

Abstract
A key cardioprotective effect of high-density lipoprotein involves the interaction of its major protein, apolipoprotein A-I (apoA-I) with ATP-binding cassette transporter A1 (ABCA1), a macrophage cholesterol exporter. ApoA-I is thought to remove cholesterol from macrophages by a cascade of events. First it binds directly to ABCA1, activating signaling pathways, and then it binds to and solubilizes lipid domains generated by ABCA1. HDL isolated from human atherosclerotic lesions and blood of subjects with established coronary artery disease contains elevated levels of 3-chlorotyrosine and 3-nitrotyrosine, two characteristic products of myeloperoxidase (MPO), a heme protein secreted by macrophages. Here we show that chlorination (but not nitration) of apoA-I by the MPO pathway impairs its ability to interact directly with ABCA1, to activate the Janus kinase 2 signaling pathway, and to promote efflux of cellular cholesterol. In contrast, oxidation of apoA-I has little effect on its ability to stabilize ABCA1 protein or to solubilize phospholipids. Our results indicate that chlorination of apoA-I by the MPO pathway selectively inhibits two critical early events in cholesterol efflux: (1) the binding of apoA-I to ABCA1 and (2) the activation of a key signaling pathway. Therefore, oxidation of apoA-I in the artery wall by MPO-generated chlorinating intermediates may contribute to atherogenesis by impairing cholesterol efflux from macrophages.
Keywords: atherogenesis, high-density lipoprotein, hydrogen peroxide, hypochlorous acid, Janis kinase 2, oxidation, peroxynitrite, ATP-binding cassette transporter A1
Population studies have shown an inverse relationship between plasma HDL levels and the risk for atherosclerotic disease, implying that factors associated with HDL metabolism are cardioprotective. HDL and its major protein, apolipoprotein A-I (apoA-I), are thought to protect against atherosclerosis by several diverse mechanisms, including removing excess cholesterol from arterial macrophages, reducing inflammation, and inhibiting lipoprotein oxidation (1). There is strong evidence that the inflammatory milieu of atherosclerotic tissue can promote oxidation of HDL apolipoproteins and impair their cardioprotective activities (2).
One oxidative pathway in the human artery wall involves myeloperoxidase (MPO), a phagocyte heme protein that colocalizes with arterial macrophages (3, 4). MPO uses hydrogen peroxide (H2O2) and chloride to generate hypochlorous acid (HOCl), a powerful chlorinating oxidant (5). MPO also uses H2O2 and nitrite (NO2−) to generate reactive species that nitrate proteins (6, 7). HDL isolated from human atherosclerotic lesions contains high levels of 3-chlorotyrosine and 3-nitrotyrosine (8–10). In mouse models of acute inflammation, generation of both of these oxidized amino acids is markedly impaired when the mice are deficient in MPO (7, 11). Moreover, levels of 3-chlorotyrosine and 3-nitrotyrosine are markedly higher in HDL isolated from plasma of subjects with established coronary artery disease than in HDL from healthy subjects (8–10). These findings indicate that MPO targets HDL for oxidation in humans and support the concept that reactions mediated by the enzyme help generate dysfunctional HDL in atherosclerotic lesions.
One cardioprotective pathway associated with HDL metabolism is controlled by ATP-binding cassette transporter A1 (ABCA1) (12). This membrane protein exports cholesterol and phospholipids from cells to apoA-I and other lipid-poor HDL apolipoproteins. ABCA1 is induced by intracellular cholesterol through the liver X receptor transcription pathway, and thus is highly expressed in cholesterol-loaded cells, including the foam cells that characterize early atherosclerotic lesions (13). Mutations that make ABCA1 nonfunctional strongly associate with an increased risk of cardiovascular disease in humans (14). Moreover, ablation of ABCA1 in mouse macrophages promotes atherosclerosis in hypercholesterolemic mice (15, 16), indicating that cholesterol efflux by the ABCA1 pathway of macrophages is of central importance in the cardioprotective effects of HDL.
Removal of cholesterol and phospholipids from cells through the ABCA1 pathway requires lipid-poor apolipoproteins to bind directly to ABCA1 (17, 18). It also requires the apolipoproteins to solubilize lipid domains that ABCA1 creates in the plasma membrane (19–22). In addition, the interaction of apolipoproteins with cells that express ABCA1 elicits several other responses, including stabilization of ABCA1 protein (23–25) and activation of Janus kinase 2 (JAK2) and other intracellular signaling pathways (20, 26–30). However, it is unclear how these different responses are related and whether they all require apolipoproteins to bind to ABCA1.
Chlorination of apoA-I by the MPO pathway impairs its ability to remove excess cellular cholesterol by the ABCA1 pathway (8, 10, 31, 32). Mass spectrometric analysis of chlorinated apoA-I reveals that this loss of activity associates with oxidation of several methionine residues and chlorination of a single tyrosine residue Tyr192 (31, 32). Analysis of a mutated form of apoA-I and biochemical studies with methionine sulfoxide reductase suggest that chlorination of Tyr192 in concert with methionine oxidation impairs ABCA1 transport activity (32). Tryptophan residues have also been implicated in loss of the ABCA1 activity of oxidized apoA-I (33). However, the lipid-free mutant protein used for these studies exhibited a marked increase in α-helical content relative to lipid-free native protein, raising concerns that the mutations introduced major changes in the secondary and tertiary structure of the protein. Collectively, these observations indicate that MPO oxidatively damages HDL in humans and suggest that oxidation of specific amino acid residues in apoA-I contribute to atherogenesis by impairing cholesterol efflux from macrophages.
To probe the cascade of cellular events implicated in cholesterol efflux by the ABCA1 pathway, the current studies used the MPO pathway to chlorinate or nitrate apoA-I. Our observations indicated that chlorination (but not nitration) blocks the binding of apoA-I to ABCA1, preventing JAK2 activation. In contrast, neither chlorination nor nitration altered the ability of apoA-I to stabilize ABCA1 or solubilize phospholipids. Thus, oxidation of apoA-I by the MPO-H2O2-Cl− system selectively impairs the apolipoprotein interactions with ABCA1 that are required for lipid removal and signaling. In contrast, oxidation has little or no effect on ABCA1 stabilization and phospholipid solubilization.
EXPERIMENTAL PROCEDURES
Oxidation of apoA-I
HDL was prepared by sequential ultracentrifugation (density range 1.125–1.21 g/ml) and depleted of apoE and apoB by heparin-agarose chromatography (34). ApoA-I was purified from HDL by ion exchange chromatography (34). MPO (donor:hydrogen peroxide, oxidoreductase, EC 1.11.1.7) was isolated from human neutrophils by lectin affinity and size exclusion chromatographies and stored at –20°C (35). Enzyme concentration (A430/A280 > 0.8) was determined spectrophotometrically (ϵ430 = 178 mM−1cm−1). ONOO− was synthesized by reacting nitrite and H2O2 under acidic conditions followed by rapid quenching with an excess of sodium hydroxide (36, 37). Concentrations of ONOO−, HOCl, and H2O2 were determined spectrophotometrically (ϵ302 = 1,670 M−1 cm−1, ϵ292 = 350 M −1 cm−1, and ϵ240 = 39.4 M−1cm −1, respectively) (36, 37).
Oxidation reactions were carried out at 37°C in phosphate buffer (20 mM sodium phosphate, 100 μM diethylenetriaminepentaacetic acid (DTPA), pH 7.4) containing 10 μM apoA-I. To create the MPO-H2O2-Cl− system, we supplemented the reaction mixture with 75 nM myeloperoxidase, 100 mM NaCl, and 250 μM H2O2. For the MPO-H2O2-NO2− system, the reaction mixture was supplemented with 75 nM myeloperoxidase, 100 μM sodium nitrite, and 250 μM H2O2. Reactions were initiated by adding oxidant and terminated by adding 5 mM methionine.
Proteolytic digestion of oxidized apoA-I, LC-ESI-MS, and quantifying oxidized amino acids
Native or oxidized lipid-free apoA-I was digested with sequencing grade modified trypsin (Promega) or sequencing grade endoproteinase Glu-C (Roche Applied Science) and fractionated by liquid chromatography as described (31, 32, 38). MS and MS/MS analyses were performed in the positive ion mode (mass range 200–2,000 Da) with a Thermo-Finnigan LCQ Deca XP Plus instrument as previous described (38). Peptide ion currents were used to quantify modified amino acids. Product yield of oxidized peptides was determined with reconstructed ion chromatograms of product and precursor peptides, calculated as: product yield (%) = [(product ion peak area) / (precursor ion peak area + product ion peak area)] × 100. This method assumes that all precursor peptide is converted into known oxidation products and that the MS response characteristics of the product ions are similar to those of the precursor ion (38).
Cholesterol efflux
Baby hamster kidney (BHK) cells expressing mifepristone-inducible human ABCA1 were radiolabeled for 24 h by adding [3H]cholesterol to the 10% fetal bovine growth medium (39). ABCA1 expression was then induced by incubating cells for 20 h with DMEM containing 1 mg/ml BSA (DMEM/BSA) and 10 nM mifepristone (39). Efflux of [3H]cholesterol was measured after a 2-h incubation with DMEM/BSA without or with control or oxidized apoA-I, as described (31, 32, 39). Cholesterol efflux mediated by apoA-I was calculated as the percentage of total [3H]cholesterol (medium plus cell) released into the medium after the value obtained with DMEM/BSA alone was subtracted. BHK cells incubated with native, oxidized, or reduced apoA-I for up to 4 h showed no changes in morphology, and the cell protein and cholesterol content per well remained stable.
Competitive cell-surface and ABCA1 binding
ApoA-I was labeled with 125I as described previously (28) to an activity of 50,000–100,000 cpm/μg apoA-I using IODO-BEADS Iodination Reagent (Pierce) according to manufacturer's instructions. For the competitive cell-surface binding assay, mifepristone-treated BHK cells were incubated for 2 h with 1 μg/ml 125I-apoA-I minus or plus the indicated concentrations of unlabeled native or oxidized apoA-I, chilled on ice, washed twice at 0°C with PBS/BSA and twice with PBS, and digested with 0.1 N NaOH. Cell-associated radioactivity and cell protein were measured, and the results were expressed as ng apoA-I per mg cell protein. For the ABCA1 binding studies, cells were incubated for 2 h with 1–2 µg/ml 125I-apoA-I minus or plus 2 µg/ml unlabeled control or oxidized apoA-I, treated for 30 min at room temperature with PBS containing 1 mg/ml DSP (a cross-linking agent), and washed twice with cold PBS containing 20 mM glycine (25, 40). ABCA1 was isolated from detergent extracts by immunoprecipitation and reduced SDS PAGE, and 125I-apoA-I was visualized by phosphorimaging and quantified using OptiQuant (Packard Instruments) software.
ABCA1 stabilization
J774 macrophages were loaded with cholesterol by incubating them for 24 h with 50 µg/ml acetylated LDL. To induce ABCA1, they were then incubated with DMEM/BSA plus 0.5 mM 8-Br-cAMP for 20 h. The cells were washed twice with PBS/BSA, and then incubated for 4 h with DMEM/BSA with or without 8-Br-cAMP plus or minus 10 μg/ml control or oxidized apoA-I (25, 28).
Immunoblot analyses
To identify ABCA1 protein, we isolated membranes from homogenized J774 macrophages by ultracentrifugation, solubilized them in SDS buffer, and resolved the proteins by SDS-PAGE. ABCA1 was identified by immunoblotting (40). The amounts of JAK2 and phosphorylated JAK2 in cells were measured by immunoblot analyses using antibodies to JAK2 (Santa Cruz Biotechechnology, Inc., Santa Cruz, CA) and to tyrosine-phosphorylated JAK2 (Biosource International) (28, 29). Equal amounts of membrane or cell protein were added per gel lane.
Solubilization of phospholipid vesicles
The ability of control and oxidized apoA-I to solubilize multilamellar liposomes made of dimyristylpalmitylcholine (DMPC) was studied by a kinetic-turbidimetric method (41). DMPC (10 mg/ml) was dissolved in chloroform:methanol (2:1, v/v) in a glass tube, and the solution was dried under a gentle stream of nitrogen with constant rotation to deposit a thin layer of lipid on the sides of the tube. Dried DMPC was dispersed in PBS buffer (pH 7.4) at 2 mg/ml. The solution was vortexed thoroughly and sonicated in a water bath for 10 min to generate multilamellar liposomes. Control or oxidized apoA-I (200 μg in PBS buffer, pH 7.4) was added to an aliquot of the DMPC solution (400 μg DMPC) to give a final DMPC/apoA-I ratio of 2:1 (w/w) at a final protein concentration of 0.17 mg/ml. The experiment was performed at 24.5°C, and the absorbance at 325 nm was monitored at 1- or 2-min intervals, using a Varian Cary 100 Bio UV-visible spectrophotometer. The τ1/2 was defined as the time required for the initial turbidity to decrease by 50%.
Phospholipid binding of apoA-I
14C-Labeled apoA-I was prepared at a specific activity of ∼1 μCi/mg protein by reductive methylation of lysines with [14C]formaldehyde and sodium cyanoborohydride (42, 43). For each 3 mg of apoA-I (∼2.24 μmol of Lys) dissolved in 15 mM sodium phosphate buffer (pH 7.0), 22.5 μmol of NaCNBH3 in sodium phosphate buffer (pH 7), ∼4 μCi of [14C]formaldehyde in distilled water was added, and the mixture was incubated at 4°C for 18 h. Reductive methylation reaction was stopped by dialysis at 4°C against 10 mM sodium phosphate buffer (pH 7). For binding experiments, 0.28 mg/ml 14C-labeled lipid-free apoA-I (∼10 μM) was oxidized by 250 μM HOCl, ONOO−, or by 250 μM H2O2 in the MPO-H2O2-NaCl (MPO-Cl) or MPO-H2O2-NaNO2 (MPO-N) system as described above.
Small unilamellar vesicles (SUV) were prepared by dissolving 20 mg of egg L-α-phosphatidylcholine (PC) in 1 ml chloroform:methanol (2:1) in a Corex glass tube (∼10 ml) (43). After adding [3H]cholesterol, the lipid mixture was dried onto the wall of the tube using a stream of nitrogen, and residual organic solvent was removed under vacuum. Lipid was rehydrated in 5 ml of 10 mM sodium phosphate buffer (pH 7.4; final concentration, 4 mg/ml of egg PC and 170 pmol/ml of [3H]cholesterol) and sonicated on ice under nitrogen with a Misonix Sonicator 3000 until the initially cloudy lipid dispersion became translucent. After spinning for 15 min at 3,000 rpm (Eppendorf 5810R centrifuge) to remove titanium debris, the supernatant were centrifuged at 187,800 g for 245 min at 4°C (Beckman Optima L Preparative Ultracentrifuge) to get rid of any remaining large vesicles. The top layer was collected as SUV, which were stored at 4°C and used within a few days of preparation.
The binding of apoA-I to SUV was assayed by size-exclusion chromatography as previously reported (43, 44). SUV (0.2 mg egg PC/ml) were incubated with shaking for 1 h at room temperature with 25 μg/ml of 14C-labeled control or oxidized apoA-I in 0.15 M NaCl and 10 mM sodium phosphate (pH 7.4). Samples (500 μl) were then filtered, loaded onto a Superdex 200 column (60 × 1.6 cm) and eluted with 10 mM Tris buffer at a flow rate of 1 ml/min using a Pharmacia FPLC system. Levels of [3H]cholesterol and [14C]apoA-I were monitored in each fraction (1 ml) using liquid scintillation counting.
RESULTS
Many lines of evidence support the hypothesis that apolipoproteins remove lipids from cells in three steps: (1) the apolipoprotein binds directly to ABCA1;(2) it then binds to exovesiculated lipid domains formed by ABCA1; and (3) it solubilizes the lipids (12, 19, 21, 45). Any of these steps could be impaired if apoA-I were oxidized by the MPO pathway. Here, we examined the effects of MPO on the ability of apoA-I to promote cholesterol efflux, interact with ABCA1, modulate ABCA1 stability and function, and solubilize phospholipids.
We first determined whether chlorinating and nitrating intermediates generated by MPO can impair cholesterol efflux by the ABCA1 pathway. Our previous studies demonstrated that Tyr192 in apoA-I is the major target for both types of oxidizing intermediates (31). Exposing apoA-I to increasing concentrations of H2O2 in the presence of MPO and NaCl progressively and severely impaired its ability to remove cellular cholesterol from BHK cells transfected with ABCA1 (Fig. 1A). Similar results were observed when apoA-I was exposed to increasing concentrations of reagent HOCl (Fig. 1A). This impairment closely correlated with the extent of chlorination of Tyr192 (31). At a 25-fold molar excess of H2O2 or HOCl, the high-affinity, saturable component of apoA-I-mediated cholesterol efflux was nearly abolished (Fig. 1B).
Fig. 1.

ABCA1-dependent cholesterol efflux activities of apoA-I oxidized by MPO. (A) [3H]Cholesterol efflux from ABCA1-transfected BHK cells was measured after a 2-h incubation with 3 µg/ml of apoA-I, apoA-I exposed to MPO in the presence of H2O2 and either NaCl (MPO-Cl) or NaNO2 (MPO-NO2), or apoA-I exposed to reagent HOCl or ONOO−. Oxidation reactions were carried out at the indicated ratio (mol:mol) of oxidant to apoA-I. (B) [3H]Cholesterol efflux was measured during 2-h incubations with the indicated concentrations of control apoA-I or apoA-I treated with a 25:1 ratio (mol:mol) of oxidant to apoA-I. [3H]Cholesterol efflux is expressed as % of total medium and cell [3H]cholesterol that is released into the medium. Results are means ± SD of three determinations, and are representative of more than six independent experiments.
At this molar ratio of oxidants, LC-ESI-MS and MS/MS analyses of a tryptic digest of the oxidized protein confirmed that Tyr192 was the predominant site of chlorination by either HOCl or the MPO-H2O2-chloride system (∼25%) (Fig. 2A). Tyr236 and Tyr115 were the second targets (7%–10%), while a low level of chlorination was observed at Tyr18, Tyr29, Tyr100, and Tyr166 (2%–5%). We also confirmed that all three methionine residues in apoA-I were quantitatively (>95%) oxidized to methionine sulfoxide (Fig. 2C). Results from multiple experiments with both ABCA1-transfected BHK cells and cAMP-treated J774 macrophages showed that these chlorinating reactions impaired apoA-I function by 60%–85%. In contrast, exposing apoA-I to a 25-fold molar excess of H2O2 in the presence of MPO and NO2 or to reagent ONOO− had little or no effect on cholesterol efflux (Fig. 1A, B).
Fig. 2.

Product yields of (A) chlorotyrosine, (B) nitrotyrosine, (C) methionine sulfoxide, and (D) hydroxytryptophan in apoA-I exposed to HOCl, MPO-H2O2-chloride system, ONOO−, or MPO-H2O2-nitrite system. ApoA-I (10 μM) was exposed to HOCl (solid bars), ONOO− (empty bars), H2O2 in the MPO-chloride system (single-line shaded bars), or MPO-nitrite system (crossing-line shaded bars) at molar ratio of 25:1 (oxidant/apoA-I) for 60 min at 37°C in phosphate buffer (100 μM DTPA, 20 mM sodium phosphate, pH 7.4). After the reaction was terminated with L-methionine, a tryptic or Glu-C digest of oxidized apoA-I was analyzed by LC-ESI-MS and MS/MS, and the oxidized peptides were detected and quantified using reconstructed ion chromatograms of precursor and product peptides as described in the Methods section. Peptide sequences were confirmed using MS/MS. Results are from three independent experiments (mean ± SD).
Using LC-ESI-MS/MS analysis, we also confirmed that all tyrosine residues had been targeted for nitration and that Tyr192 was the predominant site of nitration when apoA-I was exposed to either the MPO-H2O2-nitrite system or reagent ONOO− (∼50% or ∼85%, respectively) (Fig. 2B). Tyr236 (∼40%) was the second major target for nitration, and lower levels of nitration were observed at Tyr18, Tyr29, Tyr100, Tyr115, and Tyr166 (8%–20% for ONOO− and ∼25% for MPO-H2O2-nitrite system). At a 25:1 molar ratio of oxidant, ONOO− oxidized ∼18% of Met86, ∼55% of Met112, and ∼40% of Met148, while the MPO-H2O2-nitrite system oxidized ∼50%, ∼98%, and ∼75% of Met86, Met112, and Met148, respectively. These observations suggest that reactive nitrogen species are less efficient at oxidizing methionine residues in apoA-I than is HOCl. These findings are consistent with the proposal that modifying Tyr192 alone cannot account for the observed loss of function of oxidized apoA-I; oxidation of methionines is also required (32).
We used 125I-labeled apoA-I and the cross-linking agent DSP to determine if chlorination reduces the direct binding of apoA-I to ABCA1. Because chlorination and nitration modify tyrosine residues that are radiolabeled, we used a competitive rather than direct binding assay to test for changes in ABCA1 interactions. ABCA1-transfected BHK cells were incubated with 1–2 µg/ml 125I-apoA-I in the absence or presence of 2–10 µg/ml unlabeled unmodified (control) or oxidized apoA-I, and then treated with DSP. ABCA1 was isolated by immunoprecipitation and reduced SDS PAGE, and cross-linked 125I-apoA-I was detected by autoradiography and phosphorimaging (Fig. 3A). Addition of unlabeled control apoA-I or nitrated apoA-I reduced the amount of complex detected, indicating that unmodified and nitrated apoA-I compete equally with 125I-apoA-I for binding to ABCA1. In contrast, apoA-I exposed to either HOCl or the MPO-H2O2-Cl− system was unable to compete as effectively with 125I-apoA-I to cross-link to ABCA1. Quantification of data from two sets of experiments showed that chlorinated apoA-I had less ability than wild-type or nitrated apoA-I to compete for apoA-I binding to ABCA1 (Fig. 3B). These observations indicate that the binding of chlorinated apoA-I to ABCA1 is impaired compared with the binding of native apoA-I. In contrast, nitration appears to have little effect on the binding of apoA-I to ABCA1.
Fig. 3.
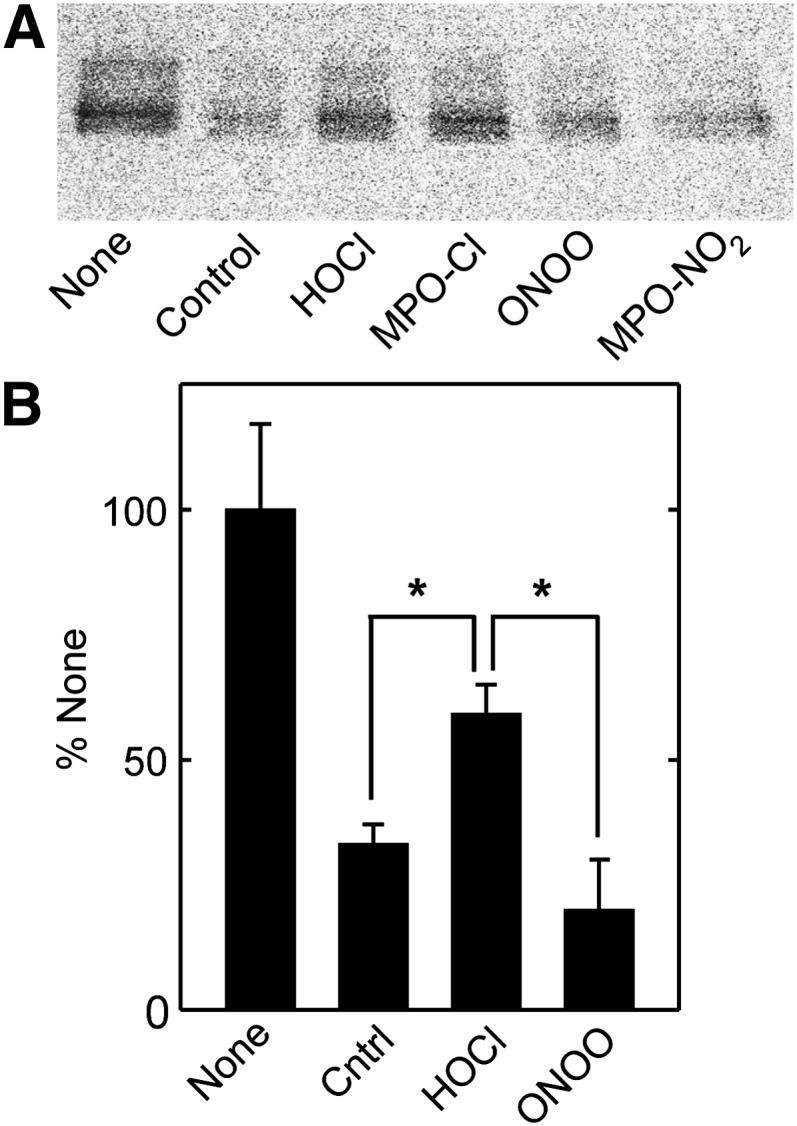
Binding of MPO-oxidized apoA-I to ABCA1. BHK cells transfected with ABCA1 were incubated for 2 h at 37°C with 1 (A) or 2 (B) µg/ml 125I-apoA-I minus (None) or plus 2 (A) or 10 (B) µg/ml unlabeled control apoA-I (Ctrl) or apoA-I treated with reagent HOCl or ONOO− or with MPO and H2O2 in the presence of either 100 mM NaCl (MPO-Cl) or 100 μM NaNO2 (MPO-NO2). Oxidation reactions were carried out at a 25:1 (A) or 50:1 (B) (mol:mol) ratio of oxidant to apoA-I. Cells were treated with the cross-linker DSP, detergent-solubilized ABCA1 was immunoprecipitated and isolated by reduced SDS PAGE, and 125I-apoA-I was visualized by phosphorimaging (A) and quantified (B). Results are representative of five similar experiments. Values in (B) are the mean ± SD of 4–6 incubations from two experiments (*P < 0.001).
The interaction of apoA-I with ABCA1 rapidly elicits intracellular signals, including autophosphorylation of JAK2 (28, 29). This signaling pathway promotes the binding of apoA-I to ABCA1 that is required for lipid removal. As previously reported (28, 29), incubating BHK cells that express ABCA1 with control apoA-I for only 15 min dramatically increases JAK2 phosphorylation (Fig. 4). We observed similar results when we nitrated apoA-I with either ONOO− or the MPO-H2O2-NO2− system. In contrast, the ability of apoA-I to stimulate JAK2 phosphorylation was greatly (though not completely) reduced by chlorination with either HOCl or MPO-H2O2-Cl− system. Thus, in addition to impairing lipid removal, chlorination of apoA-I reduces its ability to stimulate JAK2 signaling.
Fig. 4.
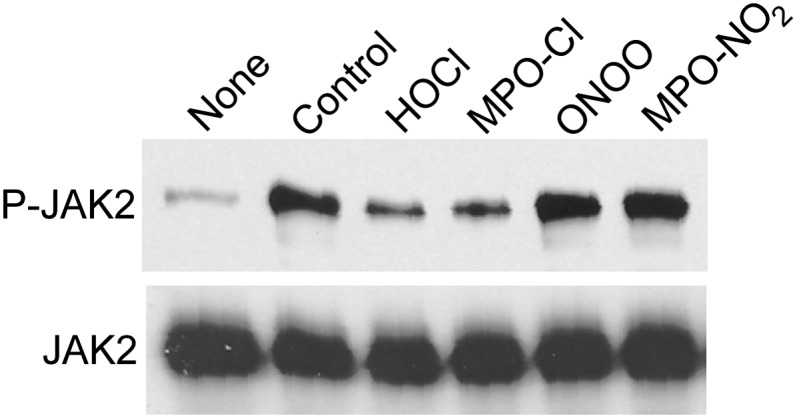
Stimulation of JAK2 autophosphorylation by MPO-oxidized apoA-I. BHK cells transfected with ABCA1 were incubated for 15 min without (None) or with 10 µg/ml apoA-I (Control), or apoA-I treated with HOCl, MPO-H2O2-NaCl (MPO-Cl), ONOO−, or MPO-H2O2-NaNO2 (MPO-NO2). Oxidation reactions were carried out at a 25:1 [mol:mol] ratio of oxidant to apoA-I. Tyrosine-phosphorylated JAK2 (P-JAK2) was detected by immunoblotting. Immunoblots were stripped of antibody, and reprobed with a total JAK2 antibody (JAK2). Results are representative of three experiments.
The majority of apoA-I binding sites on the surfaces of cells that express ABCA1 appear to be lipid domains formed by ABCA1 through exofacially directed transport of phospholipids to the outer plasma membrane (19, 21, 22, 46). We therefore determined how oxidation by the MPO pathway affected the ability of apoA-I to bind to such cells. ApoA-I exposed to a 25-fold molar excess of either HOCl or H2O2 plus MPO and NaCl lost most of its ability to compete with 125I-apoA-I for binding to BHK cells that expressed ABCA1 (Fig. 5). This was particularly evident at the lowest concentration of apoA-I, which represents high-affinity binding. In contrast, nitrating apoA-I with either ONOO− or H2O2 plus MPO and NaNO2 had no effect on its ability to compete with 125I-apoA-I. These results indicate that chlorination (but not nitration) severely impairs the high-affinity binding of apoA-I to cells that express ABCA1.
Fig. 5.

Binding of MPO-oxidized apoA-I to ABCA1-expressing cells. ABCA1-transfected BHK cells were incubated for 2 h with 1 µg/ml 125I-apoA-I minus or plus the indicated concentrations of unlabeled control apoA-I (Control) or apoA-I oxidized with HOCl or ONOO− or with MPO plus H2O2 in the presence of either NaCl (MPO-Cl) or NO2 (MPO-NO2). Cell-associated 125I-apoA-I was then measured. Oxidation reactions were carried out at a 25:1 [mol:mol] ratio of oxidant to apoA-I. Results are means ± SD of three determinations and are representative of two independent experiments.
Because apoA-I appears not to bind directly to ABCA1 when it stabilizes the transporter (28), we compared the abilities of control and oxidized apoA-I to stabilize ABCA1 protein. For these studies, we used J774 macrophages that express ABCA1, because these cells rapidly degrade ABCA1 protein in the absence of apolipoproteins and ABCA1 inducers (25, 28, 40). When the macrophages were treated with 8-Br-cAMP (to induce ABCA1) and subsequently incubated without cAMP, most of the induced ABCA1 protein disappeared within 4 h (Fig. 6, lane 2). Including control, chlorinated, or nitrated apoA-I in the 4-h chase media prevented most of this loss (Fig. 6, lanes 3–11). Thus, although chlorinated apoA-I was markedly less able to interact with ABCA1 and activate signaling and lipid removal, it was just as effective as untreated or nitrated apoA-I in stabilizing ABCA1 protein. These results provide additional evidence that the interactions of apoA-I with ABCA1 that promote JAK2 signaling and lipid removal differ from the interactions that stabilize ABCA1.
Fig. 6.

Stabilization of ABCA1 protein by MPO-oxidized apoA-I. J774 macrophages loaded with cholesterol were incubated for 20 h with 0.5 mM 8-Br-cAMP followed by a 4-h incubation with (+) or without (−) 8-Br-cAMP minus apoA-I (None) or plus 10 µg/ml apoA-I treated with the indicated molar ratios of oxidants. The membrane content of ABCA1 was measured by immunoblotting. Results represent those from three independent experiments.
We used a kinetic turbidimetric method to compare the abilities of untreated apoA-I and apoA-I that had been oxidized by MPO to solubilize phospholipids. In this assay, the rate of clearance of multilamellar DMPC liposomes was monitored by following the apolipoprotein-induced rate of decrease in the solution's absorbance. After 20 min, there was essentially no difference between the extent to which the various apoA-I preparations (control, HOCl-oxidized, MPO-chlorinated, ONOO−-oxidized, and MPO-nitrated) solubilized DMPC liposomes (Fig. 7). However, the time required for the initial turbidity to decrease by 50% (τ1/2) did differ (Table 1). As monitored by the rate constants (k1/2 [k1/2 = 1/τ1/2]) for clearance (47), both chlorinated and nitrated apoA-I converted multilamellar liposomes to SUV more rapidly than did control apoA-I. Similar results have been reported for apoA-I containing oxidized methionine residues (48). These observations suggest that oxidation of apoA-I does not reduce its ability to solubilize phospholipids as assessed by this assay.
Fig. 7.

Solubilization of phospholipid vesicles by MPO-oxidized apoA-I. Control or oxidized apoA-I samples were added to a solution of DMPC vesicles to give a final DMPC/apoA-I ratio of 2:1 (w/w) at a final protein concentration of 0.17 mg/ml. The experiment was performed at 24.5°C with monitoring of absorbance at 325 nm. Oxidation reactions were carried out at a 25:1 [mol:mol] ratio of oxidant to apoA-I. Results represent two independent experiments.
TABLE 1.
Rate of clearance of DMPC liposomes by unmodified and oxidized apoA-I
| Control | HOCl | MPO-Cl | ONOO− | MPO-NO2 | |
|---|---|---|---|---|---|
| τ1/2 (min) | 5.4 | 2.7 | 3.2 | 4.1 | 3.0 |
| k1/2 (=1/τ1/2) | 0.1852 | 0.3704 | 0.3125 | 0.2439 | 0.3333 |
| k1/2 ratio (oxided/control) | 1.00 | 2.00 | 1.69 | 1.32 | 1.80 |
The time required for the initial turbidity to decrease by 50% (τ1/2) and the rate constants (k1/2 [k1/2 = 1/τ1/2]) for clearance were calculated from the data shown in Fig. 7.
As a second approach for studying the binding properties of oxidized apoA-I to phospholipids, we incubated apoA-I or oxidized apoA-I with SUV prepared from egg phosphatidylcholine and trace amounts of radiolabeled free cholesterol, and then used size-exclusion chromatography to separate lipid-free or poorly lipidated apoA-I from apoA-I associated with SUV (44). Fig. 8 shows the elution profiles of [3H]cholesterol (SUV, peak I in Fig. 8A), lipid-free 14C-labeled apoA-I (peak II in Fig. 8A), and 14C-labeled control and MPO oxidized apoA-I (peak I and peak II in Fig. 8B) after incubation with SUV. A significant fraction of apoA-I (∼50%) coeluted with SUV after apoA-I was incubated with the phospholipid vesicles (Fig. 8B, peak I), indicating that this peak corresponds to the elution position of lipid-bound apoA-I. We next compared the binding properties of control and oxidized apoA-I proteins to SUV. The overall elution profiles of [14C]apoA-I incubated with SUV were similar for the different forms of oxidized apoA-I and control apoA-I. However, the ratio of lipid-bound to lipid-free (or lipid-poor) for HOCl and MPO-Cl oxidized apoA-I were ∼20% lower than untreated apoA-I, while ONOO− or MPO-N oxidized apoA-I were essentially no different from that of control apoA-I (Fig. 8C). These observations, in contrast to those obtained with the turbidimetric method, suggest that oxidation of apoA-I by MPO-derived chlorinating intermediates might modestly impair the ability of the protein to associate with phospholipids.
Fig. 8.

Binding of MPO-oxidized apoA-I to egg PC SUV. Oxidation reactions were carried out at a 25:1 [mol:mol] ratio of oxidant to apoA-I. SUV (0.2 mg/ml egg PC containing 170 pmol/ml of [3H]cholesterol) were incubated with 25 μg/ml control or oxidized [14C]apoA-I for 1 h at room temperature. The mixture was then subjected to size-exclusion chromatography on a Superdex 200 column that was eluted with 10 mM Tris buffer at a flow rate of 1 ml/min with collection of 1 ml fractions. Radioactivity was determined by liquid scintillation counting. A: Elution profiles of [3H]Cholesterol of SUV (empty square) and [14C]apoA-I of lipid-free apoA-I (solid square); B: elution profiles of [14C] control (empty circle) or MPO-Cl modified (solid circle) apoA-I after binding to vesicles; C: binding capacity of oxidized apoA-I to egg PC SUV. Results are the average and ranges of two independent experiments. Abbreviations: apoA-I, apolipoprotein A-I; MPO, myeloperoxidase; PC, L-α-phosphatidylcholine; SUV, small unilamellar vesicles.
DISCUSSION
MPO-mediated chlorination (but not nitration) of apoA-I markedly reduces its ability to remove cholesterol from cells by the ABCA1 pathway. This process normally involves a cascade of events that includes direct binding of apoA-I to ABCA1, activating signaling pathways, binding of apoA-I to lipid domains formed by ABCA1, and solubilizing those lipids to generate nascent HDL particles (12, 19, 21, 45). Here, we show that oxidation of apoA-I with HOCl or with MPO in the presence of NaCl and H2O2 selectively impairs the key early steps in this cholesterol export pathway.
Oxidation of apoA-I with HOCl or the MPO-H2O2-Cl− system impaired the direct binding of apoA-I to ABCA1, as determined by a cross-linking assay. This impairment was associated with a reduced ability of apoA-I to stimulate autophosphorylation of JAK2, which normally occurs within minutes of exposing ABCA1-expressing cells to apoA-I (28). Inhibition or ablation of JAK2 dramatically reduces the cross-linking of apoA-I to ABCA1 (28, 29), suggesting that optimal binding of the two proteins involves two steps, where apoA-I first activates JAK2 and then JAK2 enhances the binding of apoA-I to ABCA1. Our results show that MPO-mediated modification of apoA-I impaired this initial activation step.
Chlorination also reduced the binding of apoA-I to high-affinity binding sites on cells that expressed ABCA1. Most of those sites, however, are not ABCA1 itself, but instead appear to be phospholipid domains that ABCA1 creates by redistributing phospholipids from the cytosolic to the exofacial leafs of the plasma member (19, 21, 22). Nevertheless, several lines of evidence suggest that apolipoproteins must bind directly to ABCA1 in order to bind to these lipid domains. First, studies of apolipoprotein and ABCA1 mutants showed that binding of apolipoproteins to ABCA1 correlates closely with binding of apolipoproteins to the cell-surface and with lipid efflux (19, 45, 49). Second, inducing ABCA1 in the absence of apolipoproteins generates cholesterol domains that are accessible to the enzyme cholesterol oxidase and are subsequently depleted by apolipoproteins (39). Thus, generation of lipid domains that can be removed by apolipoproteins does not require interactions between apolipoprotein and ABCA1. Third, inhibiting or ablating JAK2 reduces direct binding of apolipoproteins to ABCA1 and to the cell-surface without affecting the ability of ABCA1 to form lipid domains on the cell surface (29). It is, therefore, likely that the reduced cell-surface binding of chlorinated apoA-I is secondary to its impaired binding to ABCA1.
The conclusion that chlorination of apoA-I impairs binding to ABCA1 rather than to the lipid domains formed by ABCA1 is supported our observation that HOCl and the MPO-H2O2-Cl− system actually improved the ability of apoA-I to solubilize DMPC vesicles. A study of structural variants of apoA-I showed that ABCA1-dependent cholesterol efflux correlated strongly with solubilization of DMPC vesicles (21), implying that chlorinated apoA-I would retain its ability to remove ABCA1-generated lipid domains if its binding to ABCA1 were normal. In contrast, an SUV assay using egg PC showed that chlorination (but not nitration) of apoA-I decreased binding capacity by 20%. Taken together, these observations suggest that oxidation of apoA-I by the MPO-H2O2-Cl− system only modestly affects the ability of the protein to interact with phospholipids.
Apolipoproteins stabilize ABCA1 by blocking its proteolysis, which occurs rapidly in their absence (23–25, 40). We showed previously that reducing the direct binding of apolipoproteins to ABCA1 by inhibiting or ablating JAK2 had no effect on protein stabilization (28). Here, we demonstrate that apoA-I that had been modified by HOCl or the MPO-H2O2-Cl−-system retained its ability to stabilize ABCA1 protein in macrophages, even though it was less able to interact with the transporter or activate JAK2. Taken together, these results indicate that stabilization of ABCA1 occurs by cellular interactions that do not involve direct binding to ABCA1 and lipid removal. Because HOCl and the MPO-H2O2-Cl− system appeared to have only modest effect on the interaction of apoA-I with phospholipids, it is possible that apolipoproteins stabilize ABCA1 by interacting with lipids that surround ABCA1, as was postulated previously (30).
We have previously shown that tryptophan residues are oxygenated by HOCl and the MPO-H2O2-chloride system (38). In the current study, we confirmed that similar oxidation patterns were observed regardless of whether apoA-I was exposed to reagent HOCl or the MPO-H2O2-chloride system. In contrast, we detected only a low level of hydroxytryptophan (∼20% for Trp72 and <10% for other three Trp residues) when we exposed lipid-free apoA-I to ONOO− or the MPO-H2O2-nitrite system (Fig. 2D), suggesting that reactive nitrogen species poorly oxidize tryptophan residues under these conditions.
Oxidation of Trp residues has been proposed to play a role in the functional inactivation of oxidized apoA-I (33). This proposal was based on an engineered form of the protein in which all four Trp residues were mutated to Phe. Analysis of the secondary structure of the lipid-free mutant protein by circular dichroism demonstrated that it contained ∼70% α-helical content (33), which is markedly increased compared with that of lipid-free apoA-I (50%–57%) (50). These observations strongly suggest that the secondary and tertiary structures of 4Trp→4Phe apoA-I are markedly different from that of the native protein, raising concerns over the physiological significance of its apparent resistance to oxidative inactivation.
The current and previous studies suggest the following model for ABCA1-dependent lipid export and its impairment by oxidative reactions. When induced by cholesterol loading, ABCA1 generates cholesterol- and phospholipid-rich domains in the plasma membrane, even in the absence of apolipoproteins (Fig. 9A) (39). Those domains bend from the plasma membrane to relieve the strain in the densely packed phospholipids (21, 46), exposing the cholesterol to added cholesterol oxidase and generating curved lipid surfaces that favor apolipoprotein interactions. During the first several minutes of exposure to apolipoprotein, JAK2 is activated by autophosphorylation (Fig. 9B), which in turn increases the binding of apolipoprotein to ABCA1 (Fig. 9C) (28, 29). This binding facilitates the interaction of apoA-I with lipid domains that protrude from the cell surface, promoting their solubilization and release into the external milieu (Fig. 9D). The interaction of apoA-I with cells also stabilizes ABCA1 protein, but this interaction occurs mostly at binding sites distinct from those that promote lipid export.
Fig. 9.

Model for lipid export by the ABCA1 pathway and its impairment by MPO. A:ABCA1 has intrinsic lipid translocase activity that generates phospholipid- (PL) and cholesterol-rich domains that protrude from the plasma membrane. B: The initial interaction of apoA-I with ABCA1 or other sites stimulates autophosphorylation of JAK2. C: Activation of JAK2 increases the binding of apoA-I to sites on ABCA1 that facilitate interactions with the lipid domains that the transporter generates. D: These lipid domains are solubilized when they interact with apoA-I, liberating nascent, discoidal particles of HDL. Inset: Schematic structure of lipid-free apoA-I showing the location of tyrosine 192 (Y192) and 2 of the 3 methionine (M86 and M148) residues in the 4-helix bundle in the N-terminal domain. Previous studies (31, 32) demonstrated that MPO selectively targets those residues for chlorination (Y192) and oxidation (M86, M148). Abbreviations: ABCA1, ATP-binding cassette transporter A1; apoA-I, apolipoprotein A-I; JAK2, Janus kinase 2; MPO, myeloperoxidase.
An important issue is whether enough oxidative modification of apoA-I occurs in the artery wall to significantly impair the ABCA1 pathway. It is likely that the apoA-I in the pericellular environment of the macrophage is exposed to high levels of oxidants. Macrophages produce MPO and have membrane-associated NADPH oxidase, which generates H2O2. It is therefore feasible that a large fraction of apoA-I in contact with macrophages has undergone extensive oxidative damage and has lost its ability to interact with ABCA1. The extent of this damage could not be assessed from total apoA-I isolated from atherosclerosis lesions, because most of the lesion apoA-I colocalizes with proteoglycans in noncellular regions (51).
Exposing apoA-I to the MPO-H2O2-Cl− system or HOCl chlorinates Tyr192 and oxidizes methionines 86, 112, and 148 (31, 32). Mutating tyrosine 192 to phenylalanine and reducing the oxidized methionines back to their native state almost completely restores apoA-I-mediated lipid efflux (32), indicating that modifications of this tyrosine and one or more methionines are largely responsible for impairing function. Biophysical studies indicate that the N-terminal domain of lipid-free apoA-I exists as a 4-helix bundle (Fig. 9, insert) (50, 52, 53). Unfolding this bundle has been proposed to generate an intermediate that can interaction with ABCA1 (Fig. 9B, C) (52). It is noteworthy that Tyr192 and two of the three methionines (Met86 and Met148) modified by the MPO-H2O2-Cl− system are in or adjacent to loops between helical regions (Fig. 9, insert). Modifying amino acids in the hinge domains of the helical bundles, therefore, could interfere with the remodeling that enables apoA-I to bind to ABCA1. As a result, apoA-I loses its ability to remove lipids from cholesterol-loaded cells. The MPO-H2O2-Cl− oxidation pathway is highly active in atherosclerotic lesions (3, 4, 8, 10), which are inflamed and contain large numbers of phagocytes. The operation of this pathway in the artery wall could promote the transition from macrophages to foam cells, a key event in atherogenesis.
Footnotes
Abbreviations:
- apoA-I
- apolipoprotein A-I
- BHK
- baby hamster kidney
- DMPC
- dimyristylpalmitylcholine
- HOCl
- hypochlorous acid
- H2O2
- hydrogen peroxide
- JAK2
- Janus kinase 2
- MPO
- myeloperoxidase
- MPO-Cl
- MPO-H2O2-Cl− system
- MPO-NO2
- MPO-H2O2-NO2− system
- ONOO−
- peroxynitrite PC, L-α-phosphatidylcholine
- SUV
- small unilamellar vesicles
This work was supported by National Institutes of Health Grants HL-055362, HL-085437, HL-030086, HL-078527, and HL-086798. This work was also supported by K99/R00 Award K99HL091055 from the National Heart, Lung, and Blood Institute (B.S.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or other granting agencies.
REFERENCES
- 1.Kontush A., Chapman M. J. 2006. Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 58: 342–374. [DOI] [PubMed] [Google Scholar]
- 2.Vaisar T., Shao B., Green P. S., Oda M. N., Oram J. F., Heinecke J. W. 2007. Myeloperoxidase and inflammatory proteins: pathways for generating dysfunctional high-density lipoprotein in humans. Curr. Atheroscler. Rep. 9: 417–424. [DOI] [PubMed] [Google Scholar]
- 3.Daugherty A., Dunn J. L., Rateri D. L., Heinecke J. W. 1994. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Invest. 94: 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leeuwenburgh C., Rasmussen J. E., Hsu F. F., Mueller D. M., Pennathur S., Heinecke J. W. 1997. Mass spectrometric quantification of markers for protein oxidation by tyrosyl radical, copper, and hydroxyl radical in low density lipoprotein isolated from human atherosclerotic plaques. J. Biol. Chem. 272: 3520–3526. [DOI] [PubMed] [Google Scholar]
- 5.Hurst J. K., Barrette W. C., Jr 1989. Leukocytic oxygen activation and microbicidal oxidative toxins. Crit. Rev. Biochem. Mol. Biol. 24: 271–328. [DOI] [PubMed] [Google Scholar]
- 6.Eiserich J. P., Cross C. E., Jones A. D., Halliwell B., van der Vliet A. 1996. Formation of nitrating and chlorinating species by reaction of nitrite with hypochlorous acid. A novel mechanism for nitric oxide-mediated protein modification. J. Biol. Chem. 271: 19199–19208. [DOI] [PubMed] [Google Scholar]
- 7.Gaut J. P., Byun J., Tran H. D., Lauber W. M., Carroll J. A., Hotchkiss R. S., Belaaouaj A., Heinecke J. W. 2002. Myeloperoxidase produces nitrating oxidants in vivo. J. Clin. Invest. 109: 1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergt C., Pennathur S., Fu X., Byun J., O'Brien K., McDonald T. O., Singh P., Anantharamaiah G. M., Chait A., Brunzell J., et al. 2004. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc. Natl. Acad. Sci. USA. 101: 13032–13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pennathur S., Bergt C., Shao B., Byun J., Kassim S. Y., Singh P., Green P. S., McDonald T. O., Brunzell J., Chait A., et al. 2004. Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J. Biol. Chem. 279: 42977–42983. [DOI] [PubMed] [Google Scholar]
- 10.Zheng L., Nukuna B., Brennan M. L., Sun M., Goormastic M., Settle M., Schmitt D., Fu X., Thomson L., Fox P. L., et al. 2004. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Invest. 114: 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaut J. P., Yeh G. C., Tran H. D., Byun J., Henderson J. P., Richter G. M., Brennan M. L., Lusis A. J., Belaaouaj A., Hotchkiss R. S., et al. 2001. Neutrophils employ the myeloperoxidase system to generate antimicrobial brominating and chlorinating oxidants during sepsis. Proc. Natl. Acad. Sci. USA. 98: 11961–11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oram J. F., Heinecke J. W. 2005. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 85: 1343–1372. [DOI] [PubMed] [Google Scholar]
- 13.Passarelli M., Tang C., McDonald T. O., O'Brien K. D., Gerrity R. G., Heinecke J. W., Oram J. F. 2005. Advanced glycation end product precursors impair ABCA1-dependent cholesterol removal from cells. Diabetes. 54: 2198–2205. [DOI] [PubMed] [Google Scholar]
- 14.Singaraja R. R., Brunham L. R., Visscher H., Kastelein J. J., Hayden M. R. 2003. Efflux and atherosclerosis: the clinical and biochemical impact of variations in the ABCA1 gene. Arterioscler. Thromb. Vasc. Biol. 23: 1322–1332. [DOI] [PubMed] [Google Scholar]
- 15.Aiello R. J., Brees D., Francone O. L. 2003. ABCA1-deficient mice: insights into the role of monocyte lipid efflux in HDL formation and inflammation. Arterioscler. Thromb. Vasc. Biol. 23: 972–980. [DOI] [PubMed] [Google Scholar]
- 16.van Eck M., Bos I. S., Kaminski W. E., Orso E., Rothe G., Twisk J., Bottcher A., Van Amersfoort E. S., Christiansen-Weber T. A., Fung-Leung W. P., et al. 2002. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc. Natl. Acad. Sci. USA. 99: 6298–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fitzgerald M. L., Morris A. L., Chroni A., Mendez A. J., Zannis V. I., Freeman M. W. 2004. ABCA1 and amphipathic apolipoproteins form high-affinity molecular complexes required for cholesterol efflux. J. Lipid Res. 45: 287–294. [DOI] [PubMed] [Google Scholar]
- 18.Wang N., Silver D. L., Costet P., Tall A. R. 2000. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 275: 33053–33058. [DOI] [PubMed] [Google Scholar]
- 19.Hassan H. H., Denis M., Lee D. Y., Iatan I., Nyholt D., Ruel I., Krimbou L., Genest J. 2007. Identification of an ABCA1-dependent phospholipid-rich plasma membrane apolipoprotein A-I binding site for nascent HDL formation: implications for current models of HDL biogenesis. J. Lipid Res. 48: 2428–2442. [DOI] [PubMed] [Google Scholar]
- 20.Vaughan A. M., Tang C., Oram J. F. 2009. ABCA1 mutants reveal an interdependency between lipid export function, apoA-I binding activity, and Janus kinase 2 activation. J. Lipid Res. 50: 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vedhachalam C., Duong P. T., Nickel M., Nguyen D., Dhanasekaran P., Saito H., Rothblat G. H., Lund-Katz S., Phillips M. C. 2007. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J. Biol. Chem. 282: 25123–25130. [DOI] [PubMed] [Google Scholar]
- 22.Vedhachalam C., Ghering A. B., Davidson W. S., Lund-Katz S., Rothblat G. H., Phillips M. C. 2007. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler. Thromb. Vasc. Biol. 27: 1603–1609. [DOI] [PubMed] [Google Scholar]
- 23.Arakawa R., Yokoyama S. 2002. Helical apolipoproteins stabilize ATP-binding cassette transporter A1 by protecting it from thiol protease-mediated degradation. J. Biol. Chem. 277: 22426–22429. [DOI] [PubMed] [Google Scholar]
- 24.Wang N., Chen W., Linsel-Nitschke P., Martinez L. O., Agerholm-Larsen B., Silver D. L., Tall A. R. 2003. A PEST sequence in ABCA1 regulates degradation by calpain protease and stabilization of ABCA1 by apoA-I. J. Clin. Invest. 111: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y., Oram J. F. 2002. Unsaturated fatty acids inhibit cholesterol efflux from macrophages by increasing degradation of ATP-binding cassette transporter A1. J. Biol. Chem. 277: 5692–5697. [DOI] [PubMed] [Google Scholar]
- 26.Haidar B., Denis M., Marcil M., Krimbou L., Genest J., Jr 2004. Apolipoprotein A-I activates cellular cAMP signaling through the ABCA1 transporter. J. Biol. Chem. 279: 9963–9969. [DOI] [PubMed] [Google Scholar]
- 27.Nofer J. R., Remaley A. T., Feuerborn R., Wolinnska I., Engel T., von Eckardstein A., Assmann G. 2006. Apolipoprotein A-I activates Cdc42 signaling through the ABCA1 transporter. J. Lipid Res. 47: 794–803. [DOI] [PubMed] [Google Scholar]
- 28.Tang C., Vaughan A. M., Anantharamaiah G. M., Oram J. F. 2006. Janus kinase 2 modulates the lipid-removing but not protein-stabilizing interactions of amphipathic helices with ABCA1. J. Lipid Res. 47: 107–114. [DOI] [PubMed] [Google Scholar]
- 29.Tang C., Vaughan A. M., Oram J. F. 2004. Janus kinase 2 modulates the apolipoprotein interactions with ABCA1 required for removing cellular cholesterol. J. Biol. Chem. 279: 7622–7628. [DOI] [PubMed] [Google Scholar]
- 30.Yamauchi Y., Hayashi M., Abe-Dohmae S., Yokoyama S. 2003. Apolipoprotein A-I activates protein kinase C alpha signaling to phosphorylate and stabilize ATP binding cassette transporter A1 for the high density lipoprotein assembly. J. Biol. Chem. 278: 47890–47897. [DOI] [PubMed] [Google Scholar]
- 31.Shao B., Bergt C., Fu X., Green P., Voss J. C., Oda M. N., Oram J. F., Heinecke J. W. 2005. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. J. Biol. Chem. 280: 5983–5993. [DOI] [PubMed] [Google Scholar]
- 32.Shao B., Oda M. N., Bergt C., Fu X., Green P. S., Brot N., Oram J. F., Heinecke J. W. 2006. Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J. Biol. Chem. 281: 9001–9004. [DOI] [PubMed] [Google Scholar]
- 33.Peng D. Q., Brubaker G., Wu Z., Zheng L., Willard B., Kinter M., Hazen S. L., Smith J. D. 2008. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler. Thromb. Vasc. Biol. 28: 2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mendez A. J., Oram J. F., Bierman E. L. 1991. Protein kinase C as a mediator of high density lipoprotein receptor-dependent efflux of intracellular cholesterol. J. Biol. Chem. 266: 10104–10111. [PubMed] [Google Scholar]
- 35.Heinecke J. W., Li W., Daehnke H. L., 3rd, Goldstein J. A. 1993. Dityrosine, a specific marker of oxidation, is synthesized by the myeloperoxidase-hydrogen peroxide system of human neutrophils and macrophages. J. Biol. Chem. 268: 4069–4077. [PubMed] [Google Scholar]
- 36.Beckman J. S., Chen J., Ischiropoulos H., Crow J. P. 1994. Oxidative chemistry of peroxynitrite. Methods Enzymol. 233: 229–240. [DOI] [PubMed] [Google Scholar]
- 37.Nelson D. P., Kiesow L. A. 1972. Enthalpy of decomposition of hydrogen peroxide by catalase at 25 degrees C (with molar extinction coefficients of H 2 O 2 solutions in the UV). Anal. Biochem. 49: 474–478. [DOI] [PubMed] [Google Scholar]
- 38.Shao B., Heinecke J. W. 2008. Using tandem mass spectrometry to quantify site-specific chlorination and nitration of proteins: model system studies with high-density lipoprotein oxidized by myeloperoxidase. Methods Enzymol. 440: 33–63. [DOI] [PubMed] [Google Scholar]
- 39.Vaughan A. M., Oram J. F. 2003. ABCA1 redistributes membrane cholesterol independent of apolipoprotein interactions. J. Lipid Res. 44: 1373–1380. [DOI] [PubMed] [Google Scholar]
- 40.Oram J. F., Lawn R. M., Garvin M. R., Wade D. P. 2000. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J. Biol. Chem. 275: 34508–34511. [DOI] [PubMed] [Google Scholar]
- 41.Pownall H. J., Massey J. B., Kusserow S. K., Gotto A. M., Jr 1978. Kinetics of lipid–protein interactions: interaction of apolipoprotein A-I from human plasma high density lipoproteins with phosphatidylcholines. Biochemistry. 17: 1183–1188. [DOI] [PubMed] [Google Scholar]
- 42.Jentoft N., Dearborn D. G. 1983. Protein labeling by reductive alkylation. Methods Enzymol. 91: 570–579. [DOI] [PubMed] [Google Scholar]
- 43.Saito H., Dhanasekaran P., Nguyen D., Deridder E., Holvoet P., Lund-Katz S., Phillips M. C. 2004. Alpha-helix formation is required for high affinity binding of human apolipoprotein A-I to lipids. J. Biol. Chem. 279: 20974–20981. [DOI] [PubMed] [Google Scholar]
- 44.Vaughan A. M., Oram J. F. 2006. ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J. Lipid Res. 47: 2433–2443. [DOI] [PubMed] [Google Scholar]
- 45.Chroni A., Liu T., Fitzgerald M. L., Freeman M. W., Zannis V. I. 2004. Cross-linking and lipid efflux properties of apoA-I mutants suggest direct association between apoA-I helices and ABCA1. Biochemistry. 43: 2126–2139. [DOI] [PubMed] [Google Scholar]
- 46.Lin G., Oram J. F. 2000. Apolipoprotein binding to protruding membrane domains during removal of excess cellular cholesterol. Atherosclerosis. 149: 359–370. [DOI] [PubMed] [Google Scholar]
- 47.Pownall H., Pao Q., Hickson D., Sparrow J. T., Kusserow S. K., Massey J. B. 1981. Kinetics and mechanism of association of human plasma apolipoproteins with dimyristoylphosphatidylcholine: effect of protein structure and lipid clusters on reaction rates. Biochemistry. 20: 6630–6635. [DOI] [PubMed] [Google Scholar]
- 48.Panzenbock U., Kritharides L., Raftery M., Rye K. A., Stocker R. 2000. Oxidation of methionine residues to methionine sulfoxides does not decrease potential antiatherogenic properties of apolipoprotein A-I. J. Biol. Chem. 275: 19536–19544. [DOI] [PubMed] [Google Scholar]
- 49.Fitzgerald M. L., Morris A. L., Rhee J. S., Andersson L. P., Mendez A. J., Freeman M. W. 2002. Naturally occurring mutations in the largest extracellular loops of ABCA1 can disrupt its direct interaction with apolipoprotein A-I. J. Biol. Chem. 277: 33178–33187. [DOI] [PubMed] [Google Scholar]
- 50.Davidson W. S., Thompson T. B. 2007. The structure of apolipoprotein A-I in high density lipoproteins. J. Biol. Chem. 282: 22249–22253. [DOI] [PubMed] [Google Scholar]
- 51.O'Brien K. D., Olin K. L., Alpers C. E., Chiu W., Ferguson M., Hudkins K., Wight T. N., Chait A. 1998. Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: colocalization of biglycan with apolipoproteins. Circulation. 98: 519–527. [DOI] [PubMed] [Google Scholar]
- 52.Ajees A. A., Anantharamaiah G. M., Mishra V. K., Hussain M. M., Murthy H. M. 2006. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc. Natl. Acad. Sci. USA. 103: 2126–2131. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Lagerstedt J. O., Budamagunta M. S., Oda M. N., Voss J. C. 2007. Electron paramagnetic resonance spectroscopy of site-directed spin labels reveals the structural heterogeneity in the N-terminal domain of apoA-I in solution. J. Biol. Chem. 282: 9143–9149. [DOI] [PubMed] [Google Scholar]