

Abstract
Previous findings suggest that estradiol has a suppressive effect on TNF-α, but the mechanism by which estradiol regulates TNF-α expression in primary human macrophages is unknown. Herein, we demonstrate that pretreatment of human macrophages with estradiol attenuates LPS-induced TNF-α expression through suppression of NF-κB activation. Furthermore, we show that activation of macrophages with LPS decreases expression of κB-Ras2, an inhibitor of NF-κB signaling. Estradiol pre-treatment abrogates this decrease, leading to enhanced expression of κB-Ras2 with LPS stimulation. Additionally, we have identified two miRNAs, let-7a and miR-125b, that target the κB-Ras2 3′UTR. LPS induces let-7a and inhibits miR-125b expression in human macrophages and pre-treatment with estradiol abrogates these effects. 3′UTR reporter assays demonstrate that let-7a destabilizes the κB-Ras2 3′UTR while miR-125b enhances its stability, resulting in decreased κB-Ras2 in response to LPS. Our data suggest that pre-treatment with estradiol reverses this effect. We propose a novel mechanism for estradiol inhibition of LPS-induced NF-κB signaling in which κB-Ras2 expression is induced by estradiol via regulation of let-7a and miR-125b. These findings are significant in that they are the first to demonstrate that estradiol represses NF-κB activation through induction of κB-Ras2, a key inhibitor of NF-κB signaling.
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
Introduction
To limit microbial pathogenesis, mammalian host cells rely on both innate and adaptive immune mechanisms. The innate immune system deploys rapid antimicrobial responses to pathogenic challenge and simultaneously instructs the adaptive immune system regarding the nature and context of the infectious threat. As key phagocytes, macrophages provide both early recognition of pathogens and a crucial bridge between innate and adaptive immunity. This is mediated through detection of distinct molecules that are present on a broad diversity of microorganisms. These conserved products, termed pathogen associated molecular patterns (PAMPs), are recognized by a repertoire of invariant pattern recognition receptors, including the TLRs. TLR4, in association with accessory molecules MD-2 and CD14, is the signal transduction receptor for Gram-negative bacterial LPS. Engagement of TLR4 by LPS initiates a cascade of signaling events that culminates in the production of inflammatory cytokines, including TNF-α (1).
TNF-α is a potent pro-inflammatory cytokine produced by activated macrophages and has pleiotropic effects on immune cell survival, activation and differentiation. During an infection, pathogen clearance is dependent on proper regulation of TNF-α but aberrant expression of TNF-α can lead to significant morbidity and mortality (2). Deregulated TNF-α production has been correlated with disease activity in numerous autoimmune disorders, including rheumatoid arthritis and systemic lupus erythematosus, which are much more prevalent in women than men (3). Intriguingly, serum TNF-α levels are lower in healthy pre-menopausal females compared with both males and post-menopausal females (4) and TNF-α levels are attenuated in female patients during septic shock compared with males (5). Previous work suggests that 17β-estradiol (estradiol) has a suppressive effect on TNF-α production in cell lines and murine macrophages (6, 7), although no studies to date have addressed the mechanism by which estradiol regulates TNF-α expression in primary human macrophages.
LPS-induced TNF-α expression is regulated by NF-κB activation. In unstimulated cells, the NF-κB transcription factors p65 and p50 are bound to IκB and are inactive. LPS binding to TLR4 leads to activation of the IκB kinase (IKK) complex that subsequently phosphorylates IκB, targeting it for degradation. The p65 and p50 NF-κB subunits are then free to translocate to the nucleus where they modulate transcription of NF-κB regulated genes.
Activation of NF-κB can be regulated through the binding of proteins that modulate IκB degradation. κB-Ras2 is a member of the Ras family of proteins that negatively regulates NF-κB signaling. κB-Ras2 binds directly to IκBα and inhibits IKK-dependent phosphorylation and subsequent degradation, thus inhibiting NF-κB signaling (8, 9). In this study, we demonstrate that pretreatment of human macrophages with estradiol attenuates LPS-induced TNF-α expression through inhibition of NF-κB activation. Furthermore, we show that activation of macrophages with LPS inhibits expression of κB-Ras2, but that pretreatment with estradiol abrogates this inhibition, leading to enhanced expression of κB-Ras2.
Because microRNAs (miRNAs) have been increasingly implicated in the regulation of immune function, we hypothesized that miRNAs might be involved in the mechanism by which estradiol regulates expression of κB-Ras2. miRNAs are short ~22nt regulatory RNAs that bind to the 3′UTRs of target mRNAs and modulate stability and translation. Using a miRNA prediction binding program (10), we identified let-7a and miR-125b as miRNAs predicted to interact with the κB-Ras2 3′UTR. Intriguingly, we now show that LPS induces let-7a and inhibits miR-125b expression in primary human macrophages and that pre-treatment of these cells with estradiol abrogates the regulation of these miRNAs by LPS. Therefore, we propose a novel mechanism for estradiol inhibition of LPS-induced NF-κB signaling in which κB-Ras2 expression is induced by estradiol via the coordinated inhibition of let-7a and induction of miR-125b. Our model for this mechanism is depicted in Figures 7A and 7B. These findings are significant in that they are the first to demonstrate that estradiol represses NF-κB activation through induction of κB-Ras2, a key inhibitor of NF-κB signaling.
Figure 7. Proposed mechanism of estradiol regulation of NF-κB signaling in primary human macrophages.

(A) LPS binding to TLR4 induces expression of let-7a and decreases expression of miR-125b, leading to decreased expression of κB-Ras2 to enable complete activation of NF-κB signaling and TNF-α expression. (B) When macrophages are treated with estradiol prior to LPS stimulation, changes in expression of let-7a and miR-125b in response to 29LPS are abrogated. This results in up-regulation of κB-Ras2 and inhibition of NF-κB signaling, culminating in decreased expression of TNF-α.
Materials and methods
Isolation and culture of human peripheral blood monocytes and macrophages
PBMCs were obtained by leukapheresis of normal, healthy pre-menopausal female donors following informed consent. Written consent was obtained from all subjects in accordance with the human experimentation guidelines established by Dartmouth College’s Committee for the Protection of Human Subjects (CPHS), protocol #17011. To preclude confounding results associated with exogenous hormone use, individuals using hormonal contraceptives were excluded from these studies. Mononuclear cells were separated on Ficoll-Hypaque and enriched for monocytes using cold aggregation (11). Monocyte purity was >95% as determined by CD14 expression using flow cytometry (data not shown). To generate macrophages, monocytes were cultured in the presence of 10 μg/ml GM-CSF (Peprotech) for 7 days. Macrophages were cultured in complete HEPES-buffered RPMI 1640 (Cellgro) supplemented with 10% FBS (Hyclone) and 50 μg/ml gentamicin sulfate (Sigma-Aldrich).
Estradiol, LPS and inhibitor treatments
Forty eight hours prior to hormone treatment, culture media was replaced with HEPES-buffered phenol red-free RPMI 1640 (Cellgro) supplemented with 10% charcoal dextran-stripped FBS (Hyclone) and 50 μg/ml gentamicin sulfate. All hormone treatments were preformed using this media. 17β-estradiol (Calbiochem) was resuspended in ethanol immediately prior to treatment. Cells were treated with 100 nM estradiol, unless otherwise indicated, or ethanol as a vehicle control. For LPS stimulation experiments, cells were pre-treated with estradiol for 24 hours followed by administration of 10 ng/ml ultrapure E. coli LPS (Sigma-Aldrich) for 12 hours unless otherwise noted. Culture supernatants were analyzed for TNF-α production using the human TNF-α Quantikine ELISA kit (R&D Systems) according to the manufacturer’s protocol. For estrogen receptor inhibitor experiments, cells were treated with 1 μM ICI 182,780 (Tocris) for 1 hour prior to hormone treatment. In previous work, we determined that incubation of macrophages with 1 μM of the ER-antagonist ICI 182, 780 optimally blocks binding of estradiol (10−7M) to the ER (12, 13). For NF-κB inhibition experiments, macrophages were treated with 10 μM 6-Amino-4-(4-phenoxyphenylethylamino)quinazoline (Calbiochem) for 1 hour prior to LPS stimulation. As we have previously reported, none of the hormone or antagonist doses affected macrophage viability (12, 14).
Total RNA extraction and Real-Time PCR
Total RNA, including miRNA, was extracted from cells using the miRNeasy Mini kit with on-column DNase I treatment (Qiagen). RNA integrity and concentration were determined using the RNA6000 Nano LabChip kit (Agilent). For mRNA analysis, 500 ng of RNA were reverse transcribed using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer’s protocol. miRNA reverse transcription was performed using the TaqMan microRNA Reverse Transcription Kit and miRNA specific primers (Applied Biosystems). Real-time TaqMan PCR was used to quantify mRNA and miRNA expression using TaqMan Master Mix and validated primer/probe sets (Applied Biosystems). Amplification was carried out using the Applied Biosystems 7300 Real-Time PCR system. Threshold cycle number was determined using Opticon software. mRNA levels were normalized to β-actin and miRNA levels were normalized to U6 expression using the formula 2−(Et−Rt) where Rt is the mean cycle threshold for the control gene and Et is the mean threshold for the experimental gene. Cycling conditions for TaqMan PCR consisted of an initial incubation at 50°C for 2 minutes and 95°C for 10 minutes followed by 40 cycles of 95°C for 15 sec and 60°C for 1 minutes. Product accumulation was measured during the extension phase and all samples were run in triplicate.
Preparation of lysates and immunoblot analysis
Whole cell lysates were prepared using passive lysis buffer (Promega). Lysates were analyzed for total protein concentration using a Micro BCA Protein Assay kit (Pierce). 25 μg of each lysate were separated on 10% acrylamide gels and electrotransferred to nitrocellulose membrane in Tris-glycine buffer with 20% methanol. Membranes were blocked with 5% milk in 1X TBS and 0.1% Tween-20 for 1h at room temperature. Blots were then probed with primary detection antibodies for IKKβ (AbCam), IκBα (Cell Signaling), phospho-IκBα (Cell Signaling) or κB-Ras2 (AbNova), followed by HRP-conjugated secondary antibody (BioRad). To control for protein loading, blots were probed for GAPDH expression using mouse mAb 6C5 (American Research Products). Incubations with the primary antibody were overnight at 4°C with rocking and incubations with secondary antibody were performed at room temperature for 45 minutes following thorough washing. Blots were visualized using Supersignal chemiluminesence substrate (Pierce) exposed to film (Kodak).
ELISA
Macrophages were treated with 100 nM estradiol for 24 hours prior to stimulation with LPS (10 ng/ml) for 1 hour. Nuclear protein lysates were generated using the NE-PER kit (Pierce). 50 μg of nuclear protein lysate was used according to the manufacturer’s protocol to analyze NF-κB p65 expression by ELISA (Biosource).
Culture supernatants were analyzed for TNF-α expression by ELISA (R and D Systems) according to the manufacturer’s protocol. Samples stimulated with LPS were diluted 100 fold and unstimulated samples were run undiluted.
Luciferase Assay
RAW 264.7 cells were plated 24 hours prior to transfection at a density of 2 × 105 cells per well in 24-well tissue-culture dishes. Cells were transfected with 100 nM pre-miRNA or negative control pre-miRNA (Ambion) and 800 ng of κB-Ras2 3′ UTR luciferase plasmid or control plasmid (Switchgear genomics) using lipofectamine 2000 transfection reagent (Invitrogen). Transfection efficiency was normalized to Renilla luciferase activity by co-transfection of 100 ng pRL-TK expression vector (Promega). Cells were lysed 48 hours post-transfection using passive lysis buffer (Promega). Luciferase activity was determined using the dual-luciferase reporter assay system (Promega) and the Berthold Centro LB960 luminometer. Data are the mean of at least three independent experiments and are represented as normalized relative light units (RLUs).
Transient transfection of pre-miRNAs
U937 cells (obtained from ATCC) were differentiated with PMA (6 ng/ml, Sigma) for two days. Cells were plated at a density of 8×105 cells per well in 6-well culture dishes 24 hours prior to transfection. Cells were transfected with 100 nM pre-miR-125b, pre-let-7a or negative control pre-miRNA (Ambion) using FugeneHD transfection reagent (Roche). 48hrs after transfection, total RNA was harvested and analyzed for κB-Ras2 expression. Data are representative of three independent transfections.
Statistical analysis
All experiments were performed using N=3–4 individual donors unless otherwise noted. Results are presented as mean +/− standard error. Statistical analysis was performed using a paired Student’s t- test and significance was achieved at p<0.05.
Results
Estradiol attenuates TNF-α production in human macrophages
Although previous reports have shown that estradiol affects TNF-α production in murine macrophages (6, 7); it was unknown whether estradiol influences TNF-α production in human macrophages. To determine the effect of estradiol on TNF-α production in macrophages, cells were pre-treated with estradiol at concentrations ranging from 0.001–100 nM for 24 hours. Following estradiol treatment, cells were then stimulated with LPS for an additional 12 hours and soluble TNF-α levels were measured in the culture supernatants. As demonstrated in Figure 1A, estradiol pre-treatment dose-dependently attenuated LPS-induced TNF-α production. TNF-α protein levels were undetectable in unstimulated cells both in the presence and absence of estradiol (data not shown). Similarly, estradiol treatment decreased TNF-α mRNA levels in response to LPS (Figure 1B). Data are presented as percent of control to permit optimal appreciation of estradiol-mediated effects on TNF-α expression. Although the magnitude of the TNF-α response to LPS stimulation varied among donors, the percent by which estradiol pretreatment attenuated TNF-α production was consistent between individuals. This is in accordance with other reports of inter-individual differences in LPS-induced TNF-α production (15–17).
Figure 1. Estradiol attenuates LPS-induced TNF-α.
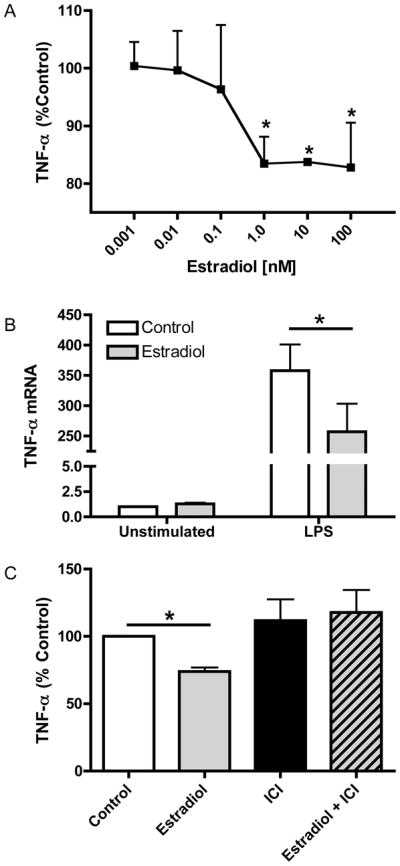
(A) ELISA analysis of TNF-α levels in culture supernatants from macrophages treated with estradiol or ethanol control for 24 hours prior to stimulation with LPS (10 ng/ml) for 12 hours. Data are presented as percent of TNF-α produced by LPS-stimulated cells not receiving estradiol treatment. (B) Total RNA was extracted from macrophages treated with or without estradiol (100 nM) followed by stimulation with LPS for 5 hours. TNF-α mRNA was measured by TaqMan real-time PCR. (C) Macrophages were treated with the estrogen receptor antagonist ICI 182,780 (1 μM) for 1 hour prior to estradiol treatment. Hormone and LPS treatments were the same as in A. TNF-α levels were measured by ELISA and are presented as percent of control treated cells stimulated with LPS. *p<0.05 vs. control cells stimulated with LPS.
To determine whether this effect was mediated by signaling through the estrogen receptor, cells were either pre-treated with the estrogen receptor antagonist ICI 182,780 alone or in combination with estradiol prior to LPS stimulation. In accordance with our earlier observations, pretreatment with estradiol attenuated LPS-induced TNF-α expression. However, TNF-α levels were not inhibited in macrophages that had been pre-treated with ICI 182,780 and estradiol (Figure 1C). Incubation with the ER antagonist alone had no effect on TNF-α expression. These data demonstrate that estradiol attenuates LPS-induced TNF-α secretion by human macrophages in an estrogen receptor-dependent manner.
Estradiol treatment attenuates LPS induced NF-κB signaling
NF-κB plays a central role in the regulation of inflammation and is a major downstream target of nuclear steroid receptors and signal transduction pathways. Stimulus-induced activation of NF-κB contributes to the enhanced production of many pro-inflammatory cytokines, including TNF-α. Macrophages treated with an NF-κB activation inhibitor produced significantly less TNF-α in response to LPS, confirming the importance of this signaling pathway in the TLR4/TNF-α signaling axis (Figure 2A).
Figure 2. Estradiol inhibits NF-κB signaling.

(A) Cells were treated with NF-κB activation inhibitor (10 μM) for 1 hour followed by LPS stimulation for 5 hours. TNF-α levels in culture supernatants were measured by ELISA. N.D., not detected. (B) Whole cell lysates were prepared from macrophages pre-treated with estradiol and then stimulated with LPS for 1 hour. IKKβ, IκBα and phospho-IκBα levels were measured by immunoblot and GAPDH served as a loading control. (C) Cells were treated as in B, and nuclear lysates were prepared and analyzed for p65 by ELISA. Data are normalized to control treated, unstimulated lev28ls. *p<0.01 vs. LPS stimulation alone. **p<0.05 vs. control treated and unstimulated.
Given the importance of NF-κB in the regulation of inflammation and our observation that estradiol pretreatment attenuated LPS-induced TNF-α production, we hypothesized that estradiol modulates activation of NF-κB in human macrophages. To test this hypothesis, we pretreated macrophages with estradiol prior to stimulation with LPS and assessed estradiol effects on phosphorylation of IκBα. As demonstrated in Figure 2B, estradiol pretreatment inhibited LPS-induced IκBα phosphorylation, suggesting that estradiol reduces LPS-induced nuclear translocation of NF-κB. Importantly, estradiol treatment inhibited the degradation of total IκBα in response to LPS, resulting in enhanced expression of total IκBα following LPS stimulation. In the absence of stimulation, total and phospho-IκBα were unaltered by estradiol treatment and expression of IKKβ was similarly unaffected (Figure 2B). Collectively, these data suggest that estradiol treatment attenuates NF-κB signaling through decreased IκBα phosphorylation and degradation.
To determine if reduced IκBα phosphorylation and degradation results in decreased p65 nuclear translocation, macrophages were pre-treated with or without estradiol and stimulated with LPS for 1 hour. Nuclear lysates were prepared and analyzed for p65 expression by ELISA. As expected, nuclear p65 levels were low but detectable in unstimulated cells and were significantly increased with LPS treatment (Figure 2C). Notably, pretreatment with estradiol markedly diminished LPS-induced nuclear p65 levels. These data demonstrate that estradiol attenuates NF-κB signaling through reduced IκBα phosphorylation and degradation, resulting in inhibited LPS-induced nuclear translocation of NF-κB.
Estradiol abrogates LPS-mediated inhibition of κB-Ras2
κB-Ras2 associates with IκBα and inhibits its degradation, resulting in decreased activation of NF-κB signaling (8, 9). Since estradiol inhibited NF-κB signaling at the levels of IkBα phosphorylation and degradation, we decided to test whether κB-Ras2 expression was modulated by estradiol in the context of LPS activation. Expression of κB-Ras2 has been demonstrated in a variety of tissues including the heart, placenta, liver and lung (8). However, it was unknown whether human macrophages express this protein and whether its expression was regulated by LPS. As demonstrated in Figure 3A, κB-Ras2 mRNA expression decreases in response to LPS, reaching a nadir within 3–8 hours of stimulation, and returns to baseline after 12 hours of LPS treatment. Similarly, κB-Ras2 protein expression is maximally inhibited after 12 hours of stimulation and expression returns to unstimulated levels following 24 hours of LPS treatment (Figure 3B). Data shown are representative of 4 experiments. Although the magnitude of inhibition varied between 55% and 15%, in each instance tested LPS attenuated κB-Ras2 expression.
Figure 3. κB-Ras2 is up-regulated by estradiol.

(A) Total RNA was extracted from macrophages treated with LPS at indicated times. κB-Ras2 mRNA was measured by TaqMan real-time PCR. Values are relative to unstimulated controls. (B) Immunoblot analysis of κB-Ras2 protein levels in response to LPS. (C–D) mRNA and protein levels, respectively, of κB-Ras2. Macrophages were treated with estradiol prior to LPS stimulation for 8 hours (C) or 12–24 hours (D). Data shown are representative of 4 experiments. *p< 0.05 vs. unstimulated control
We next asked whether LPS-induced changes in κB-Ras2 expression were modulated by estradiol. Intriguingly, estradiol pretreatment abrogated the LPS-induced inhibition of κB-Ras2 mRNA in human macrophages (Figure 3C). As shown in Figure 3C–D, κB-Ras2 mRNA levels were slightly increased by estradiol in the absence of LPS, but protein levels were unaltered by estradiol in unstimulated cells. To determine how estradiol affects expression of κB-Ras2 protein following activation with LPS, immunoblot analysis was performed. Figure 3D demonstrates that estradiol-treated cells not only abrogated the LPS-induced decrease in κB-Ras2 expression, but also augmented κB-Ras2 expression compared with unstimulated cells after both 12 and 24 hours of LPS stimulation. These data demonstrate for the first time that estradiol modulates LPS-regulated expression of κB-Ras2, a key negative regulator of NF-κB activation.
miR-125b and let-7a are coordinately regulated by LPS and estradiol
Since miRNAs have been identified as important regulators of NF-κB signaling (18–20), we next investigated whether κB-Ras2 is regulated by miRNAs. Using TargetScan analysis (10), we identified several putative miRNA binding sites in the κB-Ras2 3′UTR. Let-7a and miR-125b were among the predicted interacting miRNAs. Let-7a directly targets the 3′UTRs of other RAS protein family members (21) and let-7e, another member of the let-7 family, is modulated by LPS in monocytes (22). miR-125b is also an LPS-regulated miRNA (23–25) and directly targets TNF-α mRNA in mice (25). However, there are no seed binding regions in the 3′UTR of human TNF-α mRNA for miR-125b.
To determine if let-7a and miR-125b regulate κB-Ras2, we first examined their expression patterns in response to estradiol and LPS by TaqMan real-time PCR. As depicted in Figure 4A, let-7a expression is approximately 17 fold higher than miR-125b in resting macrophages. We next performed a time course to determine how expression of let-7a and miR-125b is altered by activation with LPS. Results of this study showed that let-7a was induced and conversely, miR-125b expression was decreased in response to LPS treatment (Figure 4B–C). While let-7a induction did not occur until after 12 hours of LPS stimulation, miR-125b expression decreased more rapidly. These data suggest that the effects of these miRNAs occur sequentially and are not likely contemporaneous. To determine whether the regulation of let-7a and miR-125b is dependent on NF-κB activation, cells were treated with an NF-κB activation inhibitor for 1 hour prior to stimulation with LPS. As demonstrated in Figure 4D, inhibition of NF-κB activation blocks the induction of let-7a by LPS, indicating that this pathway is necessary for let-7a expression. In contrast, miR-125b regulation by LPS occurs independent of NF-κB signaling, since inhibition of NF-κB activation did not reverse the decrease in miR-125b expression (Figure 4E). When macrophages were treated with estradiol prior to stimulation, the LPS-induced increase in let-7a expression was abrogated (Figure 5A). Estradiol treatment also blocked the LPS-induced decrease in miR-125b expression (Figure 5B). These data indicate that estradiol pretreatment reversed the effects of LPS on expression of these miRNAs. Although estradiol had no effect on miR-125b levels in unstimulated cells, estradiol significantly induced let-7a expression (Figure 5A–B).
Figure 4. Let-7a and miR-125b are regulated by LPS.

(A) Total RNA was extracted from macrophages using Qiagen miRNeasy kits. Let-7a and miR-125b expression were measured using Taqman miRNA assay system. Data are normalized to U6 expression levels. (B) let-7a and (C) miR-125b expression levels in response to LPS stimulation. Data were normalized to U6 and are presented as relative to unstimulated levels. (D–E) Macrophages from a representative donor were treated with NF-κB activation inhibitor (10 μM) for 1 hour prior to LPS stimulation for (D) 12 hours or (E) 3 hours. *p<0.05 vs. unstimulated controls.
Figure 5. Estradiol inhibits LPS effect on let-7a and miR-125b.

(A) let-7a levels in macrophages pre-treated with estradiol and stimulated with LPS for 12 hours. (B) miR-125b expression in cells treated with estradiol and stimulated with LPS for 3 hours. *p<0.05 vs. unstimulated controls.
The predicted interactions of let-7a and miR-125b with the κB-Ras2 3′UTR are shown in Figure 6A, and the secondary structure of miRNA/mRNA interactions as predicted by the RNAhybrid program (26) are shown in Figure 6B. To verify that miR-125b and let-7a directly target the κB-Ras2 3′UTR, we used a luciferase reporter system containing the 3′UTR of κB-Ras2. Transfection of let-7a decreased luciferase expression, indicating that let-7a suppresses κB-Ras2 3′UTR stability (Figure 6C). Intriguingly, cells transfected with miR-125b expressed increased levels of luciferase in comparison with those transfected with control miRNA, demonstrating that miR-125b enhances the stability of the κB-Ras2 3′UTR (Figure 6C). In agreement with this finding, macrophages transfected with pre-let-7a had reduced levels of endogenous κB-Ras2, while overexpression of pre-miR-125b led to enhanced κB-Ras2 expression (Figure 6D). These findings indicate a coordinated regulation of let-7a and miR-125b by estradiol and LPS for the net effect of increasing κB-Ras2 expression in the presence of estradiol. We have included a model depicting this mechanism of regulation in Figures 7A and 7B. Significantly, these data demonstrate estradiol regulation of miRNA in the context of inflammation in human macrophages.
Figure 6. let-7a and miR-125b directly target kB-Ras2 3′UTR.

(A) Alignment of let-7a and miR-125b with κB-Ras2 3′UTR. Solid lines indicate Watson-Crick base pairs. Dotted line indicates GU wobble pairs. The gray background denotes the seed binding region. (B) Secondary structure as predicted by RNAHybrid with κB-Ras2 3′UTR shown in black and miRNA in gray. (C) RAW 264.7 cells were transfected with luciferase expression vector containing the κB-Ras2 3′UTR or the control vector and synthetic pre-miR-125b, pre-let-7a or a negative control pre-miRNA. The cells were also transfected with a vector containing Renilla luciferase to serve as a transfection efficiency control. After 48 hours, cells were lysed and analyzed for luciferase expression using a dual-luciferase assay system. (D) PMA-differentiated U937 cells were transfected with pre-miR-125b, pre-let-7a or pre-miRNA negative control and analyzed for κB-Ras2 expression after 48 hours. *p<0.05 vs. negative control miRNA.
Discussion
This study demonstrates for the first time that estradiol attenuates LPS-induced TNF-α production in human macrophages through modulation of NF-κB activation. We show that estradiol reduces NF-κB activation through inhibition of IκBα phosphorylation and subsequent degradation, resulting in decreased p65 nuclear translocation. Although estradiol does not affect basal NF-κB signaling, treatment of macrophages with estradiol prior to LPS activation leads to an increase in the negative regulator of NF-κB signaling, κB-Ras2. Our data indicate that estradiol modulates expression of two microRNAs, let-7a and miR-125b, that regulate the stability of the κB-Ras2 3′UTR. As depicted in Figure 7A, in the absence of estradiol, LPS induces expression of let-7a and inhibits expression of miR-125b. This provides a net instability to the κB-Ras2 3′UTR. Estradiol reverses the effects of LPS on let-7a and miR-125b expression, for a net increase in κB-Ras2 stability (Figure 7B). These data demonstrate a novel mechanism by which estradiol modulates NF-κB signaling and provide the first account of estradiol modulation of miRNA expression in human macrophages.
Numerous human and animal studies have shown that estradiol regulates pro-inflammatory cytokine production (12, 27–29). It is notable that males have a higher mortality rate due to sepsis versus pre-menopausal females, and this can be attributed at least in part to elevated serum TNF-α level (5). These observed differences in TNF-α expression implicate a role for sex hormones in the regulation of TNF-α expression. In this regard, plasma TNF-α values are inversely correlated with estradiol levels in female trauma patients (30). In addition, monocytes from pre-menopausal females who underwent oophorectomy produce elevated levels of TNF-α, concordant with reduction in serum estradiol concentration. Treatment with exogenous estrogens abrogates this effect (31). Previous reports have also shown that estradiol inhibits TNF-α in the murine macrophage cell line RAW 264.7 (7, 32) and in human THP-1 cells (33). Here we demonstrate that estradiol attenuates LPS-induced TNF-α in primary human monocyte-derived macrophages in a dose-dependent manner. These effects were mediated through the estrogen receptor, as indicated by a lack of effect with receptor blockade.
As demonstrated in Figure 1, estradiol modulates LPS-induced TNF-α expression in a dose-dependent manner. We chose to perform the remainder of our studies with 10−7M estradiol because it demonstrated the maximal effect on TNF-α induction in macrophages. This concentration is physiologically relevant because it is consistent with levels present in the human female reproductive tract during ovulation. Estradiol levels in the ovarian vein, which drains directly into the human female reproductive tract, are 14–80 fold higher during ovulation than levels measured in peripheral blood, and more than 100-fold higher than during the early proliferative phase of the menstrual cycle (34). Estrogen has been shown to accumulate in the cycling human uterine endometrium and vagina to at least 37 and 11 times that seen in plasma, respectively (35). Therefore, macrophages at these sites are routinely exposed to this concentration of estradiol. Intriguingly, 10−7M estradiol also has physiologic relevance during pregnancy (36, 37), and thus our studies may have significant immunologic relevance at this time.
LPS binding to TLR4 activates the NF-κB signaling cascade, resulting in transcriptional up-regulation of pro-inflammatory genes, including TNF-α. Figure 2 demonstrates that estradiol treatment prior to LPS challenge results in a reduction in NF-κB signaling through inhibition of IκBα phosphorylation and degradation. These findings are in agreement with the observed decrease in LPS-induced IKKβ activity by estradiol in endothelial cells (38) as well as the estradiol-mediated reduction of IκBα phosphorylation in cerebral ischemia (39). Decreased IκBα degradation impairs translocation of the NF-κB transcription factors to the nucleus. Indeed, our data show that estradiol pretreatment inhibits LPS-induced increases in nuclear p65 levels. These data are in agreement with observations by Ghisletti et al. that estradiol also inhibits p65 nuclear translocation in RAW 264.7 cells (32). However in contrast to our findings, these authors found that estradiol reduced p65 translocation through inhibition of non-genomic pI3K signaling, which was independent of IκBα degradation. The disparity in these results is most likely attributable to timing. The duration of estradiol treatment prior to LPS stimulation differs between their study, in which macrophages were treated with estradiol for 10 minutes prior to stimulation, and our study where cells were treated for 24 hours prior to LPS treatment. The longer estradiol treatment duration used in our study allows for transcriptional up-regulation of genes that may be necessary for the effect of estradiol on IκBα, whereas estradiol pre-treatment for 10 minutes is likely to mediate mainly non-genomic effects. While estradiol has been reported to inhibit NF-κB signaling in various cell types (40), the mechanisms by which this occurs are largely unknown. κB-Ras2 is a Ras-like protein that inhibits NF-κB signaling. Unlike the classic Ras proteins, κB-Ras2 lacks a C-terminal membrane attachment sequence and is constitutively bound to GTP. κB-Ras2 inhibits NF-κB signaling through direct binding to IκBα, decreasing its phosphorylation and consequent degradation (8). Our observation that estradiol inhibited NF-κB signaling at the level of IκBα prompted us to examine how κB-Ras2 expression is modulated by estradiol and LPS. Although relatively little is known about the regulation of κB-Ras2, it was recently identified as a heat-shock responsive gene in THP-1 cells (41) and has been shown to be regulated transcriptionally by FOXO3a in HUVECs (42). Figure 3 demonstrates that κB-Ras2 is expressed in human macrophages and is down regulated by LPS at both the mRNA and protein levels. Given these findings, we propose that κB-Ras2 may serve as a basal inhibitor of NF-κB signaling in the absence of TLR stimulation. In this study, we have demonstrated that estradiol pre-treatment not only abrogates the LPS-induced decrease in κB-Ras2 expression, but also augments expression of κB-Ras2 when compared with unstimulated cells. We believe that estradiol-mediated up-regulation of LPS-modulated κB-Ras2 represents a novel mechanism for inhibition of NF-κB signaling in human macrophages.
Activation of macrophages with LPS leads to changes in the expression of miRNAs that regulate molecules involved in TLR signaling. Taganov et al. describe negative feedback on NF-κB signaling through the suppression of IRAK-1 and TRAF-6, which is mediated by miR-146a (20). In addition, miR-199a targets IKKβ in epithelial ovarian cancer cells (18) and IkB ζ expression is suppressed by miR-124a in Hep2G cells (19). Notably, miR-9 suppresses NFKB1 expression in both monocytes and neutrophils and is induced in response to LPS (22). To determine the role of miRNAs in regulation of kB-Ras2 by estradiol and LPS, we used TargetScan analysis of the 3′UTR of κB-Ras2 mRNA and identified let-7a and miR-125b as potentially interacting miRNAs. Intriguingly, the 3′UTRs of the Ras family members HRAS, KRAS and NRAS are also direct targets of let-7a (21). Figure 4 illustrates the reciprocal regulation of let-7a and miR-125b in response to LPS stimulation in human macrophages. Our observation that miR-125b expression is decreased by LPS in human cells corroborates the recent finding that miR-125b is down-regulated in activated mouse macrophages (24). The changes in let-7a and miR-125b expression occur with different kinetics, suggesting that the effects of these miRNAs on κB-Ras2 are not additive but sequential. Since κB-Ras2 appears to serve as a basal inhibitor of TLR signaling that prevents NF-κB activation in the absence of stimulation, regulation of miR-125b early following LPS stimulation and let-7a later, allows for multiple phases of κB-Ras2 regulation. Our data demonstrate that whereas let-7a induction is dependent on NF-κB signaling, miR-125b regulation in response to LPS occurs independently of NF-κB activation (Figure 4D–E). In addition to the NF-κB pathway, TLR ligation leads to activation of several other signaling pathways including MAPK and pI3K. Therefore, one or more of these mechanisms likely regulates the miR-125b response to LPS. In support of this, Androulidaki et al. have recently shown that the Akt signaling is necessary for the LPS- mediated decrease in miR-125b expression in murine peritoneal and bone marrow derived macrophages (24).
While recent work implicates a role for estradiol in regulation of miRNAs (43), the effect of estradiol on miRNA expression in the context of inflammation is largely unknown. Here we demonstrate that estradiol pre-treatment of human macrophages inhibits both the LPS-mediated induction of let-7a and inhibition of miR-125b expression. Given our observation that κB-Ras2 expression is also decreased by LPS stimulation, we hypothesized that let-7a and miR-125b have opposing effects on the stability of the κB-Ras2 3′UTR. We confirmed this hypothesis using the κB-Ras2 3′UTR luciferase reporter system and showed that let-7a over-expression suppressed the κB-Ras2 3′UTR while miR-125b enhanced its stability. miRNAs modulate post-transcriptional gene expression by inducing changes in rates of mRNA decay and/or translation of target genes. Our results demonstrate that let-7a and miR-125b regulate mRNA expression of κB-Ras2 (Figure 6D). These findings suggest that these miRNAs alter κB-Ras2 expression through modulation of mRNA stability; however, it is also possible that these miRNAs regulate κB-Ras2 expression through changes in rates of translation as well. Although miRNAs are mainly thought to suppress gene expression either though mRNA degradation or translational repression, recent studies have shown that miRNAs can also stabilize their target messages (25, 44, 45).
miR-125b dysregulation is observed in various types of cancer, including acute myeloid leukemia (46) and breast cancer (47). In addition, let-7a is a bona fide tumor suppressor miRNA and is dysregulated in cancers of the lung and breast (21, 48) and in primary effusion lymphoma (49). Intriguingly, both miR-125b-1 and let-7a-1 localize to the 11q24.1 locus (www.atlasgeneticsoncology.org). Given that 11q24.1 is a susceptibility locus for chronic lymphocytic leukemia (50) and that deletions in the 11q23–11q24 region are common in cervical, breast, and ovarian cancer (51–55), it is tempting to speculate that dysregulation of miR-125b and let-7a together contribute to the pathogenesis of disease.
We now report, for the first time, regulation of miRNAs by estradiol in LPS-activated human macrophages. We show that LPS down-regulation of κB-Ras2 expression occurs in association with reciprocal regulation of let-7a and miR-125b. These studies implicate estradiol-mediated regulation of LPS-modulated κB-Ras2 expression as a novel mechanism for NF-κB inhibition and suggest that estradiol regulation of miRNAs serves as an important factor in controlling inflammation.
Acknowledgments
We thank Allan Munck, Mark Yeager and Jane Collins for helpful discussions and critical review of the manuscript.
This work was supported by NIH grant RO1AI051547 (PMG) and the Centers of Biomedical Research Excellence P20 RR 016437 (PMG and PAP). Prouty Pilot Project funds were provided to PAP by the Norris Cotton Cancer Center at Dartmouth-Hitchcock Medical Center. AJM received support from an NIH Autoimmunity and Connective Tissue Training Grant (T32AR007576).
Abbreviations in this manuscript
- Estradiol
17β-estradiol
- IKKβ
IkB kinaseβ
- miRNA
microRNA
References
- 1.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 2.Billiau A, Vandekerckhove F. Cytokines and their interactions with other inflammatory mediators in the pathogenesis of sepsis and septic shock. Eur J Clin Invest. 1991;21:559–573. doi: 10.1111/j.1365-2362.1991.tb01410.x. [DOI] [PubMed] [Google Scholar]
- 3.Zandman-Goddard G, Peeva E, Shoenfeld Y. Gender and autoimmunity. Autoimmunity Reviews. 2007;6:366–372. doi: 10.1016/j.autrev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Vural P, Akgul C, Canbaz M. Effects of hormone replacement therapy on plasma pro-inflammatory and anti-inflammatory cytokines and some bone turnover markers in postmenopausal women. Pharmacological Research. 2006;54:298–302. doi: 10.1016/j.phrs.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Schröder J, Kahlke V, Staubach KH, Zabel P, Stüber F. Gender differences in human sepsis. Archives of surgery (Chicago, Ill: 1960) 1998;133:1200–1205. doi: 10.1001/archsurg.133.11.1200. [DOI] [PubMed] [Google Scholar]
- 6.Deshpande R, Khalili H, Pergolizzi RG, Michael SD, Chang MD. Estradiol down-regulates LPS-induced cytokine production and NFkB activation in murine macrophages. Am J Reprod Immunol. 1997;38:46–54. doi: 10.1111/j.1600-0897.1997.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 7.Srivastava S, Weitzmann MN, Cenci S, Ross FP, Adler S, Pacifici R. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. Journal of Clinical Investigation. 1999;104:503–513. doi: 10.1172/JCI7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fenwick C, Na SY, Voll RE, Zhong H, Im SY, Lee JW, Ghosh S. A subclass of Ras proteins that regulate the degradation of IkappaB. Science. 2000;287:869–873. doi: 10.1126/science.287.5454.869. [DOI] [PubMed] [Google Scholar]
- 9.Huxford T, Ghosh G. Inhibition of Transcription Factor NF-κB Activation by κB-Ras. Methods in Enzymology. 2006;407:527–534. doi: 10.1016/S0076-6879(05)07042-4. [DOI] [PubMed] [Google Scholar]
- 10.Research, W. I. f. B. TargetScanHuman Release 5.1: April 2009.
- 11.Mentzer SJ, Guyre PM, Burakoff SJ, Faller DV. Spontaneous aggregation as a mechanism for human monocyte purification. Cell Immunol. 1986;101:312–319. doi: 10.1016/0008-8749(86)90144-9. [DOI] [PubMed] [Google Scholar]
- 12.Pioli PA, Jensen AL, Weaver LK, Amiel E, Shen Z, Shen L, Wira CR, Guyre PM. Estradiol attenuates lipopolysaccharide-induced CXC chemokine ligand 8 production by human peripheral blood monocytes. J Immunol. 2007;179:6284–6290. doi: 10.4049/jimmunol.179.9.6284. [DOI] [PubMed] [Google Scholar]
- 13.Pioli PA, Weaver LK, Schaefer TM, Wright JA, Wira CR, Guyre PM. Lipopolysaccharide-induced IL-1 beta production by human uterine macrophages up-regulates uterine epithelial cell expression of human beta-defensin 2. J Immunol. 2006;176:6647–6655. doi: 10.4049/jimmunol.176.11.6647. [DOI] [PubMed] [Google Scholar]
- 14.Murphy AJ, Guyre PM, Wira CR, Pioli PA. Estradiol regulates expression of estrogen receptor ERalpha46 in human macrophages. PLoS ONE. 2009;4:e5539. doi: 10.1371/journal.pone.0005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bakiyeva LT, Brooks RA, Rushton N. Inter-individual and intra-individual variability in TNF-alpha production by human peripheral blood cells in vitro. Cytokine. 2005;30:35–40. doi: 10.1016/j.cyto.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 16.van der Linden MW, Huizinga TW, Stoeken DJ, Sturk A, Westendorp RG. Determination of tumour necrosis factor-alpha and interleukin-10 production in a whole blood stimulation system: assessment of laboratory error and individual variation. Journal of immunological methods. 1998;218:63–71. doi: 10.1016/s0022-1759(98)00108-2. [DOI] [PubMed] [Google Scholar]
- 17.Wurfel MM, Park WY, Radella F, Ruzinski J, Sandstrom A, Strout J, Bumgarner RE, Martin TR. Identification of high and low responders to lipopolysaccharide in normal subjects: an unbiased approach to identify modulators of innate immunity. J Immunol. 2005;175:2570–2578. doi: 10.4049/jimmunol.175.4.2570. [DOI] [PubMed] [Google Scholar]
- 18.Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, Leiser A, Schwartz PE, Rutherford T, Mor G. Regulation of IKKβ by miR-199a affects NF-κ B activity in ovarian cancer cells. Oncogene. 2008;27:4712–4723. doi: 10.1038/onc.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindenblatt C, Schulze-Osthoff K, Totzke G. IkappaBzeta expression is regulated by miR-124a. Cell Cycle. 2009;8:2019–2023. doi: 10.4161/cc.8.13.8816. [DOI] [PubMed] [Google Scholar]
- 20.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 22.Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA. 2009;106:5282–5287. doi: 10.1073/pnas.0810909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, Pierre P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci USA. 2009;106:2735–2740. doi: 10.1073/pnas.0811073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN, Tsatsanis C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 2009;31:220–231. doi: 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 26.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. Rna. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh YC, Frink M, Thobe BM, Hsu JT, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. 17Beta-estradiol downregulates Kupffer cell TLR4-dependent p38 MAPK pathway and normalizes inflammatory cytokine production following trauma-hemorrhage. Mol Immunol. 2007;44:2165–2172. doi: 10.1016/j.molimm.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Messingham KA, Heinrich SA, Kovacs EJ. Estrogen restores cellular immunity in injured male mice via suppression of interleukin-6 production. J Leukoc Biol. 2001;70:887–895. [PubMed] [Google Scholar]
- 29.Suzuki T, Yu HP, Hsieh YC, Choudhry MA, Bland KI, Chaudry IH. Estrogen-mediated activation of non-genomic pathway improves macrophages cytokine production following trauma-hemorrhage. J Cell Physiol. 2008;214:662–672. doi: 10.1002/jcp.21255. [DOI] [PubMed] [Google Scholar]
- 30.Gee AC, Sawai RS, Differding J, Muller P, Underwood S, Schreiber MA. The influence of sex hormones on coagulation and inflammation in the trauma patient. Shock. 2008;29:334–341. doi: 10.1097/shk.0b013e3181506ee5. [DOI] [PubMed] [Google Scholar]
- 31.Pacifici R, Brown C, Puscheck E, Friedrich E, Slatopolsky E, Maggio D, McCracken R, Avioli LV. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci USA. 1991;88:5134–5138. doi: 10.1073/pnas.88.12.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghisletti S, Meda C, Maggi A, Vegeto E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol Cell Biol. 2005;25:2957–2968. doi: 10.1128/MCB.25.8.2957-2968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.An J, Ribeiro RC, Webb P, Gustafsson JA, Kushner PJ, Baxter JD, Leitman DC. Estradiol repression of tumor necrosis factor-alpha transcription requires estrogen receptor activation function-2 and is enhanced by coactivators. Proc Natl Acad Sci USA. 1999;96:15161–15166. doi: 10.1073/pnas.96.26.15161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baird DT, Burger PE, Heavon-Jones GD, Scaramuzzi RJ. The site of secretion of androstenedione in non-pregnant women. J Endocrinol. 1974;63:201–212. doi: 10.1677/joe.0.0630201. [DOI] [PubMed] [Google Scholar]
- 35.Thijssen JH, Wiegerninck MA, Donker GH, Poortman J. Uptake and metabolism of oestriol in human target tissues. J Steroid Biochem. 1984;20:955–958. doi: 10.1016/0022-4731(84)90003-7. [DOI] [PubMed] [Google Scholar]
- 36.Adeyemo O, Jeyakumar H. Plasma progesterone, estradiol-17 beta and testosterone in maternal and cord blood, and maternal human chorionic gonadotropin at parturition. African journal of medicine and medical sciences. 1993;22:55–60. [PubMed] [Google Scholar]
- 37.Sarda IR, Gorwill RH. Hormonal studies in pregnancy. I. Total unconjugated estrogens in maternal peripheral vein, cord vein, and cord artery serum at delivery. American journal of obstetrics and gynecology. 1976;124:234–238. [PubMed] [Google Scholar]
- 38.Simoncini T, Maffei S, Basta G, Barsacchi G, Genazzani AR, Liao JK, De Caterina R. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ Res. 2000;87:19–25. doi: 10.1161/01.res.87.1.19. [DOI] [PubMed] [Google Scholar]
- 39.Wen Y, Yang S, Liu R, Perez E, Yi KD, Koulen P, Simpkins JW. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. 2004;1008:147–154. doi: 10.1016/j.brainres.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 40.Kalaitzidis D, Gilmore T. Transcription factor cross-talk: the estrogen receptor and NF-κB. Trends in Endocrinology and Metabolism. 2005;16:46–52. doi: 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Wong HR, Odoms K, Sakthivel B. Divergence of canonical danger signals: The genome-level expression patterns of human mononuclear cells subjected to heat shock or lipopolysaccharide. BMC Immunol. 2008;9:24. doi: 10.1186/1471-2172-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HY, Youn SW, Kim JY, Park KW, Hwang CI, Park WY, Oh BH, Park YB, Walsh K, Seo JS, Kim HS. FOXO3a turns the tumor necrosis factor receptor signaling towards apoptosis through reciprocal regulation of c-Jun N-terminal kinase and NF-kB. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;28:112–120. doi: 10.1161/ATVBAHA.107.153304. [DOI] [PubMed] [Google Scholar]
- 43.Dai R, Phillips RA, Zhang Y, Khan D, Crasta O, Ahmed SA. Suppression of LPS-induced Interferon-gamma and nitric oxide in splenic lymphocytes by select estrogen-regulated microRNAs: a novel mechanism of immune modulation. Blood. 2008;112:4591–4597. doi: 10.1182/blood-2008-04-152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Molecular Cell. 2008;30:460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 46.Bousquet M, Quelen C, Rosati R, Mansat-De Mas V, La Starza R, Bastard C, Lippert E, Talmant P, Lafage-Pochitaloff M, Leroux D, Gervais C, Viguie F, Lai JL, Terre C, Beverlo B, Sambani C, Hagemeijer A, Marynen P, Delsol G, Dastugue N, Mecucci C, Brousset P. Myeloid cell differentiation arrest by miR-125b-1 in myelodysplasic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. Journal of Experimental Medicine. 2008;205:2499–2506. doi: 10.1084/jem.20080285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA gene expression deregulation in human breast cancer. Cancer Research. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 48.Sempere LF, Christensen M, Silahtaroglu A, Bak M, Heath CV, Schwartz G, Wells W, Kauppinen S, Cole CN. Altered microRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Research. 2007;67:11612–11620. doi: 10.1158/0008-5472.CAN-07-5019. [DOI] [PubMed] [Google Scholar]
- 49.O’hara AJ, Wang L, Dezube BJ, Harrington WJ, Damania B, Dittmer DP. Tumor suppressor microRNAs are underrepresented in primary effusion lymphoma and Kaposi sarcoma. Blood. 2009;113:5938–5941. doi: 10.1182/blood-2008-09-179168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Bernardo MC, Crowther-Swanepoel D, Broderick P, Webb E, Sellick G, Wild R, Sullivan K, Vijayakrishnan J, Wang Y, Pittman AM, Sunter NJ, Hall AG, Dyer MJS, Matutes E, Dearden C, Mainou-Fowler T, Jackson GH, Summerfield G, Harris RJ, Pettitt AR, Hillmen P, Allsup DJ, Bailey JR, Pratt G, Pepper C, Fegan C, Allan JM, Catovsky D, Houlston RS. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2008;40:1204–1210. doi: 10.1038/ng.219. [DOI] [PubMed] [Google Scholar]
- 51.Pulido HA, Fakruddin MJ, Chatterjee A, Esplin ED, Beleno N, Martinez G, Posso H, Evans GA, Murty VVVS. Identification of a 6-cM minimal deletion at 11q23.1–23.2 and exclusion of PPP2R1B gene as a deletion target in cervical cancer. Cancer Res. 2000;60:6677–6682. [PubMed] [Google Scholar]
- 52.Skomedal H, Helland, Kristensen GB, Holm R, Børresen-Dale AL. Allelic imbalance at chromosome region 11q23 in cervical carcinomas. European Journal of Cancer. 1999;35:659–663. doi: 10.1016/s0959-8049(98)00413-4. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Z, Gerhard DS, Nguyen L, Li J, Traugott A, Huettner PC, Rader JS. Fine mapping and evaluation of candidate genes for cervical cancer on 11q23. Genes Chromosom Cancer. 2005;43:95–103. doi: 10.1002/gcc.20151. [DOI] [PubMed] [Google Scholar]
- 54.Davis M, Hitchcock A, Foulkes WD, Campbell IG. Refinement of Two Chromosome 11q Regions of Loss of Heterozygosity in Ovarian Cancer. Cancer Res. 1996;56:741–744. [PubMed] [Google Scholar]
- 55.Florence L, Philippe B, Ivan B, Keltouma D, Hugues de T, Kamel H, Marc E, Michel M, Rosette L. Evidence of chromosome regions and gene involvement in inflammatory breast cancer. International Journal of Cancer. 2002;102:618–622. doi: 10.1002/ijc.10729. [DOI] [PubMed] [Google Scholar]