

Abstract
Auditory steady-state auditory responses (ASSRs), in which the evoked potential entrains to stimulus frequency and phase, are reduced in magnitude in patients with schizophrenia, particularly at 40 Hz. While the neural mechanisms responsible for ASSR generation and its perturbation in schizophrenia are unknown, it has been hypothesized that the GABAA receptor subtype may have an important role. Using an established rat model of schizophrenia, the neonatal ventral hippocampal lesion (NVHL) model, 40-Hz ASSRs were elicited from NVHL and sham rats to determine if NVHL rats show deficits comparable to schizophrenia, and to examine the role of GABAA receptors in ASSR generation. ASSR parameters were found to be stable across time in both NVHL and sham rats. Manipulation of the GABAA receptor by muscimol, a GABAA agonist, yielded a strong lesion × drug interaction, with ASSR magnitude and synchronization decreased in NVHL and increased in sham rats. The lesion × muscimol interaction was blocked by a GABAA receptor antagonist when given prior to muscimol administration, confirming the observed interaction was GABAA mediated. Together, these data suggest an alteration involving GABAA receptor function, and hence inhibitory transmission, in the neuronal networks responsible for ASSR generation in NVHL rats. These findings are consistent with prior evidence for alterations in GABA neurotransmitter systems in the NVHL model and suggest the utility of this animal modelling approach for exploring neurobiological mechanisms that generate or modulate ASSRs.
Keywords: Auditory steady-state responses, GABA, 40 Hz, neonatal ventral hippocampal lesion, schizophrenia
Introduction
Schizophrenia is a chronic neuropsychiatric disorder affecting 1% of the population worldwide. The core pathophysiology of schizophrenia may involve disrupted integration of neural processes and synchronization, which are vital for many information-processing tasks compromised in schizophrenia (Lee et al. 2003) and probably result in sensory information-processing abnormalities (Uhlhaas et al. 2008). The electroencephalogram (EEG) allows for direct examination of electrical activity produced by large populations of neurons with high temporal resolution. Further, EEG synchronization can be evoked by periodic external stimuli in which EEG entrains to the frequency and phase of the eliciting stimulus (Lins & Picton, 1995; Regan, 1989). EEG entrainment to repetitive auditory stimulation, or auditory steady-state responses (ASSRs), can serve as a useful tool in testing the functional state of the networks supporting neural synchrony (Lins & Picton, 1995; Regan, 1989). Measures of power and phase synchronization are disturbed in patients with schizophrenia in the gamma frequency range (Brenner et al. 2003; Krishnan et al. 2009; Kwon et al. 1999; Light et al. 2006; Spencer et al. 2008), particularly at 40 Hz. However, ASSRs have not been characterized in animal models of mental illness. Characterization of the ASSR in an established rat model of schizophrenia could allow for testing potential neurochemical mechanisms associated with the deficit and may contribute to the development of novel treatment targets.
The neonatal ventral hippocampal lesion (NVHL) rat model of schizophrenia is a phenomenologically comprehensive and replicable model of the disorder (Lipska, 2004; Tseng et al. 2008a). Neonatal insult to the ventral hippocampus alters frontal cortical and subcortical circuit function in adulthood, probably related to developmental disruption of ventral hippocampal projections to these regions (Tseng et al. 2008a). NVHL rats exhibit many schizophrenia-like phenomena, including: behavioural changes, pharmacological responses, and molecular changes in the prefrontal cortex (Lipska & Weinberger, 2000). Moreover, using the NVHL model, our group has recently shown that these rats have evoked potential abnormalities like those observed in schizophrenia (Vohs et al. in press). Specifically, NVHL rats have reduced sensory activation and low-frequency sensory gating, as measured by phase locking across trials, suggesting they may have increased temporal variability in evoked responses. To our knowledge there are no studies of ASSR in rat models of schizophrenia, despite the extensive use of this measure in clinical studies of schizophrenia. The present study addresses this gap in the literature by examining ASSRs in the NVHL model of schizophrenia.
NVH lesions may affect gamma-aminobutyric acid (GABA)ergic receptor function (Tseng et al. 2008a), which has been implicated in schizophrenia and may mediate gamma synchronization (Lewis & Gonzalez-Burgos, 2008). GABAergic transmission has been hypothesized to play a key role in the generation of synchronous oscillations, particularly ASSRs in the gamma frequency range (Lewis et al. 2005). The GABAA receptor is thought to contribute to the generation of network oscillations via both interneuron–interneuron and interneuron–pyramidal neuron, cell connections. Synchronization is hypothesized to be propagated through networks in a cycle of GABAA-mediated inhibition followed by rebound excitation and then inhibition (Lewis & Gonzalez-Burgos, 2008). While glutamate and other transmitter systems (e.g. cholinergic and monoaminergic) are also known to be important in this process, manipulation of the GABAA receptor specifically could provide important insight into mechanisms of network oscillations.
In addition to its potential role in the generation and maintenance of synchronous oscillations (Lewis et al. 2005), the GABA neurotransmitter system has gained attention for its apparent involvement in the pathophysiology of schizophrenia (Benes & Berretta, 2001; Lewis & Moghaddam, 2006; Wassef et al. 2003), with some of the neurobiological alterations observed in schizophrenia probably resulting from compensatory response to restore inhibitory synaptic efficacy (Lewis et al. 2005). It is likely that the GABAA receptor subtype, which propagates network synchronization (Lewis & Gonzalez-Burgos, 2008), is also associated with oscillatory abnormalities observed in patients with schizophrenia. Moreover, a recent study of schizophrenia suggested up-regulation, and therefore, increased GABAA receptor density in the temporal cortex of patients with schizophrenia (Deng & Huang, 2006). Given that the temporal cortex is thought to play an important role in the generation of ASSRs, these findings suggest a potential link between GABAA abnormalities and ASSR disturbances in schizophrenia.
The present study aimed to characterize 40-Hz ASSRs in the NVHL rat model of schizophrenia and, using in-vivo methods, examine the role of the GABAA receptor in ASSR generation. Specifically, stainlesssteel screw electrodes were implanted epidurally over the temporal cortex of adult NVHL and sham rats and ASSRs were recorded to (1) test if ASSRs elicited from these rats were stable across time; (2) test if NVHL rats show an ASSR deficit comparable to patients with schizophrenia; and (3) investigate the role of the GABAA receptor in the generation and maintenance of ASSRs in NVHL and sham rats.
Methods
Subjects
All procedures were in compliance with the Guide for the Care and Use of Laboratory Animals and were approved by the Indiana University Animal Care and Use Committee (IACUC). Six pregnant Sprague–Dawley rats were obtained from Harlan Laboratories (Indianapolis, USA) at the gestational age of 14–17 d. All rats were individually housed for 7–14 d until birth.
Neonatal lesion surgeries
At post-natal day (PND) 7, pups were removed from their home cage and taken into a surgical room for <2 h. There, pups were anaesthetized by hypothermia and secured with tape to a stereotaxic platform to maintain head position during surgery. According to the procedure of Lipska et al. (1993), a horizontal incision was made across the dorsal surface of the head and a 26-gauge syringe needle was lowered through the thin level skull into the ventral hippocampal formation (AP −3.0 mm, ML ±3.5 mm, VD −5.0 mm relative to bregma). Over a period of 135 s, each pup received either 3.0 μg ibotenic acid (Sigma, USA) in 0.3 μl artificial cerebral spinal fluid to produce an excitotoxic lesion to both ventral hippocampi (n = 29), or a sham lesion (n = 23) following the same procedure minus the excitotoxin. The surgical wound was closed with veterinarian adhesive and the pups were monitored and warmed before being returned to their home cages.
Rats were weaned (PND 25) and at PND 49, paired (two rats per cage) and moved to standard plastic cages. Three days prior to implantation of EEG electrodes, rats were moved to single-house Plexiglas cages. Rats were kept in a temperature-controlled room, maintained on a 12-h light/dark cycle (lights on 07:00 hours), with food and water available ad libitum. To reduce undue stress, rats were handled daily until implantation of EEG electrodes and twice weekly thereafter.
EEG electrode implantation
Upon reaching 350 ± 50 g (at around PND 70), rats underwent surgery for EEG electrode implantation. Rats were anaesthetized via isoflurane/air mixture and positioned in a stereotaxic frame (Paxinos & Watson, 1998). A midline incision exposed the dorsal surface of the skull and another incision reflected the right temporalis muscle, exposing the temporal bone. Stainless- steel screw electrodes with wire leads were implanted epidurally over the vertex (AP −4.0 mm, ML −1.0 mm, VD 0.0 mm), temporal cortex (AP −4.5 mm, ML −7.5 mm, VD −4.0 mm), cerebellum (ground) and frontal sinus (reference). Stainless-steel lead wires were connected to an Amphenol plug and the entire assembly was secured to the skull with dental cement. All ASSRs reported here were recorded from the temporal electrode, as the vertex electrode was implanted for another study.
Drugs
The GABAA agonist muscimol hydrobromide (Sigma) and antagonist (−)-bicuculline methiodide (Sigma) were dissolved in physiological saline for subcutaneous (s.c.) injection. The concentration of both drugs was adjusted to the dose of 1 mg/kg of body weight, and mixed at 1 mg/ml. The dosage of muscimol administered was selected after a pilot study (data not shown) which tested doses between 1 and 2 mg/kg of body weight and showed that the 1 mg/kg dose was sufficient to detect significant differences from saline. Further, equivalent doses of both drugs were administered because other studies have shown that a 1 mg/kg concentration of bicuculline can antagonize behavioural effects induced by 1 mg/kg muscimol (Houston et al. 2002). Physiological saline, with an altered pH of 3.5 to match the lower pH drug (muscimol), was also used alone as a control condition at a volume of 0.3 ml.
Electrophysiological recording procedure
Recording commenced following 2 wk of recovery from surgery (approximately PND 84). Rats were placed in a five-sided plastic mesh cage in an electrically shielded chamber, where they were acclimated for recording. A female connector, with flexible, insulated wires was attached to the Amphenol plug. Single trial EEG data was collected (band pass 1–300 Hz), digitized (1000 Hz), displayed on a computer monitor and written to the hard drive. Each trial epoch was 899 ms, including a 160-ms pre-stimulus baseline. Stimulus presentation was performed using Presentation® software version 0.70 (www.neurobs.com). ASSRs were elicited in blocks of 200 trials, or 500-ms trains of clicks (1-ms click duration, 80 dB, inter-stimulus interval of 1000 ms), presented through a speaker mounted on the front of the chamber. Time of testing was held constant for each rat, with either morning (07 :00–12:30 hours) or afternoon (13:00–18:30 hours) assignment.
Figure 1 illustrates the electrophysiological recording procedure. Each recording session began with the acclimation period (20 min), followed by the first injection. The first injection was pH-altered physiological saline (0.3 ml) in the saline and muscimol conditions. In the bicuculline (bicuculline effects on muscimol-mediated changes, i.e. bicuculline + muscimol) condition, the first injection was bicuculline (1 mg/kg s.c.). After a 10-min delay, a second injection of either saline (saline condition) or muscimol (1 mg/kg s.c. ; muscimol and bicuculline + muscimol conditions) was administered. The initial saline injection was included in the saline and muscimol conditions to avoid injection confounds associated with the bicuculline injection in the bicuculline + muscimol condition. After the second injection, a single block of trials was collected (block 1, time 0), followed by a 20-min drug absorption period, observed in all conditions. A second recording block (block 2), 20 min following the second injection, was then recorded. Additional blocks of ASSRs were then elicited at 60 min (block 3) and 100 min (block 4) following the second injection. The time allotted between blocks of trials was included to minimize potential habituation effects to repeated blocks of stimuli.
Fig. 1.

Electrophysiological recording procedure. The three drug conditions were performed at least 24 h apart. Each rat was allowed a 20-min acclimation period followed by the first injection. The first injection was saline (0.3 ml s.c.) in the saline and muscimol conditions and bicuculline (1.0 mg/kg s.c.) in the bicuculline + muscimol (Bic + Mus) condition. A 10-min delay was followed by a second injection of saline (0.3 ml in saline condition) or muscimol (1 mg/kg in both the muscimol and bicuculline + muscimol conditions) and then immediately by the first block of auditory steady-state responses (ASSRs) (block 1, time 0). Exactly 20 min (absorption period, indicated in saline condition) following the second injection, the second block of trials was presented. Four blocks of 200 trials of 500-ms click train stimuli were presented at 0 min (block 1), 20 min (block 2), 60 min (block 3), and 100 min (block 4) after the second injection. An average ASSR waveform is presented here to represent each block of trials. Amplitude (mV) is on the y axis and trial duration (ms) is on the x axis. The two vertical lines in each average represent onset (left, 0 ms) and offset (right, 500 ms) of the click train (labelled in saline condition).
Experiments were performed to : (1) test for reduced 40-Hz ASSRs in NVHL rats compared to sham rats, as well as the stability of 40-Hz ASSRs to ensure our measures were replicable across blocks of trials ; (2) test for potential effects of GABAA agonism on ASSRs in sham and NVHL rats ; and (3) establish that expected effects and interactions were due to manipulation of the GABAA receptor. ASSRs elicited at time 0 post-injection of saline (0.3 ml s.c.) from sham (n = 21) and NVHL (n = 18) rats were used for the 40-Hz comparison, while ASSRs at multiple time-points were used to demonstrate the stability of the ASSR response (see Fig. 1, saline condition). To investigate the role of GABAA receptors, ASSRs were elicited from sham and NVHL rats at multiple points in time following the administration of muscimol (1.0 mg/kg s.c.) (see Fig. 1, muscimol condition). Finally, a randomly selected subset of NVHL (n = 12) and sham (n = 12) rats were used to confirm GABAA receptor manipulation. At least 24 h after the muscimol recording session, rats received an injection of bicuculline (1 mg/kg s.c.), a GABAA antagonist selected to block the effect of the muscimol on GABAA receptors, followed by a muscimol (1 mg/kg s.c.) injection (see Fig. 1, bicuculline + muscimol condition).
Histology
Following completion of recordings, NVHL and sham control rats were sacrificed (deep isoflurane anaesthesia and decapitation) and brains were sectioned on a cryostat into coronal slices 40-μm thick and spaced ~400 μm apart. Once mounted and fixed with chloroform, each section was submitted to Nissl staining with 0.05% thionin. The extent of the lesions was determined by microscopic assessment of thionin-stained sections from ~3.30 mm to 5.80 mm posterior to bregma, by an experimenter (R.A.C.) blind to electrophysiological results. NVHL rats that did not meet criteria for successful hippocampal lesions and sham rats with damage to the hippocampi or surrounding regions were excluded from further analyses. Two sham lesion control rats were excluded due to needle-track damage to the surrounding cortical areas. Eleven NVHL rats were excluded due to inaccurate or unilateral lesions. Data from each of the remaining rats (sham, n = 21; NVHL, n = 18) was then analysed. Figure 2 shows histological results, with the greatest (black) and least (grey) extent of lesion damage allowed for rats included in the study.
Fig. 2.
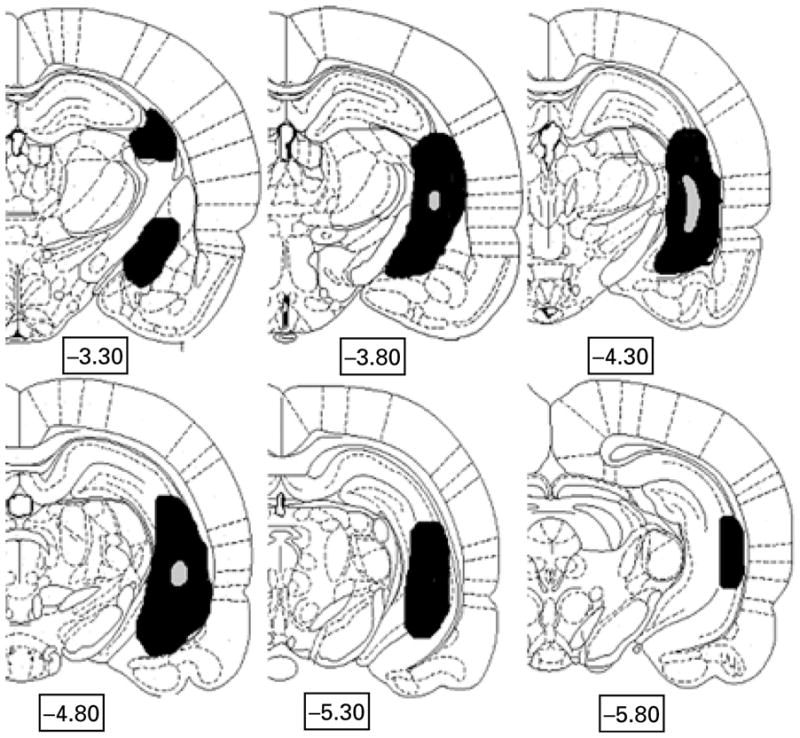
Extent of ventral hippocampal lesion (VHL) damage across neonatal VHL rats included in the study (n = 18). Hemicoronal representations showing the extent of neonatal ibotenic-acid-induced lesions of the ventral hippocampal region. Grey areas ( ) represent the smallest lesion size (on either side) of subjects accepted for analysis, and solid black areas (■) represent the largest areas. The majority of rats had lesion sizes falling between these extremes. Numbers below sections indicate distance posterior to bregma based on coordinates of Paxinos & Watson (1998).
) represent the smallest lesion size (on either side) of subjects accepted for analysis, and solid black areas (■) represent the largest areas. The majority of rats had lesion sizes falling between these extremes. Numbers below sections indicate distance posterior to bregma based on coordinates of Paxinos & Watson (1998).
Data analysis
ASSR data analysis was performed using specialized code for MatLab® (R2007a; Mathworks, USA). Data was segmented into individual trials and baseline-corrected to control for waveform polarity drift away from 0 mV. Time-frequency analyses were applied to the individual trial EEG data. Trials were first submitted to a 100% Hanning taper, a transformation commonly used when a fast Fourier transformation (FFT) is performed on a short duration time series (or sliding window). This taper is applied to the windowed EEG segments before performing FFT to reduce bias in the estimation of both power and phase (Percival & Walden, 1993). A time-frequency spectrogram, using the short-time sliding window (128 ms) FFT, was then obtained. The time-frequency spectrogram returns an output [F̃(f, t)] for each frequency (f) and time step (t) in each trial. In other words, FFT is used as a transform from time-to frequency-domain. The sliding window FFT was then moved with 10-ms time-steps to perform the spectrogram analysis. Two measures of ASSR activity, mean trial power (MTP) and phase-locking factor (PLF), were obtained from the spectrogram by averaging across trials for the normalized complex output for every time period and frequency (Delorme et al. 2002; Makeig et al. 2004; Tallon-Baudry et al. 1997). MTP was computed by determining the power, or squared amplitude, of individual trials, then subtracting the mean power of the baseline period (160 ms before stimulus onset) before averaging together the resulting power across trials. The PLF was computed by determination of phase consistency of EEG activity across trials at particular time intervals and frequencies. Therefore, while MTP provides an estimate of the average magnitude of response, PLF estimates the degree of phase synchronization across all trials presented in a block. In testing for frequency abnormalities, average MTP and PLF values were obtained for the interval between 105 and 405 ms. MTP and PLF were examined in 1-Hz windows, around the frequency of stimulation (e.g. 39–41 Hz).
Statistical analysis was performed using SPSS for Windows version 16.0 (SPSS Inc., USA). Analysis of variance (ANOVA) and repeated-measure ANOVAs were performed for each hypothesis. To test for a 40-Hz deficit in NVHL rats and to determine stability of ASSR across time, a within-subject factor time (4 : 0 min, 20 min, 60 min, 100 min) and between-subject factor lesion (2 : NVHL vs. sham) was performed. To test hypotheses regarding the effects of altered GABAA transmission, two repeated-measure ANOVAs were performed testing for lesion (2) × drug (3 : saline and muscimol or biculline + muscimol) × time (4). Effects and interactions were elucidated using univariate ANOVAs and dependent-sample t tests. Effect size, as measured by partial eta squared ( ) was calculated to reflect strength of association (Pierce et al. 2004). is the proportion of the effect variance to the effect plus error variance [ ], with higher values reflecting stronger effects. Statistical significance was considered as p<0.05.
Horizontal and vertical movement
Horizontal and vertical movements were documented during recording. Horizontal movement was defined as the number of times the rat crossed a midline point in the recording chamber. Vertical movements were defined as the animal ‘rearing’, lifting his front paws from the recording chamber floor for a minimum of 5 s.
Results
40-Hz ASSRs: NVHL and sham rat comparison and stability across time
Following the saline recording procedure (Fig. 1, saline condition), neither MTP nor PLF differed for NVHL or sham rats in the generation and maintenance of the 40-Hz ASSR at time 0 (see Fig. 3). While qualitatively, it appears that the NVHL group has higher MTP than sham rats, this difference is not significant [F(1, 38) = 0.09, , p = n.s.].
Fig. 3.

Average mean trial power (MTP) and phase-locking factor (PLF) across time after muscimol (1 mg/kg) administration in sham and neonatal ventral hippocampal lesion (NVHL) rats. The figure shows trial duration time (0–600 ms) on the x axis and frequency (0–80 Hz) on the y axis. PLF (PLF units, 0 to 0.25) and MTP (power magnitude, −0.5 to 2) indicated by colour, with hot colours (reds and oranges) indexing more activation and cool colors (blues and greens) indexing less activation. Stimuli onset occurs at 0 ms and ends at 500 ms. No difference between the sham (n = 21) and NVHL (n = 21) groups is apparent at 0-min post-injection. However, as the muscimol is absorbed, drug × lesion interactions are observed for both MTP and PLF (* p<0.05). With MTP, drug × lesion interactions were present at 60 and 100 min (* p<0.05). For PLF, a drug × lesion × time interaction was observed (*** p = 0.001). Further analysis detected drug × lesion interactions at 20 min (* p<0.05), and 60 min (** p<0.01) post-injection. The 100-min main effect of drug (p<0.05) is also demonstrated in this figure.
ASSRs were stable over time in both sham and NVHL rats. Specifically, average MTP between 105 and 495 ms post-stimulation were stable and yielded no effects of time [F(2, 62) = 1.53, , p = n.s.], lesion [F(1, 31) = 0.46, , p = n.s.] or lesion × time interaction [F(2, 62) = 1.08, , p = n.s.]. The average PLF measure, within the same time window, also reflected stability with no effects of time [F(2, 70) = 0.54, , p = n.s.], lesion [F(1, 35) = 0.07, , p = n.s.], or lesion × time interactions [F(2, 70) = 0.60, , p = n.s.].
Effects of muscimol on ASSRs at 40 Hz across time
Muscimol (1 mg/kg) produced lesion × drug interactions on ASSRs, with sham rats having increased MTP and PLF and NVHL rats having reduced MTP and PLF, following drug administration (Fig. 3). Repeated-measures ANOVA with factors time (4), drug (2), and lesion (2) was performed for each measure (MTP and PLF). The MTP measure yielded a main effect of time [F(3, 105) = 5.65, , p = 0.001], as well as time × lesion [F(3, 105) = 7.26, , p<0.001], drug × lesion [F(1, 35) = 6.05, , p = 0.02], and time × drug [F(3, 105) = 2.88, , p<0.05] interactions. Additional ANOVAs determined there were drug × lesion interactions at 60 min [F(1, 36) = 4.50, , p<0.05] and 100 min [F(1, 36) = 7.19, , p = 0.01] after muscimol administration. These effects were due to an increase in MTP for the sham group and decrease in MTP for the NVHL group after muscimol administration. The PLF ANOVA yielded main effects of time [F(3, 63) = 3.06, , p<0.05] and drug [F(1, 21) = 6.62, , p<0.05], as well as drug × lesion [F(1, 21) = 7.18, , p<0.05], time × lesion [F(3, 63) = 6.23, , p = 0.001], and time × drug × lesion [F(3, 63) = 6.23, , p = 0.001] interactions. To clarify these effects and interactions, additional ANOVAs determined that, similar to MTP, muscimol increased phase locking in sham rats and reduced phase locking in NVHL rats to 40-Hz stimuli at 20 min [F(2, 39) = 8.27, , p = 0.001] and 60 min [F(2, 40) = 5.73, , p<0.01] post-injection. A main effect of drug was then found at 100-min post-injection [F(1, 21) = 8.11, , p = 0.01]. Figure 3 illustrates detected lesion × drug interactions for both MTP and PLF measures.
Effects of bicuculline on muscimol-mediated changes
To demonstrate that manipulation of the GABAA receptor was associated with the observed muscimol effects and interactions, a third experiment tested the action of bicuculline methiodide (1 mg/kg), a GABAA receptor antagonist, on muscimol (see Fig. 4). Both MTP and PLF results suggest that the observed muscimol × lesion interactions were probably due to the agonism of the GABAA receptor by muscimol. As reported above for MTP [F(2, 23) = 5.46, , p< 0.05] and PLF [F(2, 23) = 5.46, , p<0.05], comparing saline to muscimol alone resulted in significant drug × lesion interactions. However, examining MTP [F(1, 18) = 1.29, , p = n.s.] and PLF [F(1, 18) = 1.40, , p = n.s.] with saline and bicuculline + muscimol, no effects or interactions were detected, indicating that the GABAA receptor was manipulated as bicuculline blocked the effects of muscimol agonism (Fig. 4).
Fig. 4.

Average phase-locking factor (PLF) at 40 Hz for a subset of rats, NVHL (□), sham (■), and combined groups of rats, after the administration of saline, muscimol (1 mg/kg), and bicuculline blocked (1 mg/kg) muscimol (1 mg/kg). In the muscimol alone condition, a muscimol × lesion interaction resulted, with PLF increased in sham and decreased in NVHL rats (* p<0.05). However, bicuculline antagonized the muscimol-mediated changes. Error bars reflect standard error of the mean.
Horizontal and vertical movement
No significant lesion differences in horizontal or vertical movement were detected. After the 20-min acclimation period, before injection and recording, all rats were relatively inactive and, therefore, no effect of muscimol on sham or NVHL movement was detected. Finally, because rats were freely moving during recording and movement trials were not removed prior to analysis, Pearson correlation coefficients (by lesion, drug, and collapsed across groups) were performed in order to detect any relationship between observed movement and electrophysiological measures. No correlations were detected between behaviour and ASSR measures, suggesting that movement did not produce artifacts.
Discussion
While the NVHL rat model of schizophrenia clearly demonstrates many phenotypes of the disorder, such as behavioural, pharmacological, and molecular changes (Lipska & Weinberger, 2000; Tseng et al. 2008a), the 40-Hz ASSR deficit observed in patients with schizophrenia was not observed in this study. Nevertheless, the present data suggest two important findings: (1) a lesion × drug interaction suggests potential abnormalities of the GABAA receptor in the neuronal networks responsible for ASSR generation in NVHL rats ; and (2) in-vivo GABAA agonism changes the response characteristics of 40-Hz magnitude (MTP) and synchronization (PLF), further supporting its role in ASSR generation.
Several studies have reported abnormal GABAergic transmission in NVHL rats, including reduced markers of inhibitory transmission in the prefrontal cortex (Lipska et al. 2003a, b; Tseng et al. 2008a, b) and GABAA receptor up-regulation (Endo et al. 2007; Mitchell et al. 2005). While little is known about GABAA abnormalities in most cortical regions of NVHL rats, it is possible that, like patients with schizophrenia, similar functional alterations occur in areas beyond the prefrontal cortex (Endo et al. 2007; Mitchell et al. 2005) such as the temporal cortex (Deng & Huang, 2006). Indeed, increased GABAA receptor density in the superior temporal gyrus has been shown in patients with schizophrenia (Deng & Huang, 2006). Further, neuroimaging studies point to the importance of temporal cortex in the pathophysiology of schizophrenia, as reduced temporal lobe volume is one of the most replicable findings in the disorder (Keshavan et al. 2008; Shenton, 2001), and may result in pathological phenomenon such as auditory hallucinations (Gaser et al. 2004). A recent MRI investigation showed that NVHL rats also have reduced temporal activity, as measured by decreased perfusion in the temporal cortex (Risterucci et al. 2005), suggesting altered cortical structure and function. However, there is no direct evidence of altered GABAergic transmission in NVHL temporal cortex. This is the first study to indirectly test this disturbance. Further cellular investigation will be necessary to elucidate potential temporal cortex GABAergic alterations.
Increasing inhibition, via administration of the GABAA agonist muscimol, resulted in power and phase-locking values that were increased in sham rats and greatly decreased in NVHL rats. This finding is consistent with the hypothesis that GABAA receptors have an important role in ASSR generation and maintenance (Lewis et al. 2005), with synchronization propagated through neuronal networks mediated by GABAA in cycles of inhibition and rebound excitation (Lewis & Gonzalez-Burgos, 2008). The increased synchronization observed in sham rats may reflect an inhibitory-driven increase in synchronization within a neural population, while the present finding of an interaction between GABAA agonism and lesion, suggests abnormalities involving the GABAA receptor in the NVHL temporal cortex. Decreased levels of intrinsic inhibition in NVHL temporal cortex could result in GABAA receptor up-regulation, as a compensatory mechanism for reduced transmission (Jarskog et al. 2007). Flooding the GABAA receptors with muscimol would, therefore, result in over-inhibition and a network unable to generate or maintain 40-Hz ASSRs in NVHL rats. In Fig. 3 for instance, after muscimol administration, a failure of activation at stimulus onset and maintenance of that activation throughout the trial duration window is observed.
The present findings pertaining to the GABAergic system in ASSRs and the NVHL rat model of schizophrenia further expand our understanding of normal and disturbed neural synchronization. However, unlike data from patients with schizophrenia, a 40-Hz ASSR reduction was not detected in NVHL rats. This discrepancy may be due to a number of factors. First, is the species difference; although 40-Hz ASSRs are thought to be primarily generated in temporal cortex in humans (Pastor et al. 2002; Picton et al. 2003) and rats (Franowicz & Barth, 1995), the anatomical structure of this area still differs somewhat across species. Second, there is no established evidence to date that suggests that 40 Hz is the resonant frequency, or the frequency of optimal response, for examining potential NVHL differences. Indeed, our preliminary data (Vohs et al. 2009) suggest that the frequency of maximal response in healthy rats may be around 50 Hz. Therefore, it is possible that lesion differences may have been detected with a 50-Hz stimulation frequency. This data is in line with Conti and colleagues (1999), who also found that the maximal ASSR amplitude was generated at 50 Hz, when comparing frequencies 30–60 Hz. Recently, Pèrez-Alcàzar et al. (2008) used ‘chirp’ evoked potentials to examine the optimal frequency of response in rats between 0 and 250 Hz and found two increases in response strength at 60 and 130 Hz. The 50-Hz optimal frequency of response found by Conti et al. (1999) and in our (unpublished) data may differ from Pèrez-Alcàzar et al. (2008) due to the stimulus parameters (i.e. click train vs. ‘chirp’ stimuli). Nevertheless, further investigation, using ASSR methods parallel to what is being used in human patients with schizophrenia is needed to fully address the issue of frequency response in rat models of the disorder. A third explanation for the discrepancy between the present findings and human studies in patients with schizophrenia is that, unlike patients, our subjects had no current of history of exposure to treatment medications or abused substances (see Buckley, 1998) that might alter ASSRs. In contrast to studies of patients with schizophrenia, NVHL rats were not submitted to antipsychotic medication or drugs of abuse at any point throughout the study.
There were a few issues concerning the interpretation of the present results. One potential limitation of the present investigation involves the nature of systemic injections, which were used in the pharmacological manipulations. With a systemic injection, it is unclear at what time- point, and how much of a drug is absorbed into the brain. However, this concern was addressed via repeated blocks of trials, across time, within a single recording session. Future investigations, using more exacting drug administration methods, should be performed to replicate and extend the current findings. Using direct methods of quantifying receptor number or molecular precursors in the temporal cortex of NVHL rats should be performed to more directly test the assertion that GABAA receptors are indeed up-regulated. With regards to GABAA receptor involvement in ASSR generation and maintenance, the use of deep electrode local-field potentials or single-unit recordings combined with pharmacological manipulation would provide a better understanding of this phenomenon on a cellular level. Moreover, while this study focused on the effects of GABAA agonism on ASSRs, the effect of the manipulation on baseline EEG was not determined. Given that baseline EEG may contribute to ASSR measures, effects of this pharmacological manipulation on baseline EEG should be systematically examined in future investigations. Finally, the potential role of other receptor systems, such as the glutamatergic N-methyl-D-aspartate (NMDA) receptor, which probably contributes to neuronal synchronization via a continuous cycle of excitation (NMDA) and inhibition among excitatory pyramidal cells and inhibitory interneuronal networks (Traub et al. 1996), has not been addressed in the present investigation. Therefore, examination of other such receptor systems may be a valuable future direction for research.
In conclusion, the present study showed that ASSR parameters were stable across time in both NVHL and sham rats. Moreover, the manipulation of the GABAA receptor by muscimol, a GABAA agonist, yielded decreased ASSR measures in NVHL rats and increased ASSR measures in their sham lesion counterparts. These data suggest an alteration involving GABAA receptor transmission in the temporal cortex of NVHL rats and warrant further investigation. In addition, these data are consistent with findings suggesting the importance of GABAergic transmission for ASSR generation and maintenance. Finally, the GABAA receptor subtype has properties and expression that make it well suited as a potential molecular target for novel treatment development (Möhler et al. 2008; Wassef et al. 2003), therefore, using an in-vivo animal model to characterize the role of GABAA receptors in ASSR generation has set the stage for future studies, impossible to perform in human subjects, which will advance our understanding of ASSR generation and abnormalities observed in patients with schizophrenia.
Acknowledgments
This work was supported in part by the Orvis Fund Intercampus IUMS Psychiatry Training Grant (SLM); NIDA K08 DA 019850 (RAC). J.L.V. was supported by the Research Training in Clinical Science Award, NIMH T32 MH17146 (Viken) and the Indiana CTSI Predoctoral Training Award; PHS (NCCR) grant no: TL1RR02575 (Shekhar). We express sincere gratitude to Joseph Cahall, Samuel Kaiser, Tammie Nelson, and Susan Conroy, for their technical assistance on this project.
Footnotes
Statement of Interest
None.
References
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder [Review] Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Brenner CA, Sporns O, Lysaker PH, O’Donnell BF. EEG Synchronization to modulated auditory tones in schizophrenia, schizoaffective disorder, and schizotypal personality disorder. American Journal of Psychiatry. 2003;160:2238–2240. doi: 10.1176/appi.ajp.160.12.2238. [DOI] [PubMed] [Google Scholar]
- Buckley PF. Substance abuse in schizophrenia : a review. Journal of Clinical Psychiatry. 1998;59 (Suppl 3):26–30. [PubMed] [Google Scholar]
- Conti G, Santarelli R, Grassi C, Ottaviani F, et al. Auditory steady-state response click trains from the rat temporal cortex. Clinical Neurophysiology. 1999;110:62–70. doi: 10.1016/s0168-5597(98)00045-8. [DOI] [PubMed] [Google Scholar]
- Deng C, Huang X-F. Increased density of GABAA receptors in the superior temporal gyrus in schizophrenia. Experimental Brain Research. 2006;168:587–590. doi: 10.1007/s00221-005-0290-9. [DOI] [PubMed] [Google Scholar]
- Delorme A, Makeig S, Fabre-Thorpe M, Sejnowski T. From single-trials EEG to brain area dynamics. Neurocomputing. 2002;44–46:1057–1064. [Google Scholar]
- Endo K, Hori T, Abe S, Asada T. Alterations in GABAA receptor expression in neonatal ventral hippocampal lesioned rats : comparison of prepubertal and postpubertal periods. Synapse. 2007;61:357–366. doi: 10.1002/syn.20393. [DOI] [PubMed] [Google Scholar]
- Franowicz MN, Barth DS. Comparison of evoked potentials and high-frequency (gamma-band) oscillating potentials in rat auditory cortex. Journal of Neurophysiology. 1995;74:96–112. doi: 10.1152/jn.1995.74.1.96. [DOI] [PubMed] [Google Scholar]
- Gaser C, Nenadic I, Volz H-P, Büchel C, et al. Neuroanatomy of ‘hearing voices’ : a frontotemporal brain structural abnormality associated with auditory hallucinations in schizophrenia. Cerebral Cortex. 2004;14:91–96. doi: 10.1093/cercor/bhg107. [DOI] [PubMed] [Google Scholar]
- Houston AJ, Wong JCL, Ebenezer IS. Effects of subcutaneous administration of γ-aminobutyric acidA receptor agonist muscimol on water intake in water-deprived rats. Physiology & Behavior. 2002;77:445–450. doi: 10.1016/s0031-9384(02)00876-4. [DOI] [PubMed] [Google Scholar]
- Jarskog LF, Miyamoto S, Lieberman JA. Schizophrenia : new pathological insights and therapies. Annual Review of Medicine. 2007;58:49–61. doi: 10.1146/annurev.med.58.060904.084114. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Tandon R, Boutros NN, Nasrallah HA. Schizophrenia, ‘ just the facts’ : what we know in 2008. Part 3 : Neurobiology. Schizophrenia Research. 2008;106:89–107. doi: 10.1016/j.schres.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Krishnan GP, Hetrick WP, Brenner CA, Shekhar A, O’Donnell BF. Steady state and induced auditory gamma deficits in schizophrenia. Neuroimage. 2009 doi: 10.1016/j.neuroimage.2009.03.085. Published online : 14 April 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JS, O’Donnell BF, Wallenstein GV, Greene RW, et al. Gamma frequency-range abnormalities to auditory stimulation in schizophrenia. Archives of General Psychiatry. 1999;56:1001–1005. doi: 10.1001/archpsyc.56.11.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K-H, Williams LM, Breadkspear M, Gordon E. Synchronous gamma activity : a review and contribution to an integrative neuroscience model of schizophrenia. Brain Research Reviews. 2003;41:57–78. doi: 10.1016/s0165-0173(02)00220-5. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33:141–165. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nature Review Neuroscience. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia : convergence of γ-aminobutyric acid and glutamate alterations. Neurological Review. 2006;63:1372–1376. doi: 10.1001/archneur.63.10.1372. [DOI] [PubMed] [Google Scholar]
- Light GA, Hsu JL, Hsieh MH, Meyer-Gomes K, et al. Gamma band EEG oscillations reveal neural network cortical coherence dysfunction in schizophrenia patients. Biological Psychiatry. 2006;60:1231–1240. doi: 10.1016/j.biopsych.2006.03.055. [DOI] [PubMed] [Google Scholar]
- Lins OG, Picton TW. Auditory steady-state responses to multiple simultaneous stimuli. Electroencephalography Clinical Neurophysiology. 1995;96:420–432. doi: 10.1016/0168-5597(95)00048-w. [DOI] [PubMed] [Google Scholar]
- Lipska BK. Using animal models to test a neurodevelopmental hypothesis of schizophrenia. Journal of Psychiatry and Neuroscience. 2004;29:282–286. [PMC free article] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weickert CS, et al. Gene expression in dopamine and GABA systems in an animal model of schizophrenia : effects of antipsychotic drugs. European Journal Neuroscience. 2003a;18:391–402. doi: 10.1046/j.1460-9568.2003.02738.x. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weinberger DR. The neonatal ventral hippocampal lesion model of schizophrenia : Effects on dopamine and GABA mRNA markers in the rat midbrain. European Journal Neuroscience. 2003b;18:3097–3104. doi: 10.1111/j.1460-9568.2003.03047.x. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology. 2000;23:223–239. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- Makeig S, Debener S, Onton J, Delorme A. Mining event-related brain dynamics. Trends in Cognitive Science. 2004;8:204–210. doi: 10.1016/j.tics.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Mitchell CP, Grayson DR, Goldman MB. Neonatal lesions of the ventral hippocampal formation alter GABA-A receptors subunit mRNA expression in adult rat frontal pole. Biological Psychiatry. 2005;57:49–55. doi: 10.1016/j.biopsych.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Möhler H, Rudolph U, Boison D, Singer P, et al. Regulation of cognition and symptoms of psychosis: Focus on GABA-A receptors and glycine transporter 1’. Pharmacology, Biochemistry, and Behavior. 2008;90:58–64. doi: 10.1016/j.pbb.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Pastor MA, Artieda J, Arbizu J, Marti-Climent JM, et al. Activation of human cerebral and cerebellar cortex by auditory stimulation at 40 Hz. Journal of Neuroscience. 2002;22:10501–10506. doi: 10.1523/JNEUROSCI.22-23-10501.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1998. [Google Scholar]
- Percival DB, Walden AT. Spectral Analysis for Physical Applications. Cambridge, UK: Cambridge University Press; 1993. [Google Scholar]
- Pèrez-Alcàzar M, Nicolàs MJ, Valencia M, Alegre M, et al. Chirp-evoked potentials in the awake and anesthetized rat. A procedure to assess changes in cortical oscillatory activity. Experimental Neurology. 2008;210:144–153. doi: 10.1016/j.expneurol.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Picton TW, John MS, Dimitrijevic A, Purcell D. Human auditory steady-state responses. International Journal Audiology. 2003;42:177–219. doi: 10.3109/14992020309101316. [DOI] [PubMed] [Google Scholar]
- Pierce CA, Block RA, Aguinis H. Cautionary note on reporting eta-squared values from multifactor ANOVA designs. Educational and Psychological Measurement. 2004;64:916–924. [Google Scholar]
- Regan D. Human Brain Electrophysiology : Evoked Potentials and Evoked Magnetic Fields in Science and Medicine. Amsterdam: Elsevier; 1989. [Google Scholar]
- Risterucci C, Jeanneau K, Schoppenthau S, Bielser T, et al. Functional magnetic resonance imaging reveals similar brain activity changes in two different animal models of schizophrenia. Psychopharmacology (Berlin) 2005;180:724–734. doi: 10.1007/s00213-005-2204-8. [DOI] [PubMed] [Google Scholar]
- Shenton M. A review of MRI findings in schizophrenia. Schizophrenia Research. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer KM, Salisbury DF, Shenton ME, McCarley RW. γ-band activity auditory steady-state responses are impaired in first episode psychosis. Biological Psychiatry. 2008;64:369–375. doi: 10.1016/j.biopsych.2008.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallon-Baudry C, Betrand O, Depuuech C, Permier J. Oscillatory gamma-band (30–70 Hz) activity induced by a visual search task in humans. Journal of Neuroscience. 1997;17:722–734. doi: 10.1523/JNEUROSCI.17-02-00722.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Whittington MA, Standford JG, Jefferys A. A mechanism for generation of long-range synchronous fast oscillation in the cortex. Nature. 1996;383:621–624. doi: 10.1038/383621a0. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Chambers RA, Lipska BK. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behavioural Brain Research. 2008a doi: 10.1016/j.bbr.2008.11.039. Published online : 3 December 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Lewis BL, Hashimoto T, Sesack SR, Kloc M, Lewis DL, O’Donnell P. A neonatal ventral hippocampal lesion causes functional deficits in adult prefrontal cortical interneurons. Journal of Neuroscience. 2008b;28:12691–12699. doi: 10.1523/JNEUROSCI.4166-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Haenschel C, Nikolic D, Singer W. The role of oscillations in cortical networks and their putative relevance for the pathophysiology of schizophrenia. Schizophrenia Bulletin. 2008;34:927–943. doi: 10.1093/schbul/sbn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohs JL, Chambers RA, Krishnan GP, O’Donnell BF, et al. Auditory sensory gating in the neonatal ventral hippocampal lesion model of schizophrenia. Neuropsychobiology. doi: 10.1159/000234813. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohs JL, Chambers RA, Krishnan GP, O’Donnell BF, et al. Evoked gamma band response in the neonatal ventral hippocampal lesion (NVHL) rat model of schizophrenia [Abstract 679]. Society for Biological Psychiatry: 64th Annual Scientific Convention; 14–16 May 2009; Vancouver, B.C., Canada. 2009. [Google Scholar]
- Wassef A, Baker J, Kochan LD. GABA and schizophrenia : a review of basic science and clinical studies. Journal of Clinical Psychopharmacology. 2003;23:601–640. doi: 10.1097/01.jcp.0000095349.32154.a5. [DOI] [PubMed] [Google Scholar]