

Crystallization of glycosylated human prolylcarboxypeptidase expressed in Chinese hamster ovary cells is described. The hexagonal crystals belong to space group R32 and diffract to 2.8 Å resolution.
Keywords: PrCP, prolylcarboxypeptidases, serine proteases, carboxypeptidases, angiotensinase C, dipeptidyl peptidases
Abstract
Prolylcarboxypeptidase (PrCP) is a lysosomal serine carboxypeptidase that cleaves a variety of C-terminal amino acids adjacent to proline and has been implicated in diseases such as hypertension and obesity. Here, the robust production, purification and crystallization of glycosylated human PrCP from stably transformed CHO cells is described. Purified PrCP yielded crystals belonging to space group R32, with unit-cell parameters a = b = 181.14, c = 240.13 Å, that diffracted to better than 2.8 Å resolution.
1. Introduction
Prolylcarboxypeptidase (PrCP) is a lysosomal serine carboxypeptidase that cleaves a variety of C-terminal amino acids adjacent to proline (Mallela et al., 2009 ▶). The enzyme is widely distributed, with a number of substrates. For example, angiotensin II is inactivated by PrCP, implicating a role for the enzyme in hypertension, tissue proliferation and smooth muscle growth (Mallela et al., 2009 ▶). PrCP also reportedly inactivates the anorexigenic neuromodulator α-melanocyte-stimulating hormone and PrCP activity in rodents has been implicated in a reduction in food intake (Wallingford et al., 2009 ▶).
PrCP is a member of the S28 family of proteases and is a dipeptidyl peptididase. However, PrCP shares very limited sequence identity to other proteases. The closest homologue is the serine aminopeptidase DPPII/DPPVII, with 39.6% sequence identity and 55.4% sequence similarity. The next closest human homologues are prolyl oligopeptidase, with only 8.4% sequence identity and 13.9% sequence similarity, and human DPPIV, with only 6.5% sequence identity and 11.2% sequence similarity. PrCP and DPPII/DPPVII therefore form a pair of proteases that appear to be distinct from other members of the protease family. There are currently no published structures of the S28 family of proteases.
Given the potential role of PrCP in cardiovascular and metabolic diseases and the limited sequence identity to other proteases, we have developed a robust enzyme-production protocol that produces crystallization-grade PrCP. The results lay the foundation for understanding the structural basis of PrCP activity and for the structure-guided discovery of PrCP inhibitors.
2. Materials and methods
2.1. Cloning and cell-line generation
Human cDNA (NM_005040; transcript variant 1; Tan et al., 1993 ▶) encoding residues 46–496 of PrCP was cloned with an N-terminal FLAG tag and a C-terminal heptahistidine tag under the trypsin II signal sequence in pcDNA5/FRT (Invitrogen Corp.) for expression in mammalian cells. A stable CHO cell line harboring PrCP was generated by co-transfecting the PrCP vector and pOG44 (Invitrogen Corp.), which contains FLP recombinase. Recombinant colonies were selected on the basis of hygromycin B resistance.
Baculoviruses for the expression of residues 31–496 of human PrCP with a C-terminal hexahistidine tag under either the GP64 or native signal sequences in a Gateway-adapted pVL1392 vector (Pharmingen) were prepared using previously described methods (Sunami et al., 2010 ▶), with the exception that the virus was produced using BestBAC (Expression Systems) linearized viral DNA.
2.2. Protein expression
10 l of stable CHO cells expressing recombinant human PrCP were grown in CD-CHO/DMEM/F12 growth medium supplemented with 2% FBS and 4 mM glutamine in a stirred-tank bioreactor in fed-batch mode (Kemp Biosciences). Growth was continued for 14 d with the addition of 2 l fresh medium on day 7 and glucose (1 g l−1) on days 8, 9 and 10. Cell viability was 94–99% during the entire growth. Culture supernatant was harvested on day 14 and diafiltered in 20 mM sodium phosphate pH 7.5 and 500 mM NaCl. Expression in insect cells was performed as described previously (Sunami et al., 2010 ▶).
2.3. Protein purification
Conditioned culture supernatant was concentrated tenfold and diafiltered into 20 mM sodium phosphate pH 7.5 and 500 mM NaCl. Initial purification was by Ni-affinity chromatography on a HisTrap column (GE Healthcare) equilibrated in buffer A (50 mM HEPES pH 7.5 and 300 mM NaCl) and 20 mM imidazole. Following washes with 20 and 50 mM imidazole in buffer A, PrCP was eluted with a linear imidazole gradient (50–500 mM) in buffer A. Fractions containing PrCP were pooled, diluted in a 1:2 ratio with 20 mM HEPES pH 7.5 and further purified on a HiTrap Heparin HP column (GE Healthcare) equilibrated in 20 mM HEPES pH 7.0 and 100 mM NaCl using a linear NaCl gradient (0.1–1 M). Active PrCP fractions were pooled and concentrated. Gel filtration using a Superdex 200 column (GE Healthcare) equilibrated in 20 mM HEPES pH 7.0 and 350 mM NaCl yielded purified PrCP, which was concentrated to 25 mg ml−1 for crystallization using Amicon Ultra centrifugal concentrators (Millipore, 10 000 molecular-weight cutoff, centrifuged at 1400g).
2.4. Mass spectrometry
PrCP (20 µg) was heat-denatured in 50 mM ammonium bicarbonate pH 8.0 and 5 mM DTT, alkylated with iodoacetamide for 20 min at room temperature and digested with trypsin (100 ng) for 5 h at 310 K. The digest was analyzed by reversed-phase C12 (Jupiter Proteo) chromatography using a linear water–acetonitrile gradient containing 0.02% trifluoroacetic acid followed by mass spectrometry using a Thermo LXQ system. Data were analyzed by a SEQUEST search against a database of mouse proteins supplemented with the human PrCP sequence. The intact protein mass was measured on an Applied Biosystems 4700 MALDI–TOF using a saturated sinapinic acid matrix and bovine serum albumin and apotransferrin standards.
2.5. Activity
PrCP proteolysis of MCA-Pro-Pro-Lys(DNP) (Anaspec) was monitored by fluorescence (Fluostar) using excitation and emission wavelengths of 340 and 450 nm, respectively. Assays were performed at room temperature in 20 mM MES and 0.1 M NaCl pH 5.5. In most experiments, the PrCP and substrate concentrations were 1–2 nM and 25 µM, respectively. Wells were read every 90 s for 30 min and rates were calculated from the initial linear region. Total specific activity was determined by allowing a reaction to run to completion at a high enzyme concentration and using the resultant fluorescence to calculate molar conversion for linear reactions. Z-Pro-prolinal (Z-Pro-Pro-aldehyde-dimethyl acetal) was obtained from Bachem.
2.6. Dynamic light scattering
Dynamic light-scattering measurements were performed with a DynaPro 99 (Protein Solutions, Wyatt Technology Corporation) system. Measurements were performed at protein concentrations of 14.7 and 27.4 mg ml−1 in 20 mM HEPES pH 7.0 and 350 mM NaCl. Prior to measurement, protein samples were centrifuged for 10 min at 13 000g. The hydrodynamic radius and polydispersity of purified PrCP were determined by data analysis using the DYNAMICS software package (v.6.3.40).
2.7. Crystallization
Protein solutions at 14.7 and 27.4 mg ml−1 in 20 mM HEPES pH 7.0 and 350 mM NaCl were used in crystallization experiments. Initial crystallization screening was performed at 277 and 293 K using sitting-drop vapor diffusion in 96-well MRC two-well crystallization plates (Innovaplate SD-2). A Phoenix crystallization robot (Rigaku) was used to dispense 200 nl drops containing equal parts protein and reservoir solution. For crystal optimization, systematic grid refinement and standard streak-seeding techniques were employed. For data collection, crystals were sequentially stepped through cryoprotectants consisting of reservoir solution supplemented with 5, 10, 15 and 20% glycerol for 20 s each and flash-cooled in liquid nitrogen.
2.8. X-ray diffraction analysis
Cryogenically preserved crystals were transported to Advanced Light Source beamline 5.0.2 (Lawrence Berkeley National Laboratory). Diffraction data were collected by Reciprocal Space Consulting at 100 K using an X-ray wavelength of 1.0 Å and an ADSC Q315 detector. The data were processed with HKL-2000 (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
Human PrCP has previously been expressed in Drosophilia melanogaster S2 cells (Shariat-Madar et al., 2004 ▶). In order to develop a more accessible expression system, PrCP expression was evaluated in both baculovirus Sf21 and mammalian CHO hosts. CHO-expressed PrCP was pursued for crystallization studies since the yield of PrCP from CHO cells (approximately 6 mg l−1) exceeded that from Sf21 cells (approximately 0.1 mg l−1).
PrCP expressed in CHO cells was purified to obtain crystallization-grade protein with high purity and low polydispersity (Fig. 1 ▶ a). The identity of the protein was confirmed using LC-MS analysis of a tryptic digest of purified PrCP protein. Peptide products were identified that spanned approximately 75% of the protein. Analysis of the intact protein by MALDI–TOF suggested a mass of 62 kDa, which differed from the calculated mass of 53 kDa for PrCP. The 9 kDa mass difference is likely to arise from glycosylation. Analysis of the tryptic digest LC-MS data identified two peptides containing putative glycosylation sites. The peptide containing Asn47 does not appear to be glycosylated, while the peptide containing Asn436 was found to be glycosylated with a high-mannose glycan containing glycoforms ranging from Man5GlcNAc2 to Man9GlcNAc2. Three peptides containing four other potential glycosylation sites were not identified in the analysis.
Figure 1.

Characterization of purified PrCP. (a) Coomassie-stained SDS–PAGE analysis of purified PrCP showing molecular-weight standards (lane 1; labeled in kDa) and purified PrCP (lane 2). (b) The reaction rate of PrCP is 103 min−1. (c) PrCP is inhibited by Z-Pro-prolinal with an apparent IC50 of 150 µM.
Purified PrCP was active in a fluorogenic substrate assay with a turnover rate of 103 min−1 (Fig. 1 ▶ b). The enzyme was inhibited by the product-based inhibitor Z-Pro-prolinal with an apparent IC50 of 150 µM (Fig. 1 ▶ c), in accordance with previous reports (Shariat-Madr et al., 2002 ▶, 2004 ▶).
Dynamic light scattering indicated a monodisperse state (13% polydispersity). The hydrodynamic radius was 4.6 nm, which corresponds to a mass of 122 kDa, which in turn corresponds to a dimer of the glycosylated protein. These results are consistent with previous gel-filtration results that suggested that PrCP purified from human kidney exists as a dimer in solution (Odya et al., 1978 ▶; Tan et al., 1993 ▶).
Crystallization screening was performed using automated 96-well vapor-diffusion experiments. The initial broad screen consisted of 1152 conditions from commercially available crystallization screens (Qiagen and Hampton Research). Initial crystallization trials yielded microcrystals in several conditions, the most promising of which grew from a solution consisting of 2.0 M ammonium sulfate, 0.1 M HEPES pH 7.5 and 2%(v/v) polyethylene glycol 400. Crystal growth was optimized using focused grid screens and streak-seeding. Single plate-like crystals that were amenable to crystallographic analysis were obtained using 27.4 mg ml−1 protein and a reservoir solution consisting of 1.8 M ammonium sulfate, 0.1 M HEPES pH 7.5 and 1–2% polyethylene glycol 400 mixed in a 2:1 ratio at 277 K. The crystals grew to maximum dimensions of 0.2 × 0.4 × 0.4 mm (Fig. 2 ▶ a) in 3–5 d and required freezing within 7 d for optimal diffraction.
Figure 2.

(a) Crystals of PrCP. (b) X-ray diffraction pattern of PrCP.
Synchrotron diffraction data were collected from a single crystal of PrCP (Table 1 ▶). The crystals diffracted anisotropically, exhibiting diffraction to 2.4 Å in some orientations and to 2.8 Å in others (Fig. 2 ▶ b). A 200° wedge of data was collected using 0.5° oscillations, 1 s exposures and a crystal-to-detector distance of 355 mm. The native data set was 100% complete to 2.8 Å with nearly tenfold redundancy. The data were integrated and subsequently reduced in space group R32. The hexagonal setting for this space group yielded unit-cell parameters a = b = 181.14, c = 240.13 Å. Using the glycosylated molecular weight of PrCP determined using MALDI–TOF, these unit-cell parameters suggested that two or three monomers of PrCP were present in the asymmetric unit, yielding a Matthews coefficient of 3.06 or 2.04 Å3 Da−1, respectively (Matthews, 1968 ▶). These values correspond to a solvent content of 60% (two molecules per asymmetric unit) or 40% (three molecules per asymmetric unit). PrCP appears to be a dimer in solution (see above) and therefore it is likely that the asymmetric unit contains a dimer in this crystal form.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | R32 (hexagonal setting) |
| Unit-cell parameters (Å, °) | a = b = 181.14, c = 240.13, α = β = 90, γ = 120 |
| Resolution range (Å) | 50.0–2.80 (2.87–2.80) |
| Mosaicity (°) | 0.77 |
| Observations | 2600927 |
| Unique reflections | 37814 |
| Completeness (%) | 100.0 (100.0) |
| Average redundancy | 10.9 (10.9) |
| Mean I/σ(I) | 9.9 |
| Rmerge† (%) | 9.8 (59.4) |
R
merge = 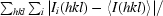
 , where I(hkl) is the integrated intensity for a given reflection.
, where I(hkl) is the integrated intensity for a given reflection.
In conclusion, we describe a robust mammalian expression system for the production of human PrCP that results in crystallization-grade PrCP. These results lay the foundation for elucidating the structural basis of the mechanism of PrCP activity and for the structure-guided discovery of PrCP inhibitors for the treatment of cardiovascular and metabolic diseases.
References
- Mallela, J., Yang, J. & Shariat-Madar, Z. (2009). Int. J. Biochem. Cell Biol.41, 477–481. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Odya, C. E., Marinkovic, D. V., Hammon, K. J., Stewart, T. A. & Erdös, E. G. (1978). J. Biol. Chem.253, 5927–5931. [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Shariat-Madar, Z., Madhi, F. & Schmaier, A. H. (2002). J. Biol. Chem.277, 17962–17969. [DOI] [PubMed]
- Shariat-Madar, Z., Mahdi, F. & Schmaier, A. H. (2004). Blood, 103, 4554–4561. [DOI] [PubMed]
- Tan, F., Morris, P. W., Skidgel, R. A. & Erdös, E. G. (1993). J. Biol. Chem.268, 16631–16638. [PubMed]
- Sunami, T., Byrne, N., Diehl, R. E., Funabashi, K., Hall, D. L., Ikuta, M., Patel, S. B., Shipman, J. M., Smith, R. F., Takahasi, I., Zugay-Murphy, J., Iwasawa, Y., Lumb, K. J., Munshi, S. K. & Sharma, S. (2010). J. Biol. Chem.285, 4587–4594. [DOI] [PMC free article] [PubMed]
- Wallingford, N., Perroud, B., Gao, Q., Coppola, A., Gyenges, E., Liu, Z., Gao, X., Diament, A., Hais, K. A., Shariat-Madar, Z., Mahdi, F., Wardlaw, S. L., Schmaier, A. H., Warden, C. H. & Diano, S. (2009). J. Clin. Invest.119, 2291–2303. [DOI] [PMC free article] [PubMed]