

The transporters of the bacterial phosphotransferase system mediate the uptake of carbohydrates with concomitant phosphorylation. Good-quality crystals have been obtained of the membrane-spanning EIICGlc domain of the E. coli glucose transporter.
Keywords: phosphotransferase systems, glucose transporters
Abstract
The glucose-import system of Escherichia coli consists of a hydrophilic EIIAGlc subunit and a transmembrane EIICBGlc subunit. EIICBGlc (UniProt P69786) contains two domains: the transmembrane EIICGlc domain (40.6 kDa) and the cytoplasmic EIIBGlc domain (8.0 kDa), which are fused by a linker that is strongly conserved among its orthologues. The EIICBGlc subunit can be split within this motif by trypsin. Here, the crystallization of the tryptic EIICGlc domain is described. A complete data set was collected to 4.5 Å resolution at 100 K.
1. Introduction
Glucose is the preferred carbon source for eukaryotic cells and bacteria. Most cells take up glucose by facilitated diffusion or cation symporters. Many bacteria, including Escherichia coli, accumulate glucose and other carbohydrates by group translocation, a mechanism that couples translocation with concomitant phosphorylation of the transported solute (Siebold et al., 2001 ▶; Deutscher et al., 2006 ▶). Phosphoenolypyruvate (PEP), an intermediate of glycolysis, is the phosphoryl donor (Kundig et al., 1964 ▶). Because one of the two PEP molecules formed from glucose is utilized again for the uptake of the next glucose, glucose transport and glycolysis are tightly coupled. The proteins coupling the latter two processes constitute the bacterial PEP:carbohydrate phosphotransferase system (PTS). The PTS consists of two general carbohydrate-nonspecific proteins, enzyme I (EI) and the histidine-containing protein (HPr), and a variable number of transporters (EIIs) of different and sometimes overlapping carbohydrate specificities. The latter are grouped into four families, which have no apparent sequence similarity but share a common functional organization of three protein subunits or domains termed EIIA, EIIB and EIIC (Tchieu et al., 2001 ▶; Barabote & Saier, 2005 ▶).
EI transfers the phosphoryl group from PEP to HPr. EIIA and EIIB sequentially transfer the phosphoryl group from HPr to the carbohydrate, being translocated by the membrane-spanning EIIC as follows:
X-ray and NMR structures of EI, HPr and EIIA as well as EIIB that are representative of all transporter families are known (Peterkofsky et al., 2001 ▶), while EIIC structures remain frustratingly elusive.
In addition to carbohydrate uptake, the PTS also controls carbon and nitrogen metabolism, chemotaxis, biofilm formation and other bacterial behaviours (Lux et al., 1995 ▶, 1999 ▶; Erni, 2006 ▶; Deutscher et al., 2006 ▶; Houot & Watnick, 2008 ▶; Poncet et al., 2009 ▶). The PTS is at the heart of a regulatory network known as carbon catabolite repression (Gorke & Stulke, 2008 ▶). Here, the EIIA and EIIB units in Gram-negative bacteria, and in addition HPr in Gram-positive bacteria, serve as sensor and signalling units. In principle, their phosphorylation state reflects the availability of carbohydrates (Hogema et al., 1998 ▶). It decreases when a PTS carbohydrate is available and transport by EIIC is limited by the supply of PEP and/or EI activity. It increases when the supply of PEP exceeds the demand by PTS carbohydrates.
The glucose transporter of E. coli consists of two subunits: EIIAGlc and EIICBGlc. EIIAGlc, a monomeric all-β protein, transfers phosphoryl groups from HPr to the EIIBGlc domain. It is also an allosteric regulator of adenylate cyclase, glycerol kinase, the lactose/proton symporter and other non-PTS transporters and as such plays a key role in E. coli catabolite repression (Deutscher et al., 2006 ▶). Homodimeric EIICBGlc consists of two domains (Buhr et al., 1994 ▶; Fig. 1 ▶). The C-terminal cytoplasmic EIIBGlc domain assumes an α/β structure (Gemmecker et al., 1997 ▶). It transfers the phosphoryl group from the EIIAGlc subunit to the carbohydrate. The EIIBGlc domain in the dephosphorylated state (for instance, in the presence of glucose) binds the transcription repressor Mlc of the ptsG gene and thereby induces (activates) expression of EIICBGlc (Plumbridge, 1998 ▶, 1999 ▶; Tanaka et al., 1999 ▶; Lee et al., 2000 ▶; Nam et al., 2001 ▶, 2008 ▶; Seitz et al., 2003 ▶; Tanaka et al., 2004 ▶). The N-terminal membrane-spanning EIICGlc domain contains the carbohydrate-binding site (Hummel et al., 1992 ▶). EIICBGlc mutants exist which retain phosphorylation activity but have reduced transport activity (Buhr et al., 1992 ▶). They are believed to assume a constrained conformation, with the carbohydrate-binding site preferentially facing the cytoplasmic side of the membrane. It is not known whether there are one or two carbohydrate-binding sites per EIICGlc homodimer. One EIICGlc domain has been predicted to span the membrane ten times (Melen et al., 2003 ▶) and there is biochemical evidence for eight of the ten membrane-spanning segments (Buhr & Erni, 1993 ▶; Beutler, Kaufmann et al., 2000 ▶; Beutler, Ruggiero et al., 2000 ▶). The monomeric EIIBGlc and homodimeric EIICGlc domains can be expressed as separate entities, which together complement carbohydrate transport and phosphorylation activities (Buhr et al., 1994 ▶). The two domains can also be circularly permuted to afford an EIIBCGlc variant which is as active as wild-type EIICBGlc. These observations suggest that the connection between the EIICGlc and EIIBGlc domains is flexible and that the full-length protein may therefore be difficult to crystallize (Gutknecht et al., 1998 ▶).
Figure 1.

The eight putative membrane-spanning sequences (I–VIII) are compatible with biochemical experiments (Buhr & Erni, 1993 ▶; Beutler, Kaufmann et al., 2000 ▶; Beutler, Ruggiero et al., 2000 ▶). The ten putative membrane-spanning sequences (I–VI and vii–x) were predicted by topology analysis (http://www.cbs.dtu.dk/services/TMHMM/). The EIIBGlc domain is shown in italics (PDB codes 1iba, 3bp3 and 1o2f; Eberstadt et al., 1996 ▶; Nam et al., 2008 ▶; Cai et al., 2003 ▶). The accessible (Lys394, Lys412, Arg424) and potential (Lys382, Arg386) trypsin cleavage sites, the residues mutated to lock the conformation (Met17 and Lys150) and the thrombin cleavage site fused to residue 400 are highlighted. The domain linker and the active-site cysteine are underlined.
EIICBGlc indeed did not crystallize. Only after several weeks did a few small rod-shaped crystals appear, which turned out to consist of the EIICGlc domain rather than full-length EIICBGlc. Edman sequencing and electrospray mass spectrometry showed that the crystals consisted of an EIICGlc domain corresponding to amino-acid residues 1–394, suggesting that this domain had the propensity to crystallize. Residue 394 is a lysine in the linker region between the EIICGlc and EIIBGlc domains.
Here, we describe the preparation, crystallization and preliminary crystallographic analysis of the membrane-spanning EIICGlc domain of the E. coli glucose transporter. The procedure affording crystals that diffracted to 4.5 Å resolution will be described in greater detail. Promising alternatives which ultimately did not improve the crystal quality will be described summarily.
2. Materials and methods
2.1. Expression and purification of EIICBGlc
E. coli BW25113ΔacrB cells (Baba et al., 2006 ▶) transformed with plasmids (Lanz & Erni, 1998 ▶) encoding EIICBGlc (UniProt P69786) and EIICGlc with a C-terminal hexahistidine tag (–FGSRSHHHHHH) were grown in 10 × 2 l Erlenmeyer flasks containing 1 l LB medium (100 µg ml−1 ampicillin) at 310 K in an orbital shaker to an OD550 of 1.2 and were induced with 0.1 mM IPTG for 4 h. Cells were harvested by centrifugation (20 min, 5000g, 277 K, Sorvall HLR6 rotor) and washed twice by resuspension/centrifugation in 2 l buffer A (50 mM Tris–HCl pH 7.5, 400 mM NaCl, 10 mM β-mercaptoethanol). Cells were resuspended in 200 ml buffer A and lysed by two passages through a French pressure cell (110 MPa). The lysate was freed of cell debris by centrifugation (12 min, 13 000g, 277 K, Sorvall SS34 rotor). Membranes were collected from the supernatant by ultracentrifugation (60 min, 300 000g, 277 K, Beckman Ti70 rotor), pooled and suspended in 36 ml buffer B (10 mM Tris–glycine pH 9.3, 10 mM β-mercaptoethanol). Peripheral membrane proteins were removed by incubation with 4 ml 10% sodium cholate in buffer B for 15 min at 277 K. The membranes were collected by ultracentrifugation as above. The pellet was resuspended in 7.5 ml buffer B, solubilized by the addition of 7.5 ml 200 mM n-dodecyl-β-d-maltoside (DDM; catalogue No. D310S, Anatrace, USA), incubated for 16 h at 277 K and freed of insoluble material by ultracentrifugation as above. The supernatant was mixed with 10 ml Ni2+–nitrilotriacetic acid (NTA) agarose (Qiagen, USA) pre-equilibrated with buffer C [50 mM Tris–HCl pH 8.0, 500 mM NaCl, 5 mM glucose, 10 mM β-mercaptoethanol, 0.2 mM DDM, 3 mM n-undecyl-β-d-maltoside (UDM; catalogue No. U300S, Anatrace)] and incubated for 30 min at room temperature (RT). The slurry was poured into a column, the flowthrough was discarded and the slurry was washed at RT with 150 ml buffer C and 100 ml buffer I20 (20 mM imidazole in buffer C). EIICBGlc was eluted with 50 ml buffer I200 (200 mM imidazole in buffer C). Fractions were analyzed by SDS–PAGE and EIICBGlc-containing fractions were pooled. Purified EIICBGlc was concentrated using an Amicon Ultra 15 50 kDa cutoff centrifugal filter (Millipore, USA) to a final volume of 1 ml (10–20 mg ml−1) and centrifuged (15 000g, 5 min, Eppendorf FA-45-24-11 rotor). The resulting supernatant was purified by gel-filtration chromatography (ÄKTAprime FPLC system, Superdex 200 16/60, Amersham Pharmacia Biotech, flow rate 1 ml min−1) pre-equilibrated with buffer GF [10 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM glucose, 0.2 mM DDM low-α (catalogue No. D310LA, Anatrace), 3 mM UDM low-α (catalogue No. U300LA, Anatrace)]. 2 ml fractions were collected and examined by SDS–PAGE. The EIICBGlc peak fractions (60% of the total) were concentrated using an Amicon Ultra 15 100 kDa cutoff centrifugal filter and used for crystallization experiments. The yield was 1.0 mg pure EIICBGlc per litre of cell culture.
2.2. Expression and purification of the EIICGlc domain
The EIICGlc domain was prepared from purified EIICBGlc by limited trypsinolysis. EIICBGlc was diluted to a final concentration of 10 µM (0.5 mg ml−1). Final concentrations of 50 mM MgCl2, 5 mM β-mercaptoethanol and 0.1 µM trypsin were added. The mixture was incubated for 12 min at 310 K. The reaction was stopped by the addition of 0.2 mM phenylmethanesulfonylfluoride (freshly prepared in 100% 2-propanol). Uncleaved EIICBGlc was removed by Ni2+–NTA affinity chromatography. The flowthrough containing EIICGlc and inactivated trypsin was concentrated to 1 ml (8–12 mg ml−1), centrifuged at 15 000g for 5 min and further purified by gel filtration as described above. Peak fractions were pooled and concentrated to 1–80 mg ml−1 using an Amicon Ultra 15 100 kDa cutoff centrifugal filter (the 100 kDa cutoff resulted in the loss of 25% of the protein, but had the advantage of not concentrating excess detergent). Prior to crystallization, the high-affinity competitive inhibitor methyl-α-d-gluco-hexodialdo-1,5-pyranoside (Garcia-Alles et al., 2002 ▶) was added to a final concentration of 5 mM and traces of precipitated material were removed by centrifugation (10 min, 15 000g).
2.3. Crystallization, dehydration and data collection
EIICBGlc and EIICGlc were crystallized in 96-well plates (CompactClover Plates, Emerald BioSystems, USA) using the sitting-drop vapour-diffusion method at 289 and 277 K. For screening purposes, 2 µl protein solution (1–80 mg ml−1) was mixed with 2 µl reservoir solution and a reservoir volume of 100 µl was used. For optimization, both the protein:precipitant ratio and reservoir volume were varied. Crystals were gently dehydrated by the addition of 60–80 µl PEG 400 to the 100 µl reservoir buffer followed by incubation for at least 48 h. The dehydrated crystals were mounted in nylon loops (Hampton Research, USA), flash-frozen and stored in liquid nitrogen.
Diffraction data were either collected in-house using an R-AXIS IV image-plate area detector on a Rigaku RU-300 X-ray generator (operated at 90 mA and 46 kV) or at a synchrotron-radiation facility, predominantly at the Swiss Light Source (SLS; Villigen, Switzerland) but also at the ESRF (Grenoble, France) and DESY (Hamburg, Germany) at 100 K. Diffraction intensities were processed and scaled with XDS (Kabsch, 2010 ▶).
3. Results and discussion
The two-domain EIICBGlc did not crystallize. However, after several weeks of incubation of EIICBGlc in 100 mM Tris–HCl pH 8.5, 100 mM NaCl, 30%(v/v) PEG 400 a few small rod-shaped crystals appeared. Edman sequencing indicated that the N-terminus was intact. An electrospray mass spectrum showed a major peak at 42 147.4 Da, corresponding to amino-acid residues 1–394, and three minor peaks corresponding to adducts with one, two and three molecules of DDM (Zurbriggen, 2008 ▶). These results indicated that the EIICGlc domain had a propensity to crystallize.
EIICGlc domains (residues 1–394, 1–412 and 1–424) were expressed with a C-terminal hexahistidine tag, none of which produced crystals. EIICGlc with an N-terminal hexahistidine tag was not well expressed and therefore was not an option. Untagged EIICGlc domains of various lengths were then prepared by two methods (Fig. 1 ▶): (i) trypsin digestion of different EIICBGlc mutants (§2) and (ii) thrombin cleavage of recombinant EIICGlc containing a C-terminal thrombin-sensitive hexahistidine tag (–RSLVPRGSHHHHHH; Fig. 1 ▶). The product of thrombin cleavage showed a single band on SDS–PAGE, while the trypsin-cleavage product was contaminated with a variable small amount of a faster migrating band (Zurbriggen, 2008 ▶). In both cases the polydispersity index of EIICGlc determined by dynamic light scattering (DynaPro, Wyatt Technology) was 10–15%. However, the thrombic and tryptic EIICGlc(1–394) fragments produced crystals of similar quality. Crystals were obtained in 100 mM MES pH 6.5, 30%(v/v) PEG 400 and in 100 mM MOPS pH 7.0, 30%(v/v) PEG 400 (MemSys 8 and 12, Molecular Dimensions Ltd). These crystals were two-dimensional plates and diffracted to 18–20 Å resolution using synchrotron radiation at DESY.
In order to obtain homogeneous tryptic products of unique length, lysines in the linker region were mutated to alanines, affording the EIICGlc variants EIICGlc(1–412,K394A) and EIICGlc(1–424,K394A,K412A) (Fig. 1 ▶). EIICGlc(1–412,K394A) formed crystals that diffracted to 16–18 Å resolution under the same conditions as used for EIICGlc(1–394).
After extensive detergent screening by exchanging the detergent on the Ni2+–NTA column, the crystal quality was further improved to obtain diffraction to 10–12 Å resolution by exchanging DDM for a mixture of 0.2 mM DDM and 3 mM UDM in the same crystallization conditions as listed above.
To stabilize one of presumably several functional conformations, previously selected point mutations (Buhr et al., 1992 ▶) that confer impaired transport but near-normal phosphorylation activity were introduced into EIICBGlc(K394A) two and three at a time in various combinations. Three of the ten mutants were not stably expressed. The remaining seven mutants could be purified in buffers containing DDM and UDM, and cleaved with trypsin as described above. The stability of the mutant EIICGlc(1–412) domains was assessed by thermal unfolding monitored by circular-dichroism spectroscopy (Jasco J-715 spectropolarimeter, 222 nm, 0.2 mg ml−1, 1 mm path length; Jasco PFD-3505 Peltier temperature controller, 1 K min−1 from 303 to 353 K). The M17T,G149S and M17T,K150E mutants had the same T m of 342 K as wild-type EIICGlc. All others were between 10 and 20 K less stable. In a high-throughput screening (HT-X, EMBL, Hamburg) EIICGlc(1–412,K394A,M17T,K150E) formed thin plates in the presence of 10 mM NiCl2, 100 mM Tris–HCl pH 8.5, 20%(v/v) PEG 2000 MME (JB Classic 1.20) that diffracted to 20 Å resolution. After sparse-matrix screening with NiCl2, EIICGlc(1–412,K394A,M17T,K150E) crystals of cubic shape and of 100 µm in all three dimensions grew in 10 mM NiCl2, 100 mM Tris–HCl pH 8.5, 30%(v/v) PEG 400 by mixing 1.5 µl protein (3.0 mg ml−1) with 3.0 µl reservoir solution (Fig. 2 ▶). These crystals diffracted to 8.0 Å resolution using synchrotron radiation at SLS.
Figure 2.

EIICGlc(1–412,K394A,M17T,K150E) crystal obtained in 10 mM Tris–HCl pH 8.5, 5 mM NiCl2, 5 mM CuCl2, 1.5 mM UDM, 31%(v/v) PEG 400 after dehydration by adding 70 µl PEG 400 to the 100 µl reservoir buffer and incubating for 48 h at 289 K.
It was found that EIICGlc(1–412,K394A,M17T,K150E) produced larger crystals in 100 mM Tris–HCl pH 8.5, 5 mM NiCl2, 5 mM CuCl2, 1.5 mM UDM low-α, 32 ± 3%(v/v) PEG 400. These crystals were gently dehydrated by the addition of 60–80 µl PEG 400 to 100 µl reservoir buffer followed by incubation for at least 48 h. Dehydration improved the diffraction from 8.0 to 4.5 Å resolution. A complete data set extending to 5.0 Å resolution was collected (Fig. 3 ▶, Table 1 ▶). The space group was determined as P212121, with unit-cell parameters a = 126.4, b = 136.0, c = 210.2 Å, α = β = γ = 90.0°. Self-rotation functions were calculated using the program MOLREP (Vagin & Teplyakov, 1997 ▶) employing different resolution ranges (25–6, 25–5 and 15–5 Å) and integration radii (15, 25 and 35 Å). They were all featureless, as were native Patterson maps calculated within the same resolution ranges. Therefore, no clues regarding multiple copies in the asymmetric unit were discovered in the diffraction data. Assuming a micellar weight of about 74 kDa (VanAken et al., 1986 ▶) and a molecular mass of 43.5 kDa for EIICGlc, a Matthews parameter of about 7.7 Å3 Da−1, corresponding to a solvent content of about 85%, is obtained assuming the presence of one monomer per asymmetric unit. Thus, despite the negative results from self-rotation and Patterson functions, the presence of more than one EIICGlc monomer per asymmetric unit appears to be likely.
Figure 3.

Typical diffraction pattern of an EIICGlc(1–412,K394A,M17T,K150E) crystal. The data were collected on the PXI beamline of the SLS at 100 K. The exposure time is 0.5 s per image without a filter. The crystal was rotated by 0.2° per image.
Table 1. Data-collection and processing statistics for EIICGlc(1–412,K394A,M17T,K150E).
Values in parentheses are for the highest resolution shell.
| No. of crystals | 1 |
| Beamline | PXI, SLS, Villigen, Switzerland |
| Wavelength (Å) | 1.1763 |
| Detector | Pilatus |
| Crystal-to-detector distance (mm) | 600 |
| Rotation range per image (°) | 0.2 |
| Total rotation range (°) | 100 |
| Exposure time per image (s) | 0.5 |
| Resolution range (Å) | 100–4.98 (5.28–4.98) |
| Space group | P212121 |
| Unit-cell parameters (Å, °) | a = 126.4, b = 136.0, c = 210.2, α = β = γ = 90.0 |
| Mosaicity (refined by XDS) (°) | 0.152 |
| Total No. of measured intensities | 58459 (9227) |
| Unique reflections | 16166 (2464) |
| Multiplicity | 3.6 (3.7) |
| Mean I/σ(I) | 13.6 (2.0) |
| Completeness (%) | 98.0 (94.0) |
| Rmerge† (%) | 4.0 (76.7) |
| Rmeas or Rr.i.m.‡ (%) | 4.7 (89.0) |
| Overall B factor from Wilson plot (Å2) | 324.7 |
R
merge = 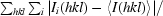
 , where I
i(hkl) is the intensity of the ith observation of reflection hkl.
, where I
i(hkl) is the intensity of the ith observation of reflection hkl.
Redundancy-independent merging statistic R
meas (also known as R
r.i.m.) = 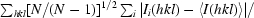 , where N is the redundancy of reflection hkl.
, where N is the redundancy of reflection hkl.
Many promising alternatives were tested which ultimately did not improve the crystal quality. Briefly, replacing Ni2+ with Cu2+ produced crystals of similar quality that diffracted to 7.5 Å resolution at best, whereas using other divalent metals did not yield any crystals (Zurbriggen, 2008 ▶). Reducing the surface entropy (Czepas et al., 2004 ▶) by mutating lysines to tyrosines did not improve diffraction. Macromolecular annealing, flash-annealing and annealing on the loop caused a deterioration in diffraction or destroyed the crystals. Monoclonal antibodies were raised against the EIICGlc domain and cocrystallization yielded crystals, but these did not diffract.
Furthermore, the following EIICGlc variants and domains of paralogues and orthologues of E. coli EIICGlc could also be purified and were screened for crystallization behaviour. EIICGlc(1–380), MalX(1–393), EIICGlcNAc(1–366) from E. coli and EIICGlcNAc from Thermoanaerobacter tengcongensis produced pseudo-crystalline spherolytes (Siebold, 2002 ▶; Navdaeva, 2006 ▶; Jeckelmann, 2009 ▶), while EIICGlc(1–439) from Deinococcus geothermalis EIICGlc(1–439), EIICGlc(1–427) from Thermosinus carboxidovorans and EIICGlc(1–399) from Vibrio shilonii produced small crystals that did not diffract beyond 15 Å resolution.
In conclusion, the best crystals were obtained with EIICGlc(1–412,M17T,K150E,K394A) as described above. Mixing 1.5 µl protein solution (10 mM Tris–HCl pH 7.5, 100 mM NaCl, 3 mM UDM, 0.2 mM DDM, 5 mM methyl-α-d-gluco-hexodialdo-1,5-pyranoside) at 3.0 mg ml−1 with 3.0 µl reservoir solution [100 mM Tris–HCl pH 8.5, 5 mM NiCl2, 5 mM CuCl2, 31%(v/v) PEG 400, 1.5 mM UDM] afforded crystals that diffracted to 8.0 Å resolution. Dehydrating these crystals by the addition of 70 µl PEG 400 to the reservoir and incubation for more than 48 h improved the diffraction to 4.5 Å resolution with >98% completeness. The next target will be to find heavy-metal derivatives in order to obtain phase information.
Acknowledgments
This research was supported by Swiss National Science Foundation Grant 3100A0-105247. We thank J. M. Jeckelmann for the synthesis of methyl-α-d-gluco-hexodialdo-1,5-pyranoside and Clemens Schulze-Briese and Takashi Tomizaki for help with data collection at the SLS, PSI Villigen.
References
- Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., Datsenko, K. A., Tomita, M., Wanner, B. L. & Mori, H. (2006). Mol. Syst. Biol.2, 2006.0008. [DOI] [PMC free article] [PubMed]
- Barabote, R. D. & Saier, M. H. Jr (2005). Microbiol. Mol. Biol. Rev.69, 608–634. [DOI] [PMC free article] [PubMed]
- Beutler, R., Kaufmann, M., Ruggiero, F. & Erni, B. (2000). Biochemistry, 39, 3745–3750. [DOI] [PubMed]
- Beutler, R., Ruggiero, F. & Erni, B. (2000). Proc. Natl Acad. Sci. USA, 97, 1477–1482. [DOI] [PMC free article] [PubMed]
- Buhr, A., Daniels, G. A. & Erni, B. (1992). J. Biol. Chem.267, 3847–3851. [PubMed]
- Buhr, A. & Erni, B. (1993). J. Biol. Chem.268, 11599–11603. [PubMed]
- Buhr, A., Flükiger, K. & Erni, B. (1994). J. Biol. Chem.269, 23437–23443. [PubMed]
- Cai, M., Williams, D. C. Jr, Wang, G., Lee, B. R., Peterkofsky, A. & Clore, G. M. (2003). J. Biol. Chem.278, 25191–25206. [DOI] [PubMed]
- Czepas, J., Devedjiev, Y., Krowarsch, D., Derewenda, U., Otlewski, J. & Derewenda, Z. S. (2004). Acta Cryst. D60, 275–280. [DOI] [PubMed]
- Deutscher, J., Francke, C. & Postma, P. W. (2006). Microbiol. Mol. Biol. Rev.70, 939–1031. [DOI] [PMC free article] [PubMed]
- Eberstadt, M., Grdadolnik, S. G., Gemmecker, G., Kessler, H., Buhr, A. & Erni, B. (1996). Biochemistry, 35, 11286–11292. [DOI] [PubMed]
- Erni, B. (2006). J. Bacteriol.188, 7036–7038. [DOI] [PMC free article] [PubMed]
- Garcia-Alles, L. F., Zahn, A. & Erni, B. (2002). Biochemistry, 41, 10077–10086. [DOI] [PubMed]
- Gemmecker, G., Eberstadt, M., Buhr, A., Lanz, R., Grdadolnik, S. G., Kessler, H. & Erni, B. (1997). Biochemistry, 36, 7408–7417. [DOI] [PubMed]
- Gorke, B. & Stulke, J. (2008). Nature Rev. Microbiol.6, 613–624. [DOI] [PubMed]
- Gutknecht, R., Manni, M., Mao, Q. & Erni, B. (1998). J. Biol. Chem.273, 25745–25750. [DOI] [PubMed]
- Hogema, B. M., Arents, J. C., Bader, R., Eijkemans, K., Yoshida, H., Takahashi, H., Alba, H. & Postma, P. W. (1998). Mol. Microbiol.30, 487–498. [DOI] [PubMed]
- Houot, L. & Watnick, P. I. (2008). J. Bacteriol.190, 311–320. [DOI] [PMC free article] [PubMed]
- Hummel, U., Nuoffer, C., Zanolari, B. & Erni, B. (1992). Protein Sci.1, 356–362. [DOI] [PMC free article] [PubMed]
- Jeckelmann, J. M. (2009). PhD thesis. University of Bern, Switzerland.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kundig, W., Gosh, S. & Roseman, S. (1964). Proc. Natl Acad. Sci. USA, 52, 1067–1074. [DOI] [PMC free article] [PubMed]
- Lanz, R. & Erni, B. (1998). J. Biol. Chem.273, 12239–12243. [DOI] [PubMed]
- Lee, S. J., Boos, W., Bouche, J. P. & Plumbridge, J. (2000). EMBO J.19, 5353–5361. [DOI] [PMC free article] [PubMed]
- Lux, R., Jahreis, K., Bettenbrock, K., Parkinson, J. S. & Lengeler, J. W. (1995). Proc. Natl Acad. Sci. USA, 92, 11583–11587. [DOI] [PMC free article] [PubMed]
- Lux, R., Munasinghe, V. R., Castellano, F., Lengeler, J. W., Corrie, J. E. & Khan, S. (1999). Mol. Biol. Cell, 10, 1133–1146. [DOI] [PMC free article] [PubMed]
- Melen, K., Krogh, A. & von Heijne, G. (2003). J. Mol. Biol.327, 735–744. [DOI] [PubMed]
- Nam, T.-W., Cho, S.-H., Shin, D., Kim, J.-H., Jeong, J.-Y., Lee, J.-H., Roe, J.-H., Peterkofsky, A., Kang, S.-O., Ryu, S. & Seok, Y.-J. (2001). EMBO J.20, 491–498. [DOI] [PMC free article] [PubMed]
- Nam, T.-W., Jung, H. I., An, Y. J., Park, Y.-H., Lee, S. H., Seok, Y.-J. & Cha, S.-S. (2008). Proc. Natl Acad. Sci. USA, 105, 3751–3756. [DOI] [PMC free article] [PubMed]
- Navdaeva, V. (2006). PhD thesis. University of Bern, Switzerland.
- Peterkofsky, A., Wang, G., Garrett, D. S., Lee, B. R., Seok, Y.-J. & Clore, G. M. (2001). J. Mol. Microbiol. Biotechnol.3, 347–354. [PubMed]
- Plumbridge, J. (1998). Mol. Microbiol.29, 1053–1063. [DOI] [PubMed]
- Plumbridge, J. (1999). Mol. Microbiol.33, 260–273. [DOI] [PubMed]
- Poncet, S., Milohanic, E., Maze, A., Abdallah, J. N., Ake, F., Larribe, M., Deghmane, A. E., Taha, M. K., Dozot, M., De, B., X, Letesson, J. J. & Deutscher, J. (2009). Contrib. Microbiol.16, 88–102. [DOI] [PubMed]
- Seitz, S., Lee, S. J., Pennetier, C., Boos, W. & Plumbridge, J. (2003). J. Biol. Chem.278, 10744–10751. [DOI] [PubMed]
- Siebold, C. (2002). PhD thesis. University of Bern, Switzerland.
- Siebold, C., Flükiger, K., Beutler, R. & Erni, B. (2001). FEBS Lett.504, 104–111. [DOI] [PubMed]
- Tanaka, Y., Itoh, F., Kimata, K. & Aiba, H. (2004). Mol. Microbiol.53, 941–951. [DOI] [PubMed]
- Tanaka, Y., Kimata, K., Inada, T., Tagami, H. & Aiba, H. (1999). Genes Cells, 4, 391–399. [DOI] [PubMed]
- Tchieu, J. H., Norris, V., Edwards, J. S. & Saier, M. H. Jr (2001). J. Mol. Microbiol. Biotechnol.3, 329–346. [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- VanAken, T., Foxall-VanAken, S., Castleman, S. & Ferguson-Miller, S. (1986). Methods Enzymol.125, 27–35. [DOI] [PubMed]
- Zurbriggen, A. (2008). PhD thesis. University of Bern, Switzerland.