

Abstract
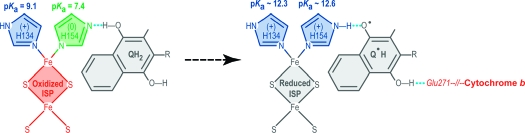
The Rieske protein component of the cytochrome bc complex contains a [2Fe−2S] cluster ligated by two cysteines and two histidines. We report here the pKa values of each of the imidazole rings of the two ligating histidines (His134 and His154) in the oxidized and reduced states of the Rieske protein from Thermus thermophilus (TtRp) as determined by NMR spectroscopy. Knowledge of these pKa values is of critical interest because of their pertinence to the mechanism of electron and proton transfer in the bifurcated Q-cycle. Although we earlier had observed the pH dependence of a 15N NMR signal from each of the two ligand histidines in oxidized TtRp (Lin, I. J.; Chen, Y.; Fee, J. A.; Song, J.; Westler, W. M.; Markley, J. L.. J. Am. Chem. Soc. 2006, 132, 10672−10673), the strong paramagnetism of the [2Fe−2S] cluster prevented the assignment of these signals by conventional methods. Our approach here was to take advantage of the unique histidine−leucine (His134−Leu135) sequence and to use residue-selective labeling to establish a key sequence-specific assignment, which was then extended. Analysis of the pH dependence of assigned 13C′, 13Cα, and 15Nε2 signals from the two histidine cluster ligands led to unambiguous assignment of the pKa values of oxidized and reduced TtRp. The results showed that the pKa of His134 changes from 9.1 in oxidized to ∼12.3 in reduced TtRp, whereas the pKa of His154 changes from 7.4 in oxidized to ∼12.6 in reduced TtRp. This establishes His154, which is close to the quinone when the Rieske protein is in the cytochrome b site, as the residue experiencing the remarkable redox-dependent pKa shift. Secondary structural analysis of oxidized and reduced TtRp based upon our extensive chemical shift assignments rules out a large conformational change between the oxidized and reduced states. Therefore, TtRp likely translocates between the cytochrome b and cytochrome c sites by passive diffusion. Our results are most consistent with a mechanism involving the coupled transfer of an electron and transfer of the proton across the hydrogen bond between the hydroquinone and His154 at the cytochrome b site.
Introduction
Thermus thermophilus Rieske protein (TtRp) is an indispensable subunit of the bacterial cytochrome bc complex, which has a counterpart in the mitochondrial cytochrome bc1 complex (complex III) (EC 1.10.2.2).1−5 Complex III consists of three catalytic subunits (with cofactors): cytochrome b (two hemes), cytochrome c(1) (heme), and the Rieske iron−sulfur protein ([2Fe−2S] cluster). Complex III is a membrane-bound protein complex that serves as a hub to accept electrons from hydroquinone and to donate the electrons to cytochrome c(1). The electron transfer mechanism is known as the bifurcated Q-cycle.6−10 In the half-reaction that takes place in the bacterium, the Rieske protein first accepts an electron from a hydromenaquinone molecule, and then donates the electron to cytochrome c.9,10 The second electron is recycled to reduce the menaquinone in the cytosolic side. The rate-limiting step occurs in the first electron transfer to the Rieske protein.9−11 The net reaction is to oxidize the hydromenaquinone to menaquinone, reduce two cytochromes c, release two protons into the periplasmic space, and transfer two protons across the membrane, against the proton gradient.6,7
Inhibitors of complex III, such as stigmatellin and myxothiazol, have been used to study the function of the complex.12−15 X-ray crystal structures show interactions among cytochrome b, stigmatellin, and the Rieske protein. The oxygen of stigmatellin (an inhibitor that mimics ubiquinone) is observed in the X-ray structure to be within hydrogen-bonding distance to the Nε2 atom of His161.12,13 (We use italics here to indicate the bovine numbering system and normal font to indicate the T. thermophilus numbering system; His161 corresponds to His154 in TtRp.) Models have been proposed to explain the catalytic mechanism, under the assumption that the ternary complex represents an intermediate in the enzymatic turnover.1,9,10,16
The redox potential of the Rieske protein is known to be coupled to the protonation states of the imidazole rings of the iron-ligated histidines.17−20 In the oxidized state, one histidine has a high pKa value (∼9.65) and the other has a value (∼7.85) near physiological pH; in the reduced state, both have very high pKa values (∼12.5).17 Therefore, the elucidation of the detailed electron and proton transfer mechanism depends critically on assigning pKa values to the individual iron-ligated histidines in the oxidized Rieske protein. If the pKa of His154 is ∼7.9, then it likely to be deprotonated before binding the hydroquinone, or before formation of the reaction complex, and then electron transfer can be coupled to proton transfer.9,16 If the pKa His154 is ∼9.7, then electron transfer would appear to be decoupled from proton transfer, and the hydroquinone deprotonation step likely occurs separately.1,2
X-ray structures identify two primary binding sites on the bc(1) complex for the Rieske protein: one, named the b-site, is close to cytochrome b, and the other, named the c-site, is close to cytochrome c(1). The b-site is the hydroquinone oxidation site.
After the proton and electron transfer at the b-site, the protein has to move and donate the electron to cytochrome c(1) at the c-site.1,2 Disruption of the translocation eliminates the enzyme activity owing to large distance between cytochromes b and c(1).21 Two major models have been advanced to explain the transfer mechanism: (i) on the basis of multiple conformations of the Rieske protein observed in different X-ray structures, Brandt et al. suggested that the protein transfers between sites by large, redox state coupled, conformational changes;1,12,22,23 (ii) others have proposed that passive diffusion between sites is more likely.8,24
Materials and Methods
Chemicals, Bacterial Strains, and Vectors
[15N]H4Cl (99%), [U-13C]-d-glucose (99%), D2O (99%), [15N]-leucine (98%), [U-13C,15N]-histidine (98%), and sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) were purchased from Cambridge Isotope Laboratories (Andover, MA). Unlabeled amino acids, BME vitamins, and corresponding antibiotics were purchased from Sigma Aldrich (St. Louis, MO). Isopropyl β-d-thiogalactoside (IPTG) was purchased from LabScientific (Livingston, NJ). Water used in all samples and buffers was purified with an Elga Maxima water purifier (Elga, Inc.; Topsfield, MA). Escherichia coli strain BL21(DE3)pLysS, Origami(DE3) (with leucine deficiency), Origami B (DE3)pLysS (with leucine deficiency), and λ-(DE3) lysogenization kit were purchased from Novagen (Madison, WI). Expression vector, pET17b carrying cDNA encoding TtRp (pET17b/TtRp), was provided by Dr. James A. Fee (Scripps Research Institute, CA). E. coli strain JM2071 (with leucine and histidine deficiencies) was purchased from the Coli Genetic Stock Center (Yale University, New Haven, CT).
Protein Expression and Labeling
[U-15N]-TtRp and [U-13C,15N]-TtRp was produced by BL21(DE3)pLysS cells containing pET17b/TtRp grown in M9 medium containing 1 g/L 15NH4Cl as described previously3 but with modifications: 0.3 mM FeCl3 and 1% (v/v) BME vitamins were added to the growth media; [U-13C]-glucose was added in two aliquots with 2 g/L added at the beginning and 2 g/L added when OD600 reached 2.5 (immediately after IPTG induction). [15N]-Leu-TtRp was produced by Origami(DE3) cells containing pET17b/TtRp in a rich synthetic medium described previously,25 with the modification that 50% of the amino acids, including the selectively labeled amino acids, was added initially, and the remaining 50% was added with the IPTG. The strain JM2071(DE3) was derived from JM2071 by using the standard protocol with a lysogenization kit (Novagen). [U-13C,15N]-His,[15N]-Leu-TtRp was produced by JM2071(DE3) cells containing pET17b/TtRp grown in a similar way to [U-15N],[NA]-Leu-TtRp, except that the histidine was enriched with 13C and 15N and the cells were grown initially in 2 L of LB medium to a high cell density (OD of ∼6) before the transfer to the synthetic medium. The yield in this medium was ∼100 mg/L culture. An auxotrophic strain of E. coli (Origami(DE3) or JM2071(DE3)) was required for leucine labeling. Details concerning other labeling patterns and the purification protocols used are presented in the Supporting Information.
NMR Sample Preparation
Amicon (Millipore, Billerica, MA) tubes were used for protein concentration and buffer exchange following protein purification. The NMR buffer was 20 mM phosphate, 20 mM Tris, and 20 mM borate, in 9% D2O at pH 8.5; the solution contained ferricyanide as the oxidizing agent and DSS as the internal chemical shift standard. Sodium dithionite was added to samples used for pH titrations or conformational analysis. Shigemi (Shigemi, Inc., Allison Park, PA) susceptibility matched NMR tubes were used for NMR samples in the oxidized state. NMR samples in the reduced state were prepared in an anaerobic chamber and transferred to NMR tubes with a J. Young valve (Wilmad, Vineland, NJ). The pH meter was calibrated before taking readings. Readings were taken before and after the data collection, and the averaged pH value was reported. In addition to the common pH standards, the pH electrode in the anaerobic chamber was calibrated against freshly prepared, saturated calcium hydroxide, as a pH 12.50 standard, and 0.05 M, 0.1 M, 0.5 M, and 1.0 M solutions of sodium hydroxide, respectively, as pH 12.67, 12.94, 13.52, and 13.73 standards.
NMR Spectroscopy
The temperature of samples of oxidized TtRp used for 13C-detection experiments was controlled at 278 K, because this temperature provided the clearest separation of the histidine 13C′ signals. The 15N-detected pH titration studies were carried out at both 278 K (to match conditions for 13C data collection) and 298 K (to match conditions used earlier).18 NMR data collection with all samples of reduced TtRp was carried out at 298 K. Technical details are presented in the Supporting Information.
Conventional triple resonance data, including 15N-HSQC, HNCACB, CBCACONH, and CCONH, were collected and used in determining spectral assignments.
NMR Data Processing and Analysis
Proton chemical shifts were referenced relative to internal DSS (taken as 0 ppm); carbon and nitrogen spectra were referenced indirectly by the canonical ratios.26 All spectra were processed by XWIN-NMR software (Bruker) with exponential line broadening. COSY spectra were apodized by applying a Gaussian window function. Additional details are provided in Supporting Information. SPARKY software27 was used for peak picking. Origin 7.0 software (OriginLab, Northampton, MA) was used to carry out nonlinear fitting of the titration data according to eq 1,28
| 1 |
in which δobs and δB represent the observed chemical shift of the signal at a certain pH and the signal at deprotonated state, respectively, Δδ represents the titration shift of the signal from the deprotonated state to the protonated state, and n is the Hill coefficient. Unless otherwise noted, all curve fitting was carried out with the Hill coefficient fixed at 1.0. The errors reported are those from fitting the experimental points.
Results and Analysis
Assignment Strategy
TtRp contains four histidine residues (Figure 1A): His120, His134, His154, and His162. Signals from the iron−sulfur cluster ligating histidines (His134 and His154, Figure 1B) were easily indentified on the basis of their paramagnetic line broadening. It has long been known that backbone signals from large proteins can be assigned through the selective labeling of 13C′i−15N(i+1) linkages.29 Upon examination of the dipeptide sequences involving the four histidines of TtRp (His120−Ala121, His134−Leu135, His154−Gly155, and His162−Gly163), it was clear that the 13C′ of His134 could be identified by incorporating [15N]-Leu and [U-13C,U-15N]-His into TtRp. Once this carbon was identified, the plan was to extend this assignment through paramagnetic optimized single-bond correlations to as much of the His134 spin system as possible. Then by comparison of the pH dependence of the assigned signals with those from the 15N NMR titration curves for the ligated histidines, the specific assignment of the two pKa values to His134 and His154 could be accomplished.
Figure 1.

Sequence and structure of TtRp. (A) Primary sequence of the water-soluble fragment of the T. thermophilus Rieske protein (TtRp) investigated here. The construct was truncated to remove the N-terminal transmembrane region (residues 1−45) and nine C-terminal residues (202−210); in addition, it contains the point mutation W142F. The sequence contains four histidine residues: two of these (His134 and His154, shown in bold) each ligates an iron atom in the Fe−S cluster; the other two (His120 and His162) are not involved in Fe−S cluster interactions. Residues identified by X-ray crystallography as hydrogen bond donors to the iron sulfur cluster are underlined. Residues with NMR assignments in both oxidized and reduced forms of the protein are highlighted in green. (B) Cross-eyed stereoscopic view of the Fe−S cluster of TtRp and surrounding amino acid residues from the X-ray structure.3 Blue dashed lines indicate hydrogen bonds to the cluster.
Assignments
In order to carry out the strategy outlined above, it was necessary first to identify the 15N NMR signal from Leu135. Because of its participation in the hydrogen-bonding network at the iron−sulfur cluster3 (Figure 1B), we expected the 15N signal to be hyperfine-shifted. Our approach was to compare directly observed 15N NMR spectra, collected with short recycling delays as optimal for paramagnetic signals, of TtRp samples containing the following labeling patterns: [U-13C,U-15N]-His,[15N]-Leu (Figure 2A,F); [15N]-Leu (Figure 2B,G); [U-15N] (Figure 2C,H); [U-15N],[NA-His] (Figure 2D); and [U-15N],[NA-His],[NA-Leu] (Figure 2E).
Figure 2.

Nitrogen-15 NMR spectra of the variously labeled samples of TtRp used in assigning signals to iron−sulfur cluster ligands. One-dimensional 15N NMR spectra of ∼10 mM oxidized T. thermophilus Rieske protein (TtRp) with different selective labeling and reverse selective labeling patterns at pH ∼ 8.5 in the oxidized state (A−E) and pH 8.0 in the reduced state (F−H): (A) 17 mM [U-13C,15N]-His,[15N]-Leu TtRp; 86 × 103 scans; (B) ∼7 mM [15N]-Leu TtRp; 1.8 × 106 scans; (C) ∼15 mM [U-15N] TtRp; 1.9 × 106 scans. (D) ∼8 mM [U-15N],[NA-Leu] TtRp; 2.1 × 106 scans; (E) ∼7 mM [U-15N],[NA-Leu],[NA-His] TtRp; 4.2 × 106 scans; (F) 14 mM [U-13C,15N]-His,[15N]-Leu TtRp; 1 × 106 scans; (G) 9 mM [15N]-Leu TtRp; 1.6 × 106 scans; the asterisk denotes the 15N14N in the air; (H) 5.5 mM [U-15N] TtRp; 2.1 × 106 scans. Technical details were presented in Supporting Information.
In spectra of selectively labeled oxidized TtRp, the 15N signal at 194.4 ppm, which was observed with all samples except for that with excess natural abundance leucine (Figure 2E) was assigned tentatively to Leu135. The same set of spectra also served to confirm the identity of 15N signals from the backbone and side chain nitrogens of His134 and 154 (Figure 2A).
Spectra of selectively labeled reduced TtRp, by comparison with previous results,18 allowed the assignment of the signal at ∼133 ppm (Figure 2F) to overlapped 15Nε2 peaks from His135 and His154. The signal at 423.6 ppm was tentatively assigned to Leu135 N (Figure 2G). The spectrum of uniformly 15N-labeled reduced TtRp (Figure 2H) was consistent with the assignment of these peaks.
The key step in assigning the iron-ligating histidines was a double resonance decoupling experiment with TtRp incorporating [U-13C,U-15N]-His,[15N]-Leu (Figure 3), which was used to identify the carbonyl carbon of His134. The directly observed undecoupled 13C NMR spectrum of this sample in the oxidized state exhibited a pair of broad doublets at ∼175.3 ppm (Figure 3A). When irradiated with 15N at 194.4 ppm (the suspected resonance frequency of Leu135), one of the doublets decreased in intensity as shown by the difference spectrum (off resonance − on resonance) (Figure 3B). This experiment identified the doublet at lower frequency as that from 13C′ of His134. The other doublet was assigned by difference to 13C′ of His154. The origin of each doublet was coupling to the adjacent 13Cα. Control experiments (Figure S1, Supporting Information) served to show that the effect observed was not an artifact.
Figure 3.

Carbon-13 [nitrogen-15] selective decoupling experiment leading to sequence-specific assignments: (A) 1D-13C-SW spectrum of 17 mM oxidized [U-13C,15N]-His,[15N]-Leu TtRp acquired at 278 K and pH 8.5; locations of the two peaks were verified by a 2D 13C-[13C] CT-COSY-SW experiment; (B) double resonance 13C−[15N] difference decoupling experiment; the decoupler was set to be on-resonance at the 15N of Leu135 in the first spectrum and off-resonance in the second spectrum; the spectrum shown is the difference between the two spectra; (C) 1D-13C-SW spectrum of 14 mM reduced [U-13C,15N]-His,[15N]-Leu TtRp acquired at 298 K and pH 7.9; (D) double resonance 13C−[15N] difference decoupling experiment of the reduced [U-13C,15N]-His,[15N]-Leu TtRp. See the Supporting Information for technical details.
We used the same approach to assign the 13C′ peaks from His134 and His154 in reduced TtRp (Figure 3C). Selective irradiation of the 15N signal tentatively assigned to Leu135N, gave rise to a sharp doublet in the difference spectrum, which served to assign the higher frequency signal to His134C′ (Figure 3D). Again, the other peak was assigned by difference to His154C′ (Figure 3C).
The decoupling experiment confirmed the tentative Leu135 15N assignments: 194.4 ppm in oxidized and 423.6 ppm in reduced state. In addition, by comparison of 13C NMR spectra taken under conditions optimized for diamagnetic signals with those optimized for paramagnetic signals, it was possible to clearly distinguish the 13C′ signals of the diamagnetic histidines (His120 and His162) from those of His134 and 154 (Figure S2, Supporting Information).
We extended the primary histidine 13C′ assignments to the 13Cα atoms of His134 and His154 (Table 1) on the basis of connectivities observed in the constant-time 2D 13C−[13C] COSY-superWEFT30 spectra of ([U-13C,U-15N]-His,[15N]-Leu) TtRp (see insets in Figure 4A,B,D). However, attempts to extend these assignments to additional atoms in the histidine spin systems proved unsuccessful.
Table 1. Nuclei Observed and Their Propertiesa.
| atom | number of bonds (H-bonds) to the nearest iron atom | distance to the closest iron atom (Å) | distance to the His134 Nε2 (Å) | distance to the His154 Nε2 (Å) |
|---|---|---|---|---|
| His134 C′ | 5 | 4.8 | 4.36 | 7.58 |
| His154 C′ | 5 | 4.8 | 7.26 | 5.96 |
| His134 Cα | 4 | 4.7 | 4.36 | 8.04 |
| His154 Cα | 4 | 4.1 | 6.66 | 4.51 |
| Leu135 N | 6 (2) | 4.1 | 4.67 | 6.80 |
Distances are the average of those in the A and B models from the X-ray structure.3
Figure 4.

Carbon-13 titration curves and analysis: (A, B) data from 2D 13C−[13C] CT-COSY-SW spectra of oxidized 21 mM [U-13C,15N]-His,[15N]-Leu TtRp collected at 278 K at various pH values; (C, D) data from 2D 13C−[13C] CT-COSY-SW spectra of reduced ∼5 mM [U-13C,15N]-His,[15N]-Leu TtRp collected at 298 K at various pH values. Open squares correspond to His134, and filled circles correspond to His154. (A) Titration data for the 13C′ NMR signals. The inset shows the 2D spectrum at pH 8.81. Fitting of the data points (curves) yielded His134 pKa = 8.68 ± 0.08 and His154 pKa = 7.89 ± 0.07. (B) Titration data for the 13Cα NMR signals. The inset shows the 2D spectrum at pH 7.85. Fitting of the data points (curves) yielded His134 pKa = 9.22 ± 0.09 and His154 pKa = 7.56 ± 0.18. (C) Titration data for the 13C′ NMR signals. Fitting of the data points yielded His134 pKa = 12.27 ± 0.03 and His154 pKa = 12.59 ± 0.03. (D) Titration data for the 13Cα NMR signals. Fitting of the data points (curves) yielded His134 pKa = 12.39 ± 0.05 and His154 pKa = 12.75 ± 0.09. The inset shows the 2D spectrum at pH 11.4. Fitting of the data points yielded His134 pKa = 12.27 ± 0.03 and His154 pKa = 12.59 ± 0.03.
pH Titrations
We collected 2D 13C−[13C] COSY-superWEFT spectra of oxidized and reduced ([U-13C,U-15N]-His,[15N]-Leu)-TtRp at multiple pH values (Figure 4). The 13C chemical shifts used as input for titration analysis were measured from the directly detected dimension, with the exception of those for 13Cα in reduced TtRp, which were measured from the indirect dimension because of its higher signal-to-noise ratios. For comparison, directly observed 1D 15N NMR spectra of oxidized (Figure 5A) and reduced (Figure 5C) [15N-His] TtRp were collected as a function of pH. The histidine 15Nε2 signals from oxidized TtRp were used to generate titration curves (Figure 5B), but the corresponding signals from reduced TtRp disappeared in spectra of samples above pH 10.48, unlike the 13C′ (Figure 4C) and 13Cα (Figure 4D) signals, which could be followed above pH 13.5. As a means of verifying that the 15N and 13C′ signals were monitoring the same titration step, we examined the linearity of their chemical shift relationship: the linear correlation coefficients were 0.998 and 0.992 for His154 and His134, respectively (Figure 5D,E). Because the carbon signals could be detected throughout the titration, were not overlapped, and were separately assigned, they provided the means for determining the pKa values of the two ligand histidines in reduced TtRp. The results of the titration studies are summarized in Table 2.
Figure 5.

Nitrogen-15 titration curves and analysis. (A, B) Data from 1D 15N NMR spectra of 12 mM oxidized [U-13C,15N]-His,[15N]-Leu TtRp collected at 278 K at various pH values. On the basis of data shown in Figures 4 and 5, the lower pKa value was assigned to His154 and the higher pKa value was assigned to His134 (Table 2). (C−E) Data from 1D 15N NMR spectra of ∼5 mM reduced [U-13C,15N]-His,[15N]-Leu TtRp collected at 298 K at various pH values. (A) Representative 15N NMR spectra of oxidized TtRp. Peak assignments are shown over the top trace. (B) The pH dependence of the chemical shifts of the peaks assigned to the 15Nε2 of His134 (open squares) and the 15Nε2 of His154 (filled circles) of oxidized TtRp. Fitting of the data points (curves) yielded His134 pKa = 9.16 ± 0.02 and His154 pKa = 7.58 ± 0.01. (C) Representative 15N NMR spectra of reduced TtRp. The 15Nε2 signals of His134 and His154 (denoted by ∗) overlapped between pH 8.14 and 10.48 and disappeared from spectra at higher pH. (D) Correlation between the His154 13C′ and 15Nε2 chemical shifts for the first six titration points. Linear regression yielded a correlation coefficient of 0.998. (E) Correlation between His134 C′ and the 15Nε2 for the first six points. Linear regression line yielded a correlation coefficient of 0.992. The inset shows the 2D 13C′−13Cα cross peak assigned to His134 at different pH values (cross peaks move to the left with increasing pH) collected at the pH values corresponding to the top 6 spectra in panel C.
Table 2. Summary of Data Used to Assign the Cluster Ligated Histidine with the Higher pKa Value to His134 and That with the Lower pKa Value to His154.
| nucleus observed | pKa | figure number | experiment |
|---|---|---|---|
| Oxidized TtRp at 278 K | |||
| His134 C′ | 8.68 ± 0.08 | 4A | 13C-[13C]-CT-COSY |
| His134 Cα | 9.22 ± 0.09 | 4B | 13C-[13C]-CT-COSY |
| His154 C′ | 7.89 ± 0.07 | 4A | 13C-[13C]-CT-COSY |
| His154 Cα | 7.56 ± 0.18 | 4B | 13C-[13C]-CT-COSY |
| His134 Nε2 | 9.16 ± 0.02 | 5B | 1D-15N rapid pulsing |
| His154 Nε2 | 7.58 ± 0.01 | 5B | 1D-15N rapid pulsing |
| Oxidized TtRp at 298 K | |||
| His134 Nε2 | 9.07 ± 0.02 | S3 | 1D-15N rapid pulsing |
| His154 Nε2 | 7.41 ± 0.01 | S3 | 1D-15N rapid pulsing |
| Reduced TtRp at 298 K | |||
| His134 C′ | 12.27 ± 0.03 | 4C | 13C-[13C]-CT-COSY |
| His134 Cα | 12.39 ± 0.05 | 4D | 13C-[13C]-CT-COSY |
| His154 C′ | 12.59 ± 0.03 | 4C | 13C-[13C]-CT-COSY |
| His154 Cα | 12.75 ± 0.09 | 4D | 13C-[13C]-CT-COSY |
Redox State Dependent Conformational Differences
Comparison of the 1H−15N HSQC spectra of oxidized and reduced TtRp indicated chemical shift changes for numerous cross peaks. We established conditions at pH 5.2 under which TtRp was half-reduced and used a 2D NMR exchange experiment to cross assign signals corresponding to specific backbone 1H−15N groups in the oxidized and reduced states.
Our next step was to carry out assignments for oxidized and reduced TtRp at pH 5.2. Conventional triple resonance NMR data sets were collected, including HNCACB, CBCACONH, 15N-HSQC, and CCONH. We supplemented these experiments with 1H−15N HSQC data from samples of TtRp labeled selectively with 15N-Leu and 15N-His. We used the PINE server31 to convert peak lists from these experiments, along with the protein sequence, into probabilistic assignments. The resulting assignments were refined by manual examination.
We succeeded in assigning signals to 122 of the 156 residues of TtRp in both oxidation states. Apart from the 18 residues belonging to the two loops (131−140 and 150−157) that surround the Fe−S cluster, which we did not expect to be able to assign, the assignments account for 87% of the residues of TtRp.
We made use of the 13Cα and 13Cβ assignments to compare the secondary structure of the oxidized and reduced forms of TtRp according to eq 2:32,33
| 2 |
where δCα,i is the 13Cα chemical shift of the ith residue of TtRp, δCβ,i is the 13Cβ chemical shift of the ith residue of TtRp, δCα,rc,i is the random coil 13Cα chemical shift for residue i, and δCβ,rc,i is the random coil 13Cβ chemical shift for residue i downloaded from BMRB Web site.34 Positive values of ΔΔδi indicate α-helices, and negative values indicate β-strands.32,33
The results of this analysis (Figure 6A) showed no evidence for any large difference in the secondary structures of oxidized and reduced TtRp. We also examined the differences (ΔΔδi,red − ΔΔδi,oxid) (Figure 6B) and found, with few exceptions, that they were small. Other than the glycine loop (Gly63−Gly65) and the residues near or between the iron−sulfur binding loop, none of the assigned carbons exhibited redox state dependent 13C NMR chemical shift differences greater than 0.5 ppm (data not shown). It appears that oxidized and reduced TtRp have very similar conformations, a result in agreement with the passive diffusion model for the translocation between b-site and c-site.24
Figure 6.

Secondary structure analysis of oxidized and reduced TtRp. Chemical shift data from 8.5 mM [U-13C,15N] TtRp at pH 5.2, 298 K, in both oxidized (orange) and reduced (blue) states are plotted as a function of residue number. Secondary structural elements derived from the X-ray structure3 are (represented at the bottom of the figure) are in agreement with the chemical shift results. (A) For each residue of TtRp, the bar represents the difference between the secondary 13Cα chemical shift (experimental shift minus random coil shift, Δδ13Cα) and the secondary 13Cβ chemical shift (experimental shift minus random coil shift, Δδ13Cβ). The symbol ∗ denotes residues that are close to the iron−sulfur binding site; # denotes the carboxyl terminus. (B) For each residue of TtRp, the bar represents the secondary shift difference (Δδ13Cα − Δδ13Cβ) in the reduced protein minus that in the oxidized protein. Large values corresponding to residues experiencing hyperfine shifts are truncated in the figure.
Discussion
NMR Assignments
In an earlier study,18 we incorporated selectively labeled histidine into the TtRp protein to distinguish between signals from the Nδ1 and Nε2 atoms of the imidazole rings. Although signals from the Nδ1 atoms of the two imidazole rings that ligate iron were unobservable, because of the strong paramagnetism, signals from the Nε2 atoms could be observed and their chemical shifts followed as a function of pH to yield distinct pKa values for the two histidine residues in the oxidized state. We speculated on the basis of solvent-accessibility arguments that the lower pKa value corresponded to His134 and outlined the mechanistic consequences of this assignment. Because of its importance, we undertook here a rigorous assignment of these signals. These assignments required the preparation of protein samples labeled selectively with 15N and 13C (Figures 2 and 3). Contrary to our earlier speculation,18 the results proved that His154 has the lower pKa value. Our current NMR assignments in oxidized and reduced TtRp include the 15Nε2, 13C′, and 13Cα atoms of each ligand histidine. These extended assignments allowed us to determine pKa values for the ligand histidines in reduced TtRp. We used conventional methods with protein labeled uniformly with 13C and 15N to carry out extensive assignments of NMR signals in both oxidized and reduced TtRp. Several of these assignments were verified by selective 15N labeling. The NMR assignments have been deposited at the BMRB under accession numbers 16787 (reduced) and 16804 (oxidized).
The similarity of the diamagnetic chemical shifts of oxidized and reduced TtRp rule out a large conformational change of the type observed by X-ray crystallography of cytochrome bc1 complexes in the presence and absence of various inhibitors.12
pH Titration Studies
Our assignments required that the oxidized protein be studied at 278 K, in order to achieve separation of the 13C′ signals of the ligand histidines. Because the earlier pH titration study was carried out at 298 K, we repeated the 15N titration study at this temperature (Figure S3, Supporting Information).
The shapes of the 15N titration curves reported here differ slightly from those reported by Lin et al.18 The midpoint broadening of the peak yielding the lower pKa observed earlier was not reproduced, and the splitting observed at high pH (attributed to interaction with titrating Tyr158)18 occurred at higher pH (11.0 instead of 10.5). These differences probably are a consequence of the presence of ferricyanide, used here but not by Lin et al., which appears to catalyze the protonation−deprotonation steps so that they become fast on the NMR time scale. The pKa values determined here from the 15N data at 298 K (7.41 ± 0.01 and 9.07 ± 0.02, Figure S3, Supporting Information) are similar to those reported by Lin et al. (7.46 ± 0.02 and 9.24 ± 0.02) at the same temperature.18
The pKa values derived from the pH dependence of NMR signals from different histidine atoms exhibited some variation (Table 2). Values derived from the chemical shifts of the imidazole atom (histidine 15Nε2) are most likely to reflect the true pKa for the residue. The pKa values derived from the 13Cα chemical shifts in 2D spectra were in good agreement with these; however, the pKa values derived from the chemical shifts of the 13C′ atoms, which are farther from the site of protonation−deprotonation, showed minor deviations (Table 2). In all cases, the higher pKa corresponded to His134 and the lower pKa to His154. The chemical shifts of the backbone nitrogens of His134, Leu135, and His154 in oxidized TtRp also were found to be pH dependent (Figure S4, Supporting Information), but these shallow titration curves were not analyzed further.
The Nε2 signals from His134 and His154 of reduced TtRp overlapped over the pH range where they were visible and disappeared from spectra of samples with pH > 10.5 (Figure 5C). However, the titration could be followed from the chemical shifts of the peaks assigned to the 13C′ and 13Cα atoms of His134 and His154 of reduced TtRp (Figure 4C,D). The fitted curves for the 13C′ signals, which exhibited larger (and hence more reliable) titration shifts than the 13Cα signals (which showed opposite pH dependence), yielded pKa values of 12.3 ± 0.3 for His134 and 12.6 ± 0.3 for His154. These values are similar to those derived earlier from electrochemical studies: pKa values ∼12.5.17
His154 exhibited a remarkably large redox state dependent shift in its pKa, from 7.6 ± 0.2 in oxidized to 12.6 ± 0.3 in reduced TtRp; this represents a 105 increase in the proton affinity of the reduced protein over the oxidized. Although the shift in the pKa of His134 is smaller, 9.2 ± 0.1 in oxidized vs 12.3 ± 0.3 in reduced TtRp, the increase represents a >103 increase in its proton affinity. Because of its high pKa in both oxidized and reduced TtRp, His134 probably remains protonated throughout the Q-cycle. The NMR data, which yield separate titration curves with Hill coefficients near unity, rule out cooperativity between protonation−deprotonation at His134 and His154. Because no other proton binding sites are within ∼9 Å of the iron−sulfur cluster, His154 is likely to be the terminal proton carrier.
Model for Proton and Electron Transfer
The model shown in Figure 7 is based the pKa values determined here, structural information about the b-site,35 and three assumptions concerning the classical semi(mena)quinone model. The assumptions are that reactions are in vitro at 298 K, that the menaquinone and ubiquinone species have the same redox potential differences as their Q/QH2 state, such that menaquinone species are all 170 mV lower than the ubiquinone species, and that pKa values of these two quinone species are the same.
Figure 7.

Model for proton and electron transfer at the Qo site on the cytochrome bc complex. Pentagons represent the imidazole rings of the histidines (green deprotonated, blue protonated). Oxidized species are depicted in red and reduced species in gray. See Discussion for details. (A) The hydro(mena)quinone hydrogen bonds to the deprotonated His154 of the oxidized ISP (TtRp, iron−sulfur protein) (top); then the electron and the proton are transferred by one of three possible pathways: proton transfer first/electron transfer second (PT/ET), A1 → A2; electron transfer first/proton transfer second (ET/PT), A′1 → A′2; or coupled electron and proton transfer (ET+PT), A′′. In each case, the intermediate formed at the end is the semi(mena)quinone radical. (B) The semiquinone radical donates the electron to oxidized cytochrome b. (C) ISP translocates between the b-site and c-site to donate an electron to oxidized cytochrome c. (D) ISP donates an electron to cytochrome c. (E) ISP translocates from site-c to site-b, and only the deprotonated ISP is capable of forming the reaction complex.
The proton-first/electron-second (PT/ET) pathway involves steps A1 (21.6 kJ mol−1; ΔpKa = 3.85; Figure 7) and A2 (−9.5 kJ mol−1; ΔE = 0.1 V). The electron-first/proton-second (ET/PT) pathway involves steps A′1 (82.8 kJ mol−1; ΔE = −0.86 V) and A′2 (−70.7 kJ mol−1, ΔpKa = −12.6). The direct mechanism A′′ involving simultaneous electron and proton transfer (ET+PT) has a free energy of 12.1 kJ mol−1. Both initial intermediates in the stepwise pathway (A1 and A′1) are energetically unstable compared with the reactants, and both represent larger energy increases than the concerted reaction A′′ (9.5 kJ mol−1 for A1; 70.7 kJ mol−1 for A′1). In addition, as we show here, adding an electron and a proton to TtRp does not involve a large conformational change. Thus, the most likely mechanism is the energetically favored concerted reaction A′′,17,36 in which electron transfer is coupled to proton transfer across the hydrogen bond between hydro(mena)quinone and His154 of the iron−sulfur protein. Theoretical arguments based on the Born−Oppenheimer approximation37 and the Hohenberg−Kohn theorem38 require concerted movement of the proton and electron. Our experimental results interpreted as shown above are inconsistent with models proposed by Link and Brandt39,40 that involve, as the first step, the deprotonation of hydro(ubi)quinone with a pKa of >11.3 in solution. The resulting high-energy semiquinone is rapidly depleted by electron transfer reaction B, thus removing the reactive semiquinone, which could contribute to oxidative stress. This, in turn, stabilizes the reduced, protonated Rieske protein from returning to the oxidized state so that it can undergo the much slower translocation from site-b to site-c (reaction C, Figure 7).
In the biological environment, however, the binding of the substrates cannot be neglected (hydromenaquinone and the oxidized, deprotonated TtRp bind to the enzyme before the reaction). In Rhodobacter sphaeroides, the binding affects the rate of the turnover and modulates the pKa of the Rieske protein and the redox potential of the hydro(ubi)quinone.16 The change is ∼0.5−1 pH unit and ∼50 mV.41 First, menaquinone binding appears to slightly lower the energy gap of the A′1 step (although it remains highly unfavored). Second, menaquinone binding makes the A1 step less favored as discussed elsewhere.16
The Proton Pathway
From the pKa assignments, it is clear that the Rieske protein is the primary proton carrier. The complete proton transfer pathway against the proton gradient involves the following steps (Figure 7). First, menaquinone accepts two electrons and two protons in the cytosolic side (this is another half-reaction to the Q-cycle in the menaquinone-reducing-site). Second, by flip−flop to the periplasmic space, it binds and forms the reaction complex there (reactant in Figure 7A). Third, hydro(mena)quinone donates one electron and one proton to TtRp (Figure 7A). TtRp carries the electron and proton until it donates the electron to the cytochrome c at the c-site and releases the proton because of the large decrease in the pKa of His154. Finally the b-site complex reforms.
Summary
Although the general features of the bifurcated Q-cycle mechanism and the proton motive gradient have been accepted, the detailed mechanism has remained controversial.42,43 Even after the confirmation of the important intermediate, ubisemiquinone,44 debates still continued over the detailed mechanisms. The primary evidence for the intermediate complex involving cytochrome b, ubihydroquinone, and the Rieske protein has come from the structure of the quinonoid inhibitor complex. Prior to this study, there was no direct evidence for the protonation state of the key histidine, His154 (corresponding to the bovine His161), of the Rieske protein. Nevertheless, models by Link, Berry, Rich, Trumpower, and Crofts, based on the X-ray structures, kinetics, and thermodynamics studies,16,39,42,43,45 agreed that the catalytic mechanism involves the formation of two hydrogen bonds as shown in Figure 7 (see the product of step A).
This study shows that His154 of oxidized TtRp is at least partially deprotonated at physiological pH. Hydrogen bonding to hydroquinone should help to stabilize the deprotonated state. The large increase in the pKa of His154 upon reduction confirms that it is the proton carrier between the b- and c-sites. Evidence also is presented here that reduction does not involve a large protein conformational change. The large conformational change in the Rieske protein observed by X-ray crystallography12 apparently occurs as part of the subsequent translocation to the c-site. These results favor the direct electron/proton transfer mechanism as the first step of the oxidation of the hydro(mena)quinone. Owing to the high degree of conservation of residues in the cluster binding domains, our results with the bacterial Rieske protein (TtRp) probably will hold for Rieske proteins of the respiratory bc1 complexes in the mitochondria of other organisms and probably also for the Rieske proteins in the photosynthetic b6f complexes.
Acknowledgments
The authors thank James A. Fee (Scripps Research Institute) for advice, encouragement, and critical comments and for supplying the plasmid encoding the Rieske protein construct. The authors thank Marco Tonelli for acquiring the triple resonance experiments. Supported by NIH Grant GM58667. NMR data were collected at the National Magnetic Resonance Facility at Madison, with support from the NIH National Center for Research Resources (Grant P41 RR02301).
Supporting Information Available
NMR related technical details (Materials and Methods, Figure 2, and Figure 3), data from controls related to the double resonance decoupling experiment (Figure S1), 1D 13C-SW spectra of oxidized [U-13C,15N]-His,[15N]-Leu TtRp collected under conditions optimized for paramagnetic and diamagnetic signals (Figure S2), 15N pH titration curves at 298 K (Figure S3), and the pH dependence of the chemical shifts of the backbone nitrogens of His134, Leu135, and His154 (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Link T. A. In Handbook of Metalloproteins; Messerschmidt A., Huber R., Poulos T. Wieghardt K., Eds.; Wiley, New York; 2001; Vol. 132, pp 518−531. [Google Scholar]
- Link T. A. In Handbook of Metalloproteins; Messerschmidt A., Huber R., Poulos T. Wieghardt K., Eds.; Wiley, New York; 2001; Vol. 132, pp 402−443. [Google Scholar]
- Hunsicker-Wang L. M.; Heine A.; Chen Y.; Luna E. P.; Todaro T.; Zhang Y. M.; Williams P. A.; McRee D. E.; Hirst J.; Stout C. D.; Fee J. A. Biochemistry 2003, 42, 7303–7317. [DOI] [PubMed] [Google Scholar]
- Mooser D.; Maneg O.; Corvey C.; Steiner T.; Malatesta F.; Karas M.; Soulimane T.; Ludwig B. Biochim. Biophys. Acta 2005, 1708, 262–274. [DOI] [PubMed] [Google Scholar]
- Mooser D.; Maneg O.; MacMillan F.; Malatesta F.; Soulimane T.; Ludwig B. Biochim. Biophys. Acta 2006, 1757, 1084–1095. [DOI] [PubMed] [Google Scholar]
- Nelson D. L.; Cox M. M.; Lehninger A. L.. Lehninger Principles of Biochemistry, 4th ed.; W.H. Freeman: New York, 2005. [Google Scholar]
- Mitchell P. J. Theor. Biol. 1976, 62, 327–367. [DOI] [PubMed] [Google Scholar]
- Crofts A. R.; Shinkarev V. P.; Kolling D. R.; Hong S. J. Biol. Chem. 2003, 278, 36191–36201. [DOI] [PubMed] [Google Scholar]
- Crofts A. R.; Holland J. T.; Victoria D.; Kolling D. R.; Dikanov S. A.; Gilbreth R.; Lhee S.; Kuras R.; Kuras M. G. Biochim. Biophys. Acta 2008, 1777, 1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry E. A.; Guergova-Kuras M.; Huang L. S.; Crofts A. R. Annu. Rev. Biochem. 2000, 69, 1005–1075. [DOI] [PubMed] [Google Scholar]
- Hong S.; Ugulava N.; Guergova-Kuras M.; Crofts A. R. J. Biol. Chem. 1999, 274, 33931–33944. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Huang L.; Shulmeister V. M.; Chi Y. I.; Kim K. K.; Hung L. W.; Crofts A. R.; Berry E. A.; Kim S. H. Nature 1998, 392, 677–684. [DOI] [PubMed] [Google Scholar]
- Hunte C.; Koepke J.; Lange C.; Rossmanith T.; Michel H. Structure 2000, 8, 669–684. [DOI] [PubMed] [Google Scholar]
- Iwata S.; Lee J. W.; Okada K.; Lee J. K.; Iwata M.; Rasmussen B.; Link T. A.; Ramaswamy S.; Jap B. K. Science 1998, 281, 64–71. [DOI] [PubMed] [Google Scholar]
- Kim H.; Xia D.; Yu C. A.; Xia J. Z.; Kachurin A. M.; Zhang L.; Yu L.; Deisenhofer J. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 8026–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofts A. R. Biochim. Biophys. Acta 2004, 1655, 77–92. [DOI] [PubMed] [Google Scholar]
- Zu Y.; Couture M. M.; Kolling D. R.; Crofts A. R.; Eltis L. D.; Fee J. A.; Hirst J. Biochemistry 2003, 42, 12400–12408. [DOI] [PubMed] [Google Scholar]
- Lin I. J.; Chen Y.; Fee J. A.; Song J.; Westler W. M.; Markley J. L. J. Am. Chem. Soc. 2006, 128, 10672–10673. [DOI] [PubMed] [Google Scholar]
- Link T. A. Adv. Inorg. Chem. 1999, 47, 83–157. [Google Scholar]
- Zu Y.; Fee J. A.; Hirst J. J. Am. Chem. Soc. 2001, 123, 9906–9907. [DOI] [PubMed] [Google Scholar]
- Xiao K.; Yu L.; Yu C. A. J. Biol. Chem. 2000, 275, 38597–38604. [DOI] [PubMed] [Google Scholar]
- Brandt U. Biochim. Biophys. Acta 1998, 1365, 261–268. [DOI] [PubMed] [Google Scholar]
- Engstrom G.; Xiao K.; Yu C. A.; Yu L.; Durham B.; Millett F. J. Biol. Chem. 2002, 277, 31072–31078. [DOI] [PubMed] [Google Scholar]
- Crofts A. R.; Lhee S.; Crofts S. B.; Cheng J.; Rose S. Biochim. Biophys. Acta 2006, 1757, 1019–1034. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Westler W. M.; Xia B.; Oh B. H.; Markley J. L. Arch. Biochem. Biophys. 1995, 316, 619–634. [DOI] [PubMed] [Google Scholar]
- Markley J. L.; Bax A.; Arata Y.; Hilbers C. W.; Kaptein R.; Sykes B. D.; Wright P. E.; Wüthrich K. J. Mol. Biol. 1998, 280, 933–952. [DOI] [PubMed] [Google Scholar]
- www.cgl.ucsf.edu/home/sparky.
- Markley J. L. Acc. Chem. Res. 1975, 8, 70–80. [Google Scholar]
- Kainosho M.; Tsuji T. Biochemistry 1982, 21, 6273–6279. [DOI] [PubMed] [Google Scholar]
- Inubushi T.; Becker E. D. J. Magn. Reson. 1983, 51, 128–133. [Google Scholar]
- Bahrami A.; Assadi A. H.; Markley J. L.; Eghbalnia H. R. PLoS Comput. Biol. 2009, 5, e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Eghbalnia H. R.; Bahrami A.; Markley J. L. J. Biomol. NMR 2005, 32, 13–22. [DOI] [PubMed] [Google Scholar]
- Wang L.; Eghbalnia H. R.; Markley J. L. J. Biomol. NMR 2006, 35, 155–165. [DOI] [PubMed] [Google Scholar]
- http://www.bmrb.wisc.edu/.
- Rich P. R. Biochim. Biophys. Acta 1984, 768, 53–79. [DOI] [PubMed] [Google Scholar]
- Brunschwig B. S.; Sutin N. J. Am. Chem. Soc. 1989, 111, 7454–7465. [Google Scholar]
- Born M.; Oppenheimer R. Ann. Phys. 1927, 84, 457–484. [Google Scholar]
- Hohenberg P.; Kohn W. Phys. Rev. 1964, 136, B864. [Google Scholar]
- Link T. A. FEBS Lett. 1997, 412, 257–264. [DOI] [PubMed] [Google Scholar]
- Brandt U. FEBS Lett. 1996, 387, 1–6. [DOI] [PubMed] [Google Scholar]
- Lhee S.; Kolling D. R.; Nair S. K.; Dikanov S. A.; Crofts A. R. J. Biol. Chem. 2010, 285, 9233–9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich P. R. Biochim. Biophys. Acta 2004, 1658, 165–171. [DOI] [PubMed] [Google Scholar]
- Berry E. A.; Huang L. S. FEBS Lett. 2003, 555, 13–20. [DOI] [PubMed] [Google Scholar]
- Cape J. L.; Bowman M. K.; Kramer D. M. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7887–7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpower B. L. Biochim. Biophys. Acta 2002, 1555, 166–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.