (unpublished document WHO/HDP/WG/HA/89.2)
- WHO (2006) Thalassaemia and other haemoglobinopathies. World Health Organization Resolutions, May 2006, EB118.R1 and WHA59.20
- WHO (2008) Joint WHO-TIF meeting on management of haemoglobin disorders (2nd: 2008: Nicosia, Cyprus) Geneva, World Health Organization. (NLM classification: WH 190)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.