

Abstract
Several cytochrome P450 (CYP) enzymes catalyze the C4-hydroxylation of retinoic acid (RA), a potent inducer of cell differentiation and an agent in the treatment of several diseases. Here, we have characterized CYP2C22, a member of the rat CYP2C family with homology to human CYP2C8 and CYP2C9. CYP2C22 was expressed nearly exclusively in hepatocytes, where it was one of the more abundant mRNAs transcripts. In H-4-II-E rat hepatoma cells, CYP2C22 mRNA was upregulated by all-trans (at)-RA, and Am580, a nonmetabolizable analog of at-RA. In comparison, in primary human hepatocytes, at-RA increased CYP2C9 but not CYP2C8 mRNA. Analysis of the CYP2C22 promoter region revealed a RA response element (5 ′ -GGTTCA-(n)5-AGGTCA-3 ′) in the distal flanking region, which bound the nuclear hormone receptors RAR and RXR and which was required for transcriptional activation response of this promoter to RA in CYP2C22-luciferase-transfected RA-treated HepG2 cells. The cDNA-expressed CYP2C22 protein metabolized [3H]at-RA to more polar metabolites. While long-chain polyunsaturated fatty acids competed, 9-cis-RA was a stronger competitor. Our studies demonstrate that CYP2C22 is a high-abundance, retinoid-inducible, hepatic P450 with the potential to metabolize at-RA, providing additional insight into the role of the CYP2C gene family in retinoid homeostasis.
Keywords: retinoic acid response element, human hepatocyte, HepG2 cell, retinoic acid oxidation, in situ hybridization, polyunsaturated fatty acid
Cytochrome P450 (CYP) genes are well recognized for their important roles in the oxidative metabolism of a large variety of endogenous compounds, including steroid hormones, cholesterol, fatty acids, and bile acids; several nutrients; and numerous drugs and xenobiotic compounds (1–4). Members of several families of the cytochrome P450 superfamily have been implicated in the metabolism of vitamin A (retinol) and its principal active metabolite, all-trans-retinoic acid (RA) (5, 6), yet the roles these enzymes play in vivo is still not well understood. Retinoic acid plays a fundamental role in embryogenesis, vertebrate development, cell proliferation, and cell differentiation (7–9). Both RA and numerous synthetic analogs are important agents in the treatment of certain cancers and other disorders (9, 10). Many of the biological effects of retinoids are mediated by their binding to two classes of nuclear receptors—RARs (α, β, γ) and RXRs (α, β, γ), which form functional RAR-RXR heterodimers and, in the presence of their principal ligand, at-RA and 9-cis-RA, respectively, and are capable of regulating the expression of numerous genes (11–13). Thus, mechanisms that control the degradation of RA are integral to the regulation of retinoid-sensitive processes in many tissues.
Cytochrome P450 genes are considered to be responsible for the metabolism of RA in many tissues. The CYP family known as CYP26 has gained attention because of the specific patterns of expression of CYP26A1, CYP26B1, and CYP26C1 genes in vertebrate embryonic tissues (7, 14), where CYP26 expression has been shown to limit RA concentrations and protect the embryo from abnormal development (15). In the liver, the potential for CYP26A1 to play an important role in retinoid homeostasis was demonstrated by its rapid, high-level induction after treatment with RA (16–18). Other cytochrome P450s that may contribute to RA metabolism include, in humans, those designated as CYP3A7, 1A1, 2C8, 2C9, 3A4, 3A5, and 4A11 (5, 19, 20). Some P450s also appear to be regulated in part by RA. In rat liver, CYP2C7, a testosterone and RA-4-hydroxylase can be modulated by retinoids (21); when rats were in a vitamin A–deficient state, dietary supplementation with retinoids restored CYP2C7 expression (21, 22). Additionally, mouse recombinant and partially purified CYP2C39 (homologous to rat CYP2C7), shown to be regulated in vivo by the aryl hydrocarbon receptor and TCDD, exhibited RA-4-hydroxylase activity (23). Thus, a variety of RA-regulated or RA-catalyzing P450 activities could potentially play roles in retinoid homeostasis, but the functionality of this group of P450s is not yet well understood.
The cDNA of the gene now referred to as CYP2C22 was first cloned in 1990 as a cytochrome P450 gene induced after isolated rat hepatocytes were plated on collagen-coated culture dishes (24, 25). Neither of these studies identified RA as a regulator of gene expression or as a substrate for the predicted product. In a study of the organization of the rat CYP2C gene locus, Wang et al. (26) identified a gene described as CYP2C70 as an uncharacterized member of the rat CYP2C family. This gene occupies the 3′-end of a cluster of 15 genes and 4 pseudogenes of the CYP2C gene family on rat chromosome 1 (26). We first became aware of the retinoid inducibility of rat CYP2C70, now CYP2C22, based on microarray studies of retinoid-inducible genes in the liver of rats. CYP2C22 was listed as a gene more highly expressed in vitamin A–sufficient than vitamin A–deficient liver (27) and induced in liver by targretin and 4-hydroxyphenylretinamide (28), analogs of RA with potential chemotherapeutic properties. However, biochemical studies to characterize the mode of regulation of CYP2C22 expression and function to determine its enzymatic activity have not been reported. The results of the present study, conducted in rodent cells and human liver cells and primary hepatocytes, suggest that rat CYP2C22 and its possible human ortholog CYP2C9 are highly abundant P450s in the liver, which are rapidly inducible by RA through a canonical RA receptor–mediated mechanism, and able to metabolize all-trans (at)-RA. Since the CYP2C family also metabolizes important drugs and lipids, our results for CYP2C22 and human CYP2C8 and CYP2C9 suggest the possibility for significant drug-retinoid and lipid-retinoid interactions.
EXPERIMENTAL PROCEDURES
Cell culture
The human hepatoma cell line, HepG2, rat hepatoma cell line H-4-II-E, and human embryonic kidney cell line HEK239T were originally obtained from American Type Culture Collection and maintained as stocks in our laboratory under standard conditions. HepG2 cells and H-4-II-E cells were cultured in EMEM supplemented with 10% FBS and antibiotics (50 units/ml penicillin and 50 µg/ml streptomycin). HEK239T cells were cultured in DMEM with 10% FBS and antibiotics. All of the cells were maintained at 37°C with 5% CO2. Primary human hepatocytes on collagen-coated 6-well plates were obtained from the Department of Surgery/Transplantation Institute, University of Pittsburgh. All-trans-RA and 9-cis-RA (Sigma, St. Louis, MO) were added to cells in DMSO so that the final DMSO concentration was <0.1%.
RNA isolation and real time PCR
Total RNA was extracted from RA-treated and untreated cells and from liver tissue using Trizol Reagent (Invitrogen). CDNA was synthesized using the M-MLV Reverse Transcriptase (Promega, Madison, WI). Fluorescence real-time PCR was performed in triplicate with iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) based on a standard curve for each gene analyzed, and the relative concentrations were normalized to the level of 18S rRNA determined in parallel. PCR reactions were run in triplicate according to a protocol which consisted of 95°C for 3 min, followed by 40 PCR cycles at 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s, using specific primers for CYP2C22 [Forward (F): 5′-CATGAAGCCCACTGTGGTGCTG, Reverse (R): 5′-GGAACACAGGCCAGAAGGAAGCT, 3′), CYP2C8 (F: 5′-GGCCACTTTCTAGATAAGAATG-3′, R: 5′-TGA-TAGCAGATCGGCAGCCA-3′), CYP2C9 (F: 5′-ACTTTCTGGATGAAGGTGGC-3′, R: 5′-GTGCAAAGATGGATAATGCC-3′) and 18S rRNA (5′-AGTCCCTGCCCTTTGTACACA-3′, 5′-CCGAGGGCCTCACTAAACC-3′]. Representative PCR products were confirmed by electrophoresis on 1.2% agarose gel.
In situ hybridization
The entire coding region of CYP2C22 cDNA was subcloned into pGEM4 plasmid vector (Promega). The plasmid clone was linearized with either HindIII or EcoR-I and then used as a template to synthesize digoxigenin (DIG)-labeled sense or antisense RNA riboprobe using either SP6 or T7 RNA polymerase, respectively, in a reaction with ribonucleotides including DIG-UTP (Roche Biotechnology). The labeled RNA was isolated, checked for size by ethidium bromide-stained agarose gel electrophoresis, and then treated in alkaline solution to cut into 100 to 150 base-polynucleotide lengths as probe (29). Liver sections, 10 µm thickness, were cut from frozen liver tissue blocks of rats that were fed a vitamin A–deficient diet (30) until plasma retinol was 0.2 µM; rats that were vitamin A–deficient followed by a 2-week repletion with retinol; and rats that received an acute treatment with single oral dose of vitamin A (200 µg) plus Am580 (30 µg, a gift of Dr. H. Kagechika, Tokyo, Japan) given 6 h prior to euthanasia. Liver sections were mounted on electrostatically charged slides. The slides were first dried in acetone and then fixed with 4% paraformaldehyde in PBS as described previously (31). Following prehybridization in 50% formamide at 60°C for 10 min, each slide was covered with 150 µl of hybridization solution containing 50% formamide, 10% dextran sulfate, 1× Denhardt's solution with 50 ng of DIG-labeled riboprobe and hybridized at 42°C overnight (29). The slides were washed in 0.2× SSC at 55°C and then treated with RNaseA as described in (29). After blocking, the slides were incubated with alkaline phosphatase–conjugated anti-DIG antibody in blocking buffer obtained from Roche Biotechnology. The slides were then washed and incubated with BCIP/NBT as synthetic substrate for alkaline phosphatase (Roche) with levamisole, overnight. The slides were then washed in TE buffer, dried, and evaluated under a digital color microscope in the Center for Quantitative Cell Analysis, Pennsylvania State University.
Gene cloning and construction of plasmids
A 2580-bp fragment of DNA covering from -2591 to -12 bp upstream from the ATG start codon of the rat CYP2C22 gene was amplified by PCR from rat genomic DNA, as described previously (32) using 5′-ataggtaccCAGCTGATAAACAACTTCAGCAAAG-3′, as the forward primer and 5′-atactcgagGAACAAACACACTGGAAAGCTTG-3′, as the reverse primer. The lowercase letters in the primers contain specific restriction digestion sequences for KpnI and XhoI, respectively. The amplified product was cloned into a pGL3-basic vector (Promega) as pGL3-CYP2C22-2580 using the KpnI and XhoI restriction sites. A site-specific mutant of the CYP2C22 promoter was generated from pGL3-CYP2C22-2580 using the Phusion Site-Directed Mutagenesis Kit (New England Biolab) and its sequence was verified. The mutation was made in the RARE region of CYP2C22 at positions located at -1990 to –1974, relative to ATG start codon. A DNA fragment of 2890 bp was amplified by PCR from the 5′-flanking region of the human CYP2C9 gene with the forward primer 5′-ataggtacc CCTACATAAACTATGAGCTTT-3′ and reverse primer 5′-atactcgag TGAAGCCTTCTCTTCTTGTT-3′ and cloned into pGL3-basic vector through the KpnI and XhoI restriction sites.
Two oligonucleotide primers were designed to amplify the entire rat CYP2C22 coding region from rat liver total RNA. The 5′ primer contained a KpnI site and a Kozak sequence, 5′-ataggtacc ACCATGGCTCTCTTCATTTTTCTG-3′, and the antisense primer, 2c22R, contained a XhoI site, 5′-atactcgag TCAGACTGGAATGAAACAGAG-3′. The amplified CYP2C22 cDNA was directionally cloned into the expression vector pcDNA3.1+ (Invitrogen) and its sequence verified.
Alignment of the amino acids of rat CYP2C22 and human CYP2C8 and CYP2C9 were performed using the ClustalW2 program (33). The rat CYP2C22 sequence was deposited in Genbank as accession #EU861213.
Transient transfection and luciferase assay
All transfected plasmids were prepared using a Midi kit (Qiagen) following standard preparation methods, and quantified by UV spectrophotometry. Cell transfections were performed with Lipofectamine 2000 (Invitrogen). HepG2 cells for transfection experiments were seeded in a 24-well plate and transfected for 24 h, with a change to fresh medium containing 10% FBS before the addition of retinoids or DMSO as vehicle for 24 h. The cells were harvested and lysed to measure the luciferase activity using a Dual Luciferase Reporter Assay System (Promega). The relative firefly luciferase activities were normalized by the Renilla luciferase activities. In RA receptor cotransfection assays, various combinations of RARα/β/γ and RXRα constructs were cotransfected into the HepG2 cells along with pGL3 reporter constructs at the following ratios: pGL3-CYP2C22:pRL-TK: RARα/β/γ: RXRα, 0.75 μg: 0.05 μg: 0.2 μg: 0.2 μg). Each experiment was repeated at least three times.
Electrophoretic mobility shift assays (EMSA)
HepG2 cell nuclear protein extract (NPE) was prepared as described (34). Oligonucleotides covering -1998 to -1966 bp from ATG start codon of the rat CYP2C22 gene with DR5, 5′-ATGGTGCTGGTTCAACTGGAGGTCACCATGGAG-3′ (the direct repeat DR5 is underlined), and 5′-CTCCATGGTGA CCTCCAGTTGAACCAGCACCAT-3′,) or mutated oligonucleotides, 5′-ATGGTGCT GGTAGTACTGGAGGAGCCCATGGAG-3′ (mutations underlined and in italics) and 5′-CTCCATGGGCTCCTCCAGTACTACC AGCACCAT-3′), were first annealed, end-labeled using [γ-32P]ATP (GE Healthcare) and incubated in binding buffer (10 mM Hepes, 100 mM KCl, 0.05 mM EDTA, 2.5 mM MgCl2, 6% glycerol) with 2 µg of NPE and poly(dI-dC) (80 μg/ml) for 15 min at room temperature. Wild-type and mutant nonradioactive competitors were added to binding reactions at the indicated concentrations without and with antibodies to RARα, RARβ, RARγ and RXRα (Santa Cruz Biotechnology, Santa Cruz, CA). Samples were loaded on 7.5% nondenaturing polyacrylamide gel. After electrophoresis, the gels were vacuum-dried and exposed to X-ray film at −70°C before development.
Analysis of RA metabolism in cultured cells
Native HEK293T cells were stably transfected with pcDNA3.1-CYP2C22 or empty vector pcDNA3.1. For studies of metabolism, stably transfected cells were seeded at a concentration of 1 × 106 cells/ml in 6-well plates and grown overnight in DMEM supplemented with 10% FBS and 1 mM 5-aminolevulinic acid. Twenty-four h postseeding, the stably transfected cells were incubated in 0.4 ml of DMEM containing 1 μM (1 μCi/ml) of [11,12-3H] at-RA (1 mCi/ml, 37 mCi/μmol, from American Radiolabeled Chemicals, St. Louis, MO). All procedures involving retinoids were conducted under subdued or yellow light. After incubation for different periods of time at 37°C, reactions were stopped by adding 20 μl of 5 M HCl, after which the culture medium and cells were extracted twice with 4 ml of hexane (16) and analyzed for the presence of at-RA metabolites as described below. Reverse-phase solid phase extraction was employed to separate the oxidized at-RA metabolites (mainly 4-hydroxy- and 4-oxo-RA) from its substrate at-RA (35, 36). Standards of at-RA and 4-oxo- at-RA, 10 nmol each, were added to 8 ml of hexane-extracted organic phase and dried completely under argon gas at 37°C. The residue was dissolved in 1.25 ml of HPLC-grade acetonitrile:water (65:35, v/v) containing 10 mM acetic acid. Solid reverse phase columns (Supelclean LC-18 SPE, Supelco) were preconditioned with 8 ml of methanol followed by 2 ml of water. After the preconditioning, the samples were loaded onto the column. Polar metabolites were first eluted by three washes, each with 4 ml of acetonitrile:water (65:35, v/v), after which the unmetabolized at-RA substrate was eluted with three applications each of 4 ml of acetonitrile:water (95:5, v/v) (36). After collection of all eluant fractions, the fractions were dried, liquid scintillation cocktail was added and 3H DPM were determined by liquid scintillation spectrometry. To assess column performance, both the major metabolite and substrate fractions were dissolved in ethanol, and then subjected to scanning UV spectrophotometry to obtain the characteristic spectra of at-RA and 4-oxo-RA in the appropriate fractions.
Statistical analysis
Data are shown as the mean ± SE. Statistical significance was determined using Student's t-test or one-way ANOVA, with P < 0.05 considered significant.
RESULTS
Tissue and cell distribution of the CYP2C22 gene transcript
To evaluate the tissue distribution of CYP2C22 mRNA, total RNA was prepared from different rat tissues and subjected to qPCR using CYP2C22-specific primers and 18S rRNA primers as a control. CYP2C22 mRNA was much more abundant in the liver than in any of the other tissues examined (Fig. 1A). We detected no appreciable differences in the level of CYP2C22 mRNA in the liver of male and female rats (data not shown).
Fig. 1.

CYP2C22 expression in different rat tissues detected by real-time PCR and localization of CYP2C22 mRNA transcripts in intact liver tissue. (A) Total RNA isolated from different rat tissues was subjected to qPCR and the expression level of CYP2C22 in each tissue was normalized to 18S rRNA. Ethidium bromide–stained agarose gel of PCR products after 40 cycles of amplification is shown below. (B) In situ hybridization from frozen liver tissue (10 µm sections) of vitamin A deficient, deficient with 2-week repletion with retinol, and deficient with 6 h treatment with retinol (200 µg) plus Am580 (30 µg). Slides were hybridized to either CYP2C22-specific digoxigenin-labeled RNA riboprobes, antisense orientation in panels a, b, c, or sense orientation in panels d, e, f.
CYP2C22 transcript was analyzed by in situ hybridization in sections of liver tissue from rats in three treatment groups: vitamin A–deficient; vitamin A–repleted (control); and vitamin A–deficient treated for 6 h with a combination of vitamin A and Am580, a stable analog of RA. The CYP2C22 transcript was barely detectable in the liver of vitamin A–deficient rats (Fig. 1B, panel a). However, upon treatment with vitamin A to restore normal vitamin A status, CYP2C22 transcript level increased significantly (panel b). Upon treatment with vitamin A combined with Am580, expression increased dramatically (panel c). In each case, CYP2C22 mRNA was nearly exclusively present in hepatocytes. Staining was evident in all zones of the liver lobule from the periportal to pericentral region, but it was absent from endothelial cells and in areas of connective tissue surrounding arteries, veins, and bile ducts. Using the CYP2C22 sense RNA probe as a control, no specific staining signal was observed (Fig. 2B, panels d–f). Thus in situ hybridization revealed that the liver-specific distribution of CYP2C22 mRNA observed in Fig. 1A represents its expression in hepatocytes and that the level of CYP2C22 mRNA increases rapidly in response to retinoid treatment in vivo.
Fig. 2.
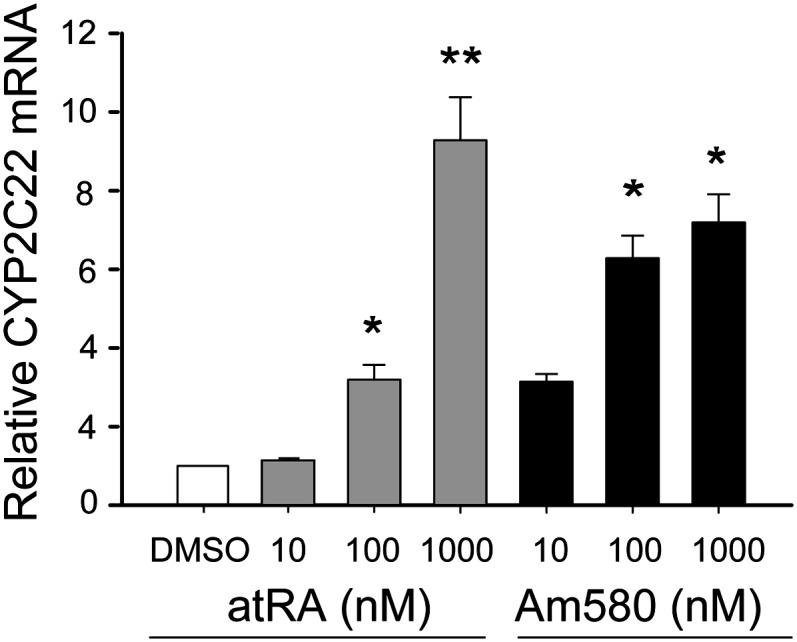
CYP2C22 mRNA is induced by retinoids in cultured H-4-II-E liver cells. H-4-II-E cells in 6-well plates were incubated in 2 ml of medium containing different concentrations of at-RA, Am580, or vehicle (DMSO) as a control. After 24 h, total RNA was extracted, and real-time PCR was utilized for CYP2C22 mRNA quantification. After normalized to the 18S rRNA internal standard, the data were expressed as the mean ± S.E; *P < 0.05; **P < 0.01 versus DMSO control.
RA and Am580 induce CYP2C22 expression in H-4-II-E cells
To test whether CYP2C22 expression is upregulated by RA in hepatocyte-like cells, the H-4-II-E rat hepatoma cell line was used as a model. When H-4-II-E cells were cultured in normal medium containing 10% fetal bovine serum in the absence of added RA, the endogenous level of CYP2C22 mRNA was almost below detection using qPCR. Upon addition of increasing concentrations of at-RA to H-4-II-E cell cultures, CYP2C22 mRNA increased concentration-dependently, with more than an 8-fold increase when 1 μM RA was added (P < 0.05 versus vehicle control; Fig. 2). As the major RAR in liver is RARα (37, 38), we also tested the response to Am580, which selectively binds to and activates RARα (39). CYP2C22 mRNA was significantly induced by 100 nM Am580, increasing up to 1 µM Am580. The low constitutive expression of CYP2C22 in the basal state compared with relatively high expression in normal liver may indicate that CYP2C22 gene expression is repressed in hepatoma cells because of tumor dedifferentiation, as a loss of retinoid metabolic functions has been ascribed previously to tumor tissue compared with normal tissue and to transformed cells compared with nontumorigenic cells (40, 41), and that a pharmacological concentration of at-RA (1 µM) may be required for strong induction of CYP2C22 in hepatoma cells.
Transcriptional activity of CYP2C22 promoter and identification of a distal RARE
Having shown that CYP2C22 expression is responsive to retinoids, we next investigated the transcriptional activity of the CYP2C22 promoter and its inducibility by RA. A 2580-bp fragment of the wild-type rat CYP2C22 promoter was amplified from genomic DNA and subcloned into the pGL3-basic luciferase reporter vector. Because very low luciferase activity was detected when the CYP2C22 promoter was transfected into H-4-II-E cells or HEK293 cells (data not shown), the human hepatoma cell line HepG2, which is also representative of hepatocytes, was used for these studies. HepG2 cells were transfected with the pGL3-CYP2C22-2580 (full length) construct or with the empty vector as a control, and then treated for 24 h with RA or Am580 (1 nM–100 nM) or DMSO vehicle as a control. Transcriptional activity, measured by the luciferase (Luc) activity of the CYP2C22 promoter-Luc construct, was increased markedly and concentration-dependently in HepG2 cells treated with either RA or Am580 (Fig. 3A). Maximum induction was achieved with concentrations of 10 nM of either RA or Am580 (P < 0.05), resulting in a 3-fold and 2.5-fold increase, respectively, in transcriptional activity.
Fig. 3.

Inducibility of CYP2C22 promoter activity by retinoids depends on a distal RARE. (A) CYP2C22 promoter construct was transfected into HepG2 cells, then luciferase activity was assessed in the presence of 1 to 100 nM at-RA, Am580, or vehicle (DMSO) only, added 24 h before the preparation of cell extracts for assay. Results were normalization to Renilla luciferase as the internal standard. *P < 0.05. (B) The sequence of RARE present in the distal region of rat CYP2C22 promoter is shown, and the consensus direct repeats of the RARE are highlighted. A deletion mutation was performed of CYP2C22-RARE (see “Experimental Procedures”) to abrogate the function of RARE. (C) Transient cotransfection assays were performed in HepG2 cells, using CYP2C22P full length (FL) and CYP2C22P DR-5M (mutations in the direct repeats; see “Experimental Procedures”) constructs described above, with and without at-RA (10 nM). The mammalian expression constructs (0.2 μg) for RARα, RARβ, RARγ, or RXRα were cotransfected where indicated, with luciferase reporter plasmid; the data were normalized and analyzed as in Fig. 3A.
Genes that respond directly to RA may possess a canonical RA response element, RARE, most often located in the region upstream of the transcription start site (13, 42), although uncommon locations have also been reported (42). The RARE motif considered canonical is composed of two direct repeats of the hexameric half-site 5′-PuG(G/T)TCA-3′), separated by two or five spacer nucleotides (DR2 and DR5) (42). Because RA rapidly induced the transcription of CYP2C22 mRNA, we examined the upstream sequence for a putative RARE within the promoter region of CYP2C22 gene. Analysis of the 5′-upstream region upstream of the ATG start codon of CYP2C22 using the TESS software (43) revealed that CYP2C22 contains a perfect RARE within the distal region, beginning at -1974 from the ATG start codon (Fig. 3B). To investigate the functionality of this putative RARE, the CYP2C22-RARE was mutated as illustrated in Fig. 3B. HepG2 cells were then transfected with either the full-length (FL) wild-type promoter construct CYP2C22P FL, or the mutated (M) promoter construct CYP2C22P DR5M, along with various combinations of expression vectors for human RARα/β/γ and RXRα. Transfection of HepG2 cells with CYP2C22P FL along with expression vectors for RA receptors resulted in more than a 6-fold increase in luciferase activity in the presence of at-RA (10 nM) compared with the activity in nontreated cells. This increase was much greater than for CYP2C22P FL in the absence either of receptors, ligands, or both (P < 0.05; Fig. 3C). In HepG2 cells transfected with the CYP2C22P DR5M promoter construct, the RA response was completed ablated, both in the presence and absence of transfected retinoid receptors. This complete loss of RA induction indicated that this distal CYP2C22-RARE contributes to the total response to at-RA.
To further confirm that the putative RARE in the CYP2C22 promoter is the target for binding of RA receptors, we performed gel electrophoretic mobility assays using HepG2 cell nuclear protein and oligonucleotides corresponding to the wild-type and mutated forms of CYP2C22-RARE as probes (Fig. 4). A protein-DNA complex was detected using the nuclear extract from HepG2 cells (lane 2). The main complex could be competed by excess unlabeled wild-type CYP2C22-RARE probe (lane 3) but not by the mutant form of CYP2C22-RARE (lane 4). A supershift assay was also performed to investigate the specific binding of RA receptors to the RARE sequence. Antibodies to RARα, RARβ, RARγ, and RXRα or their combinations RARα/RXRα, RARβ/RXRα, RARγ/RXRα were included in the coincubation along with an equal amount of nuclear extract. This supershift assay showed that the bound nuclear protein complex was retarded by antibodies to RARα, RARβ, RARγ, or RXRα (lanes 5–8). Three supershifted complexes (labeled C1, C2, and C3) were formed by inclusion of the anti-RARα antibody (lane 5, C1, C2, and C3), while only one supershifted complex was formed by inclusion of anti-RARβ/γ and anti-RXRα antibodies (lanes 6–8). A double-antibody supershift experiment showed that the simultaneous inclusion of RARα/RXRα antibodies resulted in the intensification of the three supershifted bands (lane 9), whereas the inclusion of RARβ/RXRα (lane 10) and RARγ/RXRα antibodies (lane 11) yielded a super-supershifted complex near to C2. Thus, these results agreed well with the marked increase in CYP2C22 mRNA expression by at-RA and Am580 as observed in liver tissue in vivo and in hepatocyte cell lines.
Fig. 4.

The distal RARE site in CYP2C22 promoter binds RAR and RXR. Electrophoretic mobility shift assay was performed with 32P-labeled CYP2C22-RARE–containing oligonucleotide probe bound with nuclear protein extracted from HepG2 cells. (Lane 1) Free probe only (bottom of gel omitted). (Lane 2) HepG2 cell nuclear protein extract (NPE). (Lanes 3–4) Specificity of protein-DNA binding confirmed by using 50× of unlabeled wild-type CYP2C22-RARE (DR5_WT) and mutant CYP2C22-RARE (DR5_MU) as competitors. (Lanes 5–8) Gel supershift assays conducted with individual antibodies against RARα, RARβ, RARγ, or RXRα, respectively. (Lanes 9–11) Gel supershift assays conducted with combinations of antibodies to RARα/RXRα, RARβ/RXRα or RARγ/RXRα, respectively.
CYP2C9 responds to RA in primary hepatocytes
Human CYP2C8 and CYP2C9 are two of the four members of the CYP2C family in the human genome (44, 45), and they may be involved in the metabolism of RA (5, 6). But so far, there has been no report about the regulation of expression of CYP2C8 and CYP2C9 by RA. We first compared the amino acid sequences and alignment of human CYP2C8, human CYP2C9, and rat CYP2C22 (Fig. 5). The amino acids of CYP2C22 are 60% and 62% identical to those of human CYP2C8 and CYP2C9, respectively, and ∼80% similar to both human proteins.
Fig. 5.

Amino acid comparison of rat CYP2C22 with human CYP2C8 and CYP2C9. The deduced amino acid residues of cloned rat CYP2C22 (see “Experimental Procedures”) were aligned with those of reported human CYP2C8 and CYP2C9. Rat CYP2C22 has 60% and 62% identical amino acid residues compared with human CYP2C8 and CYP2C9, respectively. Rat CYP2C22 has close to 80% amino acid similarity with both of these human CYP2C proteins.
To investigate the responsiveness of human CYP2C8 and CYP2C9 to RA, primary hepatocytes were treated with RA (10 nM) or vehicle for 10 h. CYP2C8 and CYP2C9 mRNA levels were analyzed by quantitative real-time PCR. Both CYP2C8 and CYP2C9 mRNA were detected in untreated cells, indicating that these genes are expressed constitutively in human primary hepatocytes. CYP2C8 mRNA was slightly upregulated after treatment with 10 nM RA, a low concentration. The CYP2C9 gene was significantly more responsive to RA treatment, as 10 nM RA induced CYP2C9 mRNA more than 2-fold (P < 0.05; Fig. 6A). Treatment of the primary human hepatocytes with the glucocorticoid hormone dexamethasone was included as a positive control because this steroid has been demonstrated previously to induce the expression of both CYP2C8 and CYP2C9 (46).
Fig. 6.

CYP2C9 is induced by at-RA in human hepatocytes by retinoids, and its promoter is responsive to at-RA in HepG2 cells. (A) qPCR analysis of CYP2C8 and CYP2C9 expression, normalized to 18S RNA, in primary human hepatocytes incubated with 10 nM at-RA or vehicle (DMSO) only for 10 h. *P < 0.05. (B) CYP2C9 promoter construct was transfected into HepG2 cells, then firefly luciferase activity was assessed in the presence of 10 nM to 1 μM at-RA or DMSO as control. The data were normalization by Renilla luciferase internal standard.
We also investigated the transcriptional activity of the human CYP2C9 promoter and its inducibility by at-RA or 9-cis-RA in HepG2 cells. For this analysis, a 2890-bp fragment of the human CYP2C9 promoter was amplified and subcloned into pGL3-basic luciferase reporter vector, similar to our studies on rat CYP2C22 described above. HepG2 cells were transfected with the pGL3-CYP2C9-2890 construct or with empty vector as a control. As quantified by the luciferase assay, the transcriptional activity of the CYP2C9 promoter was increased after treatment with either at-RA or 9-cis-RA. A concentration-response study testing 10 nM to 1 μM concentrations of both RA isomers showed that the addition of 1 μM of either at-RA or 9-cis-RA induced a 2-fold increase in luciferase activity (P < 0.05; Fig. 6B).
CYP2C22-transfected cells metabolize RA
The inducibility of CYP2C22 expression by at-RA prompted us to determine whether it could be a substrate for cytochrome P450. For these experiments, the full-length CYP2C22 cDNA was cloned from rat liver and inserted into a mammalian expression vector pcDNA3.1. HEK293T cells, which do not express CYP2C22 endogenously, were stably transfected with either pcDNA3.1 alone or pcDNA3.1-CYP2C22, and then incubated with 1 μM at-RA containing 0.4 μCi [11,12-3H]at-RA as tracer. After 6 h of incubation, total retinoids were extracted from each culture dish and separated on C-18 solid-phase extraction columns, after which each eluate fraction was analyzed by liquid scintillation spectrometry. By this method, polar metabolites of RA were first eluted with acetonitrile:H2O (65:35, v/v), and unmetabolized at-RA was subsequently eluted by acetonitrile:H2O (95:5, v/v). To monitor the separation, at-RA and 4-oxo-atRA were used as standards. The oxidized products mainly eluted in the first of four 3-ml eluates of acetonitrile:H2O (65:35, v/v; Fig. 7A),whereas at-RA mainly eluted in the first and the second eluates of acetonitrile:H2O (95:5, v/v). Solid-phase extraction identified more polar metabolites of [3H]RA in the medium plus cells of CYP2C22-transfected HEK293T cells, concomitant with a greater disappearance of [3H]RA substrate, compared with vector-transfected control cells (Fig. 7B). When the pcDNA3.1-CYP2C22 or pcDNA3.1-transfected HEK293T cells were incubated with [11,12-3H]at-RA for different periods of time (3, 6, and 12 h), the polar metabolites generated by CYP2C22-transfected HEK293T cells increased more rapidly than those generated by vector-transfected cells (Fig. 7C).
Fig. 7.

Analysis of CYP2C22 metabolic activity in transfected HEK293T cells. A: Reverse-phase solid-phase extraction was employed to separate the polar RA metabolites from its substrate (see “Experimental Methods”) using acetonitrile:water (65:35, v/v) and subsequently acetonitrile:water (95:5, v/v) for elution. Metabolites eluted mainly in the first 4-ml application of acetonitrile:water (65:35) eulate, and unmetabolized substrate in the first 4-ml application of the acetonitrile:water (95:5) eluate, according to the spectral properties of standards (λmax = 360 nm for 4-oxo-at-RA; λmax = 350 nm for at-RA). B: CYP2C22-transfected HEK293T cells contained more RA polar metabolites and showed a greater decrease in RA compared with pcDNA3.1-transfected control cells. C: CYP2C22 or vector-transfected HEK293T cells were incubated with [3H]at-RA for different periods of time (3, 6, and 12 h). Polar metabolites of RA were isolated as in Fig. 7A. Results in A and B are representative of N = 3 independent experiments. *P < 0.05.
Because the CYP2C family plays a role in the oxidation of long-chain polyunsaturated fatty acids (26), we further investigated the possible relationship between the RA-metabolizing activity of CYP2C22 and that of other substrates by conducting competition studies (Fig. 8A). As potential competitors, we tested at-RA itself, 9-cis-RA, retinol, arachidonic acid [(AA) n-6, 20:4], and docosahexaenoic acid [(DHA) n-3, 20:6], each added at 3 µM and assayed as described above for Fig. 7. Whereas unlabeled at-RA competed as expected, 9-cis-RA was a better competitor. Retinol had no effect, suggesting that the carboxylate group of RA is specifically recognized. Both AA and DHA [but not oleic acid (n-9,18:1) or linoleic acid (n-6,18:2) not shown] reduced the conversion of at-RA to polar metabolites, suggesting that the retinoid structure is not the only form recognized by this enzyme. Because of the marked effect of 9-cis-RA, we conducted two additional experiments. In the first, [3H]9-cis-RA was used directly in place of [3H]at-RA as a primary substrate. However, we could not detect polar metabolite formation. Second, to further examine the competitive effect of 9-cis-RA, we conducted a concentration-response study (Fig. 8B), in which [3H]at-RA was the substrate and 9-cis-RA the competitor. These results confirmed that 9-cis-RA is indeed capable of reducing the metabolism of at-RA. The reduction was half at ∼90 nM 9-cis-RA, and nearly complete at 0.5–2 µM of 9-cis-RA. These results support a role of CYP2C22 in RA oxidation, while indicating that although 9-cis-RA can inhibit the oxidation of at-RA, 9-cis-RA is unlikely to be a direct competitive inhibitor of CYP2C22.
Fig. 8.

Competition studies in transfected HEK293T cells incubated with [3H]at-RA together with unlabeled compounds as potential competitors. (A) One µM [3H]at-RA together with potential inhibitors or competitors (3 µM of at-RA, 9-cis-RA, at-retinol, arachidonic acid, or docosahexaenoic acid) or DMSO as a vehicle control, were incubated with CYP2C22-transfected HEK293T cells for 6 h. Polar metabolites of [3H]at-RA were determined as in Fig. 7. (B) Increasing concentrations of 9-cis-RA were added to cells incubated with 1 µM [3H]at-RA for 6 h.
DISCUSSION
The human CYP2C subfamily is composed of four members—CYP2C8, CYP2C9, CYP2C18, CYP2C19—that are localized within a single gene locus on chromosome 10 (44, 45). Both CYP2C8 and CYP2C9 proteins are relatively abundant, as CYP2C9 is the most highly expressed member of this family in human liver (44), and CYP2C8 constitutes about 7% of total hepatic microsomal cytochrome P450 content (47). The prototypical substrate of CYP2C8 is the anticancer drug paclitaxel (45, 48), while CYP2C9 metabolizes many clinically important drugs including the antidiabetic agent tolbutamide, the anticonvulsant phenytoin, the anticoagulant warfarin, the antihypertensive agent losartan, and nonsteroidal anti-inflammatory drugs (NSAID) (3, 49–53). It has been estimated that of 200 clinically important prescription drugs, CYP2C8 and CYP2C9 account for the metabolism of 6% and 17%, respectively (53). The functional activity of human CYP2C genes is known to be further modulated by certain common polymorphisms that have been shown to contribute to interethnic and intraethnic variability in the enzymatic function of CYP2C proteins (53, 54). Moreover, both CYP2C8 and CYP2C9 can metabolize certain endogenous compounds, such as arachidonic acid and at-RA (6, 55). In the present study, we examined a previously identified but relatively unstudied member of the rat/mouse CYP2C family, CYP2C22, which may be the ortholog of the human CYP2C9 gene, with which it shares 62% amino acid identity and 80% similarity. In the rat, the CYP2C gene cluster contains 15 genes (26), and thus the relationship of rat CYP2C genes to human CYP2C genes is not readily deduced by comparing overall gene organization. By comparison, rat CYP2C22 also shares 60% identity with rat/mouse CYP2C39 (23), which appears to be an ortholog of human CYP2C7 and is also expressed in liver. In our studies, we first tested various rat tissues for expression of CYP2C22, finding that it is predominantly expressed in the liver. The expression of CYP2C22 mRNA was readily increased in H-4-II-E cells by treatments with RA as well as Am580, a stable retinoid selective for RARα (39). The inducibility of CYP2C22 by Am580 suggests that RARα may be involved in the regulation of the CYP2C22 gene. As RARα and RXRα are the predominant forms in the liver (37, 38), and RA in liver is mainly taken up from plasma (56), CYP2C22 could be part of a sensing system through which excess RA is degraded.
Finding that CYP2C22 transcription is under the control of RA signaling prompted us to search for the mechanism through which this regulation is exerted. A RARE (5′-GGTTCAACTGGAGGTCA-3′) in the distal region of the CYP2C22 promoter corresponded to the canonical pattern of two direct repeats of the hexanucleotide motif PuG(G/T)TCA with a 5-nt spacer, optimal for binding of the RAR/RXR heterodimer (57, 58). Transfection experiments with wild-type and mutated CYP2C22 promoters confirmed that the RA inducibility was fully dependent on the presence of this RARE motif. The capacity of the CYP2C22-RARE to mediate RA response via RAR/RXR heterodimer association is further suggested by the fact that the CYP2C22-promoter responsiveness to RA was dramatically increased by cotransfection of RAR and RXR expression vectors and was supported by the results of supershift assays, which showed that CYP2C22-RARE oligonucleotide associates with an RAR/RXR heterodimer to form complexes of reduced mobility. It is likely that this DR-5 RARE is occupied by RARα/RXRα based on the results with Am580 and the observation that RARα antibody or antibodies to both RARα and RXRα generated a stronger supershifted band. However, in cotransfection studies, each of the three forms of RAR was effective in increasing CYP2C22 promoter activity in response to RA in HepG2 cells. However, we could barely detect any luciferase activity when CYP2C22 promoter was transfected into H-4-II-E cells or HEK293T cells (data not shown), although the expression of the endogenous CYP2C22 gene was induced by RA in H-4-II-E cells. These results imply that additional elements in the CYP2C22 promoter, not included in the pGL3 construct, could be important in the regulation of its expression.
In our investigation using human primary hepatocytes, CYP2C9 mRNA was upregulated by 10 nM RA, a physiological concentration similar to that present in plasma (59, 60). Primary cultured hepatocytes are thought to behave more like in vivo hepatocytes than hepatoma cells, and therefore, are often used to study the regulation of cytochrome P450 expression by xenobiotics. However, it is still difficult to assess the regulation of CYP2C genes by RA using primary cultured hepatocytes, because the vitamin A status of the liver donor from which the cells were derived is unknown, and CYP2C9 expression declines rapidly in primary cultured hepatocytes. To further investigate the CYP2C9 responsiveness to RA, we examined CYP2C9 promoter activity in HepG2 cells. These results showed that with increasing RA concentration, the luciferase activity driven by CYP2C9 promoter increased significantly. Moreover, two DR5 motifs have been found in the regulatory region of CYP2C9 gene that are considered to be the RARE and might provide a mechanistic basis for the induction of CYP2C9 expression by at-RA. Collectively, the data obtained in these two experiments provide new evidence suggesting the RA-mediated regulation of human CYP2C9 in vivo.
The metabolism of at-RA was evaluated in HEK293T cells stably transfected with CYP2C22. This cellular system is advantageous for the RA biotransformation assay, because cellular retinoic acid binding protein (CRABP), which specifically binds at-RA, is present in HEK293T cells, while the endogenous CYP26A1 is not induced on the addition of RA (61, 62). The present study demonstrated that at-RA was converted to polar metabolites in this CYP2C22-expressing cell system. These results strongly suggest that CYP2C22 is involved in the hepatic deactivation of at-RA. Under normal physiological conditions, a very low level of CYP2C22 was detected in extrahepatic tissues, which implies that CYP2C22 makes very little contribution to the metabolism of locally formed at-RA in peripheral tissues. Our results also suggest that certain fatty acids and even 9-cis-RA can act in an inhibitory manner. The mechanisms have not been elucidated, and this aspect will require in-depth studies.
The functions of CYP2C22 or its human ortholog(s) of the CYP2C family with respect to retinoid metabolism in vivo are not yet well understood. Indeed, an increasing number of cytochrome P450s from mammalian liver have been shown to be capable of catabolizing RA to its oxidized products, at least in vitro, including CYP2C, CYP3A, and CYP26 cytochrome family members (5). This apparent redundancy complicates the study of RA metabolism in vivo. However, of the P450s shown to be capable of catabolizing RA, only CYP26 is well demonstrated to be regulated in vivo, while CYP2C7 may also be regulated by RA (21, 62, 63). Here, we have characterized CYP2C22, a gene in rat liver that is induced by RA and is able to convert at-RA to its polar metabolites. Rat CYP2C22, like its apparent human ortholog CYP2C9, is highly abundant P450 in liver according to an EST counting analysis (64). Thus, the potential for CYP2C22 to make a substantial contribution to RA hydroxylation and the maintenance of RA homeostasis cannot be neglected.
Our results regarding the activity of CYP2C22 stably expressed in HEK293T cells suggest that at-RA could be a direct substrate, while also suggesting that certain fatty acids might be involved in some inhibitory manner. However, the mechanisms are unknown, and it is likely that detailed kinetic studies will be necessary to understand these effects. The reduction in metabolism of at-RA when AA or DHA was added, suggesting competition, was not surprising and appears consistent with previous studies which have shown that expressed proteins of the mouse CYP2C family are able to metabolize certain long-chain fatty acids like AA (26). We were, however, surprised that 9-cis-RA was a particularly strong inhibitor in our intact cell-based assay. 9-cis-RA, like AA, is a 20-carbon polyene, and the 9-cis double bond confers an overall shape rather similar to the side-chain of AA. We were further surprised that 9-cis-RA itself was not converted to polar metabolites by the CYP2C22-transfected HEK293T cells, suggesting it can function as an inhibitor but apparently not as a competitive substrate. While these results are intriguing, additional research, including studies of the enzymology of the expressed or purified protein as well as comparison to other CYPs such as CYP26A1, is needed to understand the enzymatic functions of this and other P450s that may be involved in RA metabolism.
CONCLUSION
Our studies on rat CYP2C22 and human CYP2C8 and CYP2C9, which included regulation of endogenous expression by diet and retinoids in vivo, examinations of human and rodent hepatocytes, promoter analysis, and functional activity assays, lend strong support for a physiologically important role of retinoids and of the RARα/RXR receptor pair as endogenous regulators of this gene family. The ability of CYP2C22 to both respond to and metabolize at-RA indicates that this system can function as a sensing-responding mechanism. By analogy to the broad metabolic profile for human CYP2C9, CYP2C22 may also be important for the metabolism of lipophilic compounds besides RA. While CYP26A1 is strongly implicated in the regulation of RA homeostasis in the liver, the potential of the CYP2C genes we studied to metabolize not only retinoids but also clinically important drugs (44, 45, 53, 54, 65), many of which have a narrow therapeutic index (54), sets the CYP2C family apart as being susceptible to drug-retinoid interactions. Thus, understanding CYP2C22 could be important for using retinoids and other common drugs to best effect in clinical practice.
Acknowledgments
The authors thank Nan-qian Li, Qiuyan Chen, Christopher Cifelli, and Yao Zhang for providing samples for analysis and helpful advice.
Footnotes
Abbreviations:
- AA
- arachidonic acid
- at-RA
- all-trans-RA
- CYP
- cytochrome P450
- DHA
- docosahexaenoic acid
- NPE
- nuclear protein extract
- RA
- retinoic acid
- RAR
- retinoid acid receptor
- RARE
- RA response element
- RXR
- retinoid X receptor
This work was supported by National Institutes of Health Grant CA-90214. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or other granting agencies. The authors have no conflicting financial interests to disclose.
REFERENCES
- 1.Gonzalez F. J., Lee Y. H. 1996. Constitutive expression of hepatic cytochrome P450 genes. FASEB J. 10: 1112–1117. [DOI] [PubMed] [Google Scholar]
- 2.Capdevila J. H., Falck J. R., Harris R. C. 2000. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J. Lipid Res. 41: 163–181. [PubMed] [Google Scholar]
- 3.Bernhardt R. 2006. Cytochromes P450 as versatile biocatalysts. J. Biotechnol. 124: 128–145. [DOI] [PubMed] [Google Scholar]
- 4.Hannemann F., Bichet A., Ewen K. M., Bernhardt R. 2007. Cytochrome P450 systems—biological variations of electron transport chains. Biochim. Biophys. Acta. 1770: 330–344. [DOI] [PubMed] [Google Scholar]
- 5.Marill J., Cresteil T., Lanotte M., Chabot G. G. 2000. Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol. Pharmacol. 58: 1341–1348. [DOI] [PubMed] [Google Scholar]
- 6.McSorley L. C., Daly A. K. 2000. Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochem. Pharmacol. 60: 517–526. [DOI] [PubMed] [Google Scholar]
- 7.Sonneveld E., van der Saag P. T. 1998. Metabolism of retinoic acid: implications for development and cancer. Int. J. Vitam. Nutr. Res. 68: 404–410. [PubMed] [Google Scholar]
- 8.Blomhoff R., Blomhoff H. K. 2006. Overview of retinoid metabolism and function. J. Neurobiol. 66: 606–630. [DOI] [PubMed] [Google Scholar]
- 9.Fields A. L., Soprano D. R., Soprano K. J. 2007. Retinoids in biological control and cancer. J. Cell. Biochem. 102: 886–898. [DOI] [PubMed] [Google Scholar]
- 10.Altucci L., Gronemeyer H. 2001. The promise of retinoids to fight against cancer. Natl. Rev Can. 1: 181–193. [DOI] [PubMed] [Google Scholar]
- 11.Chambon P. 1996. A decade of molecular biology of retinoic acid receptors. FASEB J. 10: 940–954. [PubMed] [Google Scholar]
- 12.Balmer J. E., Blomhoff R. 2002. Gene expression regulation by retinoic acid. J. Lipid Res. 43: 1773–1808. [DOI] [PubMed] [Google Scholar]
- 13.Altucci L., Leibowitz M. D., Ogilvie K. M., de Lera A. R., Gronemeyer H. 2007. RAR and RXR modulation in cancer and metabolic disease. Nat. Rev. Drug Discov. 6: 793–810. [DOI] [PubMed] [Google Scholar]
- 14.Luu L., Ramshaw H., Tahayato A., Stuart A., Jones G., White J., Petkovich M. 2001. Regulation of retinoic acid metabolism. Adv. Enzyme Regul. 41: 159–175. [DOI] [PubMed] [Google Scholar]
- 15.Ribes V., Fraulob V., Petkovich M., Dolle P. 2007. The oxidizing enzyme CYP26a1 tightly regulates the availability of retinoic acid in the gastrulating mouse embryo to ensure proper head development and vasculogenesis. Dev. Dyn. 236: 644–653. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto Y., Zolfaghari R., Ross A. C. 2000. Regulation of CYP26 (cytochrome P450RAI) mRNA expression and retinoic acid metabolism by retinoids and dietary vitamin A in liver of mice and rats. FASEB J. 14: 2119–2127. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y., Zolfaghari R., Ross A. C. 2002. Cloning of rat cytochrome P450RAI (CYP26) cDNA and regulation of its gene expression by all-trans-retinoic acid in vivo. Arch. Biochem. Biophys. 401: 235–243. [DOI] [PubMed] [Google Scholar]
- 18.Zolfaghari R., Cifelli C. J., Lieu S. O., Chen Q., Li N. Q., Ross A. C. 2007. Lipopolysaccharide opposes the induction of CYP26A1 and CYP26B1 gene expression by retinoic acid in the rat liver in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 292: G1029–G1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nadin L., Murray M. 1999. Participation of CYP2C8 in retinoic acid 4-hydroxylation in human hepatic microsomes. Biochem. Pharmacol. 58: 1201–1208. [DOI] [PubMed] [Google Scholar]
- 20.Chen H., Fantel A. G., Juchau M. R. 2000. Catalysis of the 4-hydroxylation of retinoic acids by cyp3a7 in human fetal hepatic tissues. Drug Metab. Dispos. 28: 1051–1057. [PubMed] [Google Scholar]
- 21.Westin S., Sonneveld E., van der Leede B. M., van der Saag P. T., Gustafsson J. A., Mode A. 1997. CYP2C7 expression in rat liver and hepatocytes: regulation by retinoids. Mol. Cell. Endocrinol. 129: 169–179. [DOI] [PubMed] [Google Scholar]
- 22.Fan L. Q., Brown-Borg H., Brown S., Westin S., Mode A., Corton J. C. 2004. PPARalpha activators down-regulate CYP2C7, a retinoic acid and testosterone hydroxylase. Toxicology. 203: 41–48. [DOI] [PubMed] [Google Scholar]
- 23.Andreola F., Hayhurst G. P., Luo G., Ferguson S. S., Gonzalez F. J., Goldstein J. A., De Luca L. M. 2004. Mouse liver CYP2C39 is a novel retinoic acid 4-hydroxylase. Its down-regulation offers a molecular basis for liver retinoid accumulation and fibrosis in aryl hydrocarbon receptor-null mice. J. Biol. Chem. 279: 3434–3438. [DOI] [PubMed] [Google Scholar]
- 24.Nagata K., Sasamura H., Miyata M., Shimada M., Yamazoe Y., Kato R. 1990. cDNA and deduced amino acid sequences of a male dominant P-450Md mRNA in rats. Nucleic Acids Res. 18: 4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emi Y., Chijiiwa C., Omura T. 1990. A different cytochrome P450 form is induced in primary cultures of rat hepatocytes. Proc. Natl. Acad. Sci. USA. 87: 9746–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H., Zhao Y., Bradbury J. A., Graves J. P., Foley J., Blaisdell J. A., Goldstein J. A., Zeldin D. C. 2004. Cloning, expression, and characterization of three new mouse cytochrome p450 enzymes and partial characterization of their fatty acid oxidation activities. Mol. Pharmacol. 65: 1148–1158. [DOI] [PubMed] [Google Scholar]
- 27.McClintick J. N., Crabb D. W., Tian H., Pinaire J., Smith J. R., Jerome R. E., Edenberg H. J. 2006. Global effects of vitamin A deficiency on gene expression in rat liver: evidence for hypoandrogenism. J. Nutr. Biochem. 17: 345–355. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Yao R., Maciag A., Grubbs C. J., Lubet R. A., You M. 2006. Organ-specific expression profiles of rat mammary gland, liver, and lung tissues treated with targretin, 9-cis retinoic acid, and 4-hydroxyphenylretinamide. Mol. Cancer Ther. 5: 1060–1072. [DOI] [PubMed] [Google Scholar]
- 29.Jackson D. P. 1992. In situ hybridization in plants. : Molecular Plant Pathology, a Practical Approach. Bowles D. J., Gurr S. J., McPhereson M., Oxford University Press, London: 163–174. [Google Scholar]
- 30.Sankaranarayanan S., Ma Y., Bryson M. S., Li N-Q., Ross A. C. 2007. Neonatal-age treatment with vitamin A delays postweaning vitamin A deficiency and increases the antibody response to T-cell dependent antigens in young adult rats fed a vitamin A-deficient diet. J. Nutr. 137: 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Décimo D., Georges-Labouesse E., Dollé P. 1995. In situ hybridization of nucleic acid probes to cellular RNA. : Gene Probes, a Practical Approach Book. Hames B. D., Higgins S., Oxford University Press, London: 183–210. [Google Scholar]
- 32.Zolfaghari R., Ross A. C. 2009. An essential set of basic DNA response elements is required for receptor-dependent transcription of the lecithin:retinol acyltransferase (Lrat) gene. Arch. Biochem. Biophys. 489: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.European Bioinformatics Institute. ClustalW2. (Last accessed September 30, 2009, at http://www.ebi.ac.uk/Tools/clustalw2/index.html.)
- 34.Chen Q., Ma Y., Ross A. C. 2002. Opposing cytokine-specific effects of all trans-retinoic acid on the activation and expression of signal transducer and activator of transcription (STAT)-1 in THP-1 cells. Immunology. 107: 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garrabrant T. A., End D. W. 1995. A rapid assay for measuring the metabolism of [3H]-retinoic acid in cell cultures. J. Pharmacol. Toxicol. Methods. 34: 219–223. [DOI] [PubMed] [Google Scholar]
- 36.Cifelli C. J., Ross A. C. 2006. All-trans-retinoic acid distribution and metabolism in vitamin A-marginal rats. Am. J. Physiol. Gastrointest. Liver Physiol. 291: G195–G202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato S., Mano H., Kumazawa T., Yoshizawa Y., Kojima R., Masushige S. 1992. Effect of retinoid status on α, β and γ retinoic acid receptor mRNA levels in various rat tissues. Biochem. J. 286: 755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zolfaghari R., Ross A. C. 1995. Chronic vitamin A intake affects the expression of mRNA for apolipoprotein A-I, but not for nuclear retinoid receptors, in liver of young and aging Lewis rats. Arch. Biochem. Biophys. 323: 258–264. [DOI] [PubMed] [Google Scholar]
- 39.Tamura K., Kagechika H., Hashimoto Y., Shudo K., Ohsugi K., Ide H. 1990. Synthetic retinoids, retinobenzoic acids, Am80, Am580, and Ch55 regulate morphogenesis in chick limb bud. Cell Differ. Dev. 32: 17–26. [DOI] [PubMed] [Google Scholar]
- 40.Goerner A., Coerner M. M. 1939. Vitamin A and liver cell tumors. J. Biol. Chem. 128: 559–565. [Google Scholar]
- 41.Guo X., Nanus D. M., Ruiz A., Rando R. R., Bok D., Gudas L. J. 2001. Reduced levels of retinyl esters and vitamin A in human renal cancers. Cancer Res. 61: 2774–2781. [PubMed] [Google Scholar]
- 42.Balmer J. E., Blomhoff R. 2005. A robust characterization of retinoic acid response elements based on a comparison of sites in three species. J. Steroid Biochem. Mol. Biol. 96: 347–354. [DOI] [PubMed] [Google Scholar]
- 43.Schug J. 2008. Using TESS to predict transcription factor binding sites in DNA sequence. Curr. Protoc. Bioinformatics. Chapter. 2. Unit 2.6. [DOI] [PubMed] [Google Scholar]
- 44.Goldstein J. A., de Morais S. M. 1994. Biochemistry and molecular biology of the human CYP2C subfamily. Pharmacogenetics. 4: 285–299. [DOI] [PubMed] [Google Scholar]
- 45.Gelboin H. V., Krausz K. 2006. Monoclonal antibodies and multifunctional cytochrome P450: drug metabolism as paradigm. J. Clin. Pharmacol. 46: 353–372. [DOI] [PubMed] [Google Scholar]
- 46.Gerbal-Chaloin S., Pascussi J. M., Pichard-Garcia L., Daujat M., Waechter F., Fabre J. M., Carrere N., Maurel P. 2001. Induction of CYP2C genes in human hepatocytes in primary culture. Drug Metab. Dispos. 29: 242–251. [PubMed] [Google Scholar]
- 47.Shimada T., Yamazaki H., Mimura M., Inui Y., Guengerich F. P. 1994. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 270: 414–423. [PubMed] [Google Scholar]
- 48.Rahman A., Korzekwa K. R., Grogan J., Gonzalez F. J., Harris J. W. 1994. Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2C8. Cancer Res. 54: 5543–5546. [PubMed] [Google Scholar]
- 49.Scott J., Poffenbarger P. L. 1979. Pharmacogenetics of tolbutamide metabolism in humans. Diabetes. 28: 41–51. [PubMed] [Google Scholar]
- 50.Hashimoto Y., Otsuki Y., Odani A., Takano M., Hattori H., Furusho K., Iui K. 1996. Effect of CYP2C polymorphisms on the pharmacokinetics of phenytoin in Japanese patients with epilepsy. Biol. Pharm. Bull. 19: 1103–1105. [DOI] [PubMed] [Google Scholar]
- 51.Rettie A. E., Korzekwa K. R., Kunze K. L., Lawrence R. F., Eddy A. C., Aoyama T., Gelboin H. V., Gonzalez F. J., Trager W. F. 1992. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem. Res. Toxicol. 5: 54–59. [DOI] [PubMed] [Google Scholar]
- 52.Stearns R. A., Chakravarty P. K., Chen R., Chiu S. H. 1995. Biotransformation of losartan to its active carboxylic acid metabolite in human liver microsomes. Role of cytochrome P4502C and 3A subfamily members. Drug Metab. Dispos. 23: 207–215. [PubMed] [Google Scholar]
- 53.Zanger U. M., Turpeinen M., Klein K., Schwab M. 2008. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem. 392: 1093–1108. [DOI] [PubMed] [Google Scholar]
- 54.García-Martín E., Martínez C., Ladero J. M. 2006. Interethnic and intraethnic variability of CYP2C8 and CYP2C9 polymorphisms in healthy individuals. Mol. Diagn. Ther. 10: 29–40. [DOI] [PubMed] [Google Scholar]
- 55.Rifkind A. B., Lee C., Chang T. K., Waxman D. J. 1995. Arachidonic acid metabolism by human cytochrome P450s 2C8, 2C9, 2E1, and 1A2: regioselective oxygenation and evidence for a role for CYP2C enzymes in arachidonic acid epoxygenation in human liver microsomes. Arch. Biochem. Biophys. 320: 380–389. [DOI] [PubMed] [Google Scholar]
- 56.Kurlandsky S. B., Gamble M. V., Ramakrishnan R., Blaner W. S. 1995. Plasma delivery of retinoic acid to tissues in the rat. J. Biol. Chem. 270: 17850–17857. [DOI] [PubMed] [Google Scholar]
- 57.Mangelsdorf D. J., Umesono K., Kliewer S. A., Borgmeyer U., Ong E. S., Evans R. M. 1991. A direct repeat in the cellular retinol-binding protein type II gene confers differential regulation by RXR and RAR. Cell. 66: 555–561. [DOI] [PubMed] [Google Scholar]
- 58.Kliewer S. A., Umesono K., Heyman R. A., Mangelsdorf D. J., Dyck J. A., Evans R. M. 1992. Retinoid X receptor-COUP-TF interactions modulate retinoic acid signaling. Proc. Natl. Acad. Sci. USA. 89: 1448–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Leenheer A. P., Lambert W. E., Claeys I. 1982. All-trans-retinoic acid: measurement of reference values in human serum by high performance liquid chromatography. J. Lipid Res. 23: 1362–1367. [PubMed] [Google Scholar]
- 60.Eckhoff C., Nau H. 1990. Identification and quantitation of all-trans- and 13-cis-retinoic acid and 13-cis-4-oxoretinoic acid in human plasma. J. Lipid Res. 31: 1445–1454. [PubMed] [Google Scholar]
- 61.Pfoertner S., Goelden U., Hansen W., Toepfer T., Geffers R., Ukena S. N., von Knobloch R., Hofmann R., Buer J., Schrader A. J. 2005. Cellular retinoic acid binding protein I: expression and functional influence in renal cell carcinoma. Tumour Biol. 26: 313–323. [DOI] [PubMed] [Google Scholar]
- 62.White J. A., Beckett-Jones B., Guo Y. D., Dilworth F. J., Bonasoro J., Jones G., Petkovich M. 1997. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J. Biol. Chem. 272: 18538–18541. [DOI] [PubMed] [Google Scholar]
- 63.Ray W. J., Bain G., Yao M., Gottlieb D. I. 1997. CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J. Biol. Chem. 272: 18702–18708. [DOI] [PubMed] [Google Scholar]
- 64.National Center for Biotechnology Information (NCBI). 2009. EST Profile Viewer. EST Profile (Rn. 88025 - Cyp2c22). http://www.ncbi.nlm.nih.gov/gene/171518. Last accessed April 30, 2010. [Google Scholar]
- 65.Lewis D. F., Lake B. G., Dickins M. 2004. Substrates of human cytochromes P450 from families CYP1 and CYP2: analysis of enzyme selectivity and metabolism. Drug Metabol. Drug Interact. 20: 111–142. [DOI] [PubMed] [Google Scholar]