

Abstract
Plasmodium falciparum infection is often associated with a procoagulant state. Recent identification of tissue factor (TF) in the brain endothelium of patients who died from cerebral malaria casts new light in our understanding of the coagulation disorder found in P. falciparum infection. It has also been revealed that parasitized red blood cells (pRBC) support assembly of multimolecular coagulation complexes. In this article, the role of TF expression by the endothelium and amplification of the coagulation cascade by pRBC and/or activated platelets—particularly at sequestration sites— is discussed as crucial events in mounting and sustaining a coagulation–inflammation cycle that contributes to organ dysfunction and coma in P. falciparum malaria.
Blood coagulation and malaria
Malaria caused by P. falciparum remains a highly endemic disease; it has been estimated that at least one million deaths occur every year. Children are at high risk, and often severe malaria (severe anemia or cerebral malaria [CM]) is the cause of death in this population 1–4. P. falciparum infection is associated with sequestration 5–8, endothelial cell (EC) activation 8–10, increase in inflammatory cytokines 3, 11, and activation of the coagulation cascade 12–20. Despite several papers devoted to the coagulation disorder in malaria—many of them published during the 1970s and 1980s (reviewed in Ref. 20)—the mechanism that triggers and propagates the coagulation activation in P. falciparum infection has remained elusive. These studies, however, demonstrated that bleeding and/or hemorrhage are typically not present, although laboratory tests indicate a certain degree of in vivo blood coagulation activation 12–20. Accordingly, prothrombin time (PT) and partial thromboplastin time (PTT) are close to normal values or prolonged, whereas thrombin-antithrombin complex (TAT) (which indicates in vivo thrombin formation) and D-dimers (which are a marker of in vivo fibrinolysis) are often elevated, and thrombocytopenia is a usual finding 12–21. This laboratory profile is compatible with disseminated intravascular coagulation (DIC), which is diagnosed according to a score defined by the International Society of Thrombosis and Haemostasis (ISTH)22. The scoring system takes into account i) platelet count, ii) elevated fibrin related markers, iii) prolonged prothrombin time, and iv) fibrinogen level 22 23. Depending on the score punctuation, levels < 5 are suggestive of non-overt (compensated) DIC while scores > or = 5 are compatible with overt (decompensated) DIC. Bleeding is more common in overt DIC, but it is not needed for the formal diagnosis 22 23. In other words, absence of bleeding in P. falciparum-infected patients does not exclude the presence of DIC in this same population; actually, most of these patients meet the criteria for DIC as defined above 22. Unfortunately, absence of bleeding is often mistakenly regarded as the reason why DIC is supposedly not present in malaria.
Further, some reports do not support the view that coagulation cascade/DIC play a role in malaria pathogenesis because fibrin is not always found in post-mortem studies. There has actually been studies in children* abstract and in adults where the presence of fibrin has been detected in post-mortem samples (reviewed in 20 ). While it is also correct that fibrin it not found in all autopsies, it cannot be excluded that fibrin has been formed sometime during the infection, and degraded by plasmin possibly generated by compensatory fibrinolysis 23. The main argument that supports this notion is the fact that the vast majority, perhaps all patients with mild and severe malaria, present elevated levels of D-dimers20, a specific marker of plasmin-mediated fibrin proteolysis in vivo 23. Finally, it has been reported that hemostatic alterations correlate with parasitemia, thrombocytopenia, and/or severity of P. falciparum infection in a number of studies; in addition, antiparasitic treatment halts the coagulation disorder and improves clinical outcome 12–20.Therefore, it is plausible that hemostatic alterations play a role in the disease progression and organ failure observed in malaria 16, 20.
More recently, it has been demonstrated that P. falciparum-parasitized RBC (pRBC) induce tissue factor (TF) expression in the microvascular EC in vitro, and support the assembly of multimolecular coagulation complexes 10. TF is the clotting initiator 24–26 and a structural member of the cytokine receptor family, which signifies the expansion of the adaptive immune system in vertebrates, indicating a close connection of the coagulation pathways with the host response to infection 27. Accordingly, TF has been increasingly recognized as the interface between coagulation and inflammation 27–32. Remarkably, the brain endothelium of P. falciparum-infected children who died from CM) and other causes display TF expression as determined by immunohistochemistry (IHC) studies using monoclonal anti-TF antibodies 10. These findings provide new insights in our understanding of the events associated with coagulation activation during P. falciparum infection and provide the experimental basis for the tissue factor model for malaria pathogenesis.
The tissue factor model for malaria pathogenesis
Previous models have tried to explain malaria pathogenesis including the ‘sequestration’ and the ‘cytokine’ hypotheses. The sequestration hypothesis suggests that, ‘the presence of infected erythrocytes is an essential event‘ 33. The cytokine hypothesis proposes that, ’while significant cerebral sequestration is often present and sometimes block vessels (with predictable sequelae), it is not essential for the onset of the cerebral manifestation of malaria‘ 34.
An alternative model for malaria pathogenesis, the ‘Tissue Factor Model’ (TFM) has been proposed 20, based on recently published findings, where TF has been identified in the EC exposed to pRBC in vitro and in the endothelium of patients who died from CM, and other causes 10. Late-stage pRBC support assembly of multimolecular coagulation complexes, which are crucial components of the amplification steps of the coagulation cascade 10. The TFM combines concepts evolved from both sequestration and cytokine hypotheses. In contrast to previous models, however, TFM places TF expression by the endothelium and the amplification of the coagulation cascade by pRBC and platelets—particularly at sequestration site—as crucial steps mounting and sustaining a coagulation–inflammation cycle that contributes to organ dysfunction in malaria. It also suggests that malaria is a disease with a strong vascular component, or a disease of the endothelial cells and microcirculation 20.
The TFM differs from the previous hypothesis in five respects:
Role of sequestration in tissue factor expression. According to the TFM, sequestration and/or sequestration-related events is (are) crucial step(s) leading to disease pathogenesis primarily because it is associated with EC activation 8–10, endothelium TF expression, and initiation of the coagulation cascade. Strong support for sequestration in disease pathogenesis comes from IHC and pathology studies performed in African and Asian patients 5–10, from studies that demonstrated a correlation between sequestration and disease severity using an indirect marker of sequestration 6, from clinical trials that used artesunate, which targets young and mature forms of the parasite 35, and from hemoglobinopathies (e.g., hemoglobinopathy C) that protect P. falciparum-infected and (presumably) sequestration-free patients from severe disease 36; however, it is clear that sequestration per se does not account for severe disease, as it is also found in uncomplicated cases 1–3.
Role of pRBC and activated platelets in amplification of the coagulation cascade. The TFM proposes that sequestered pRBC and activated platelets play an active role in amplifying, propagating, and consolidating the coagulation cascade initiated by endothelium TF. It is well known that amplification of coagulation cascade is supported by activated platelets that display phosphatidylserine (PS), a negatively charged phospholipid that assembles the prothrombinase and Xnase complexes 37. In fact, pRBC obtained from P. falciparum-infected patients are procoagulant ex vivo 38. Further, the presence of pRBC sequestered in vessels of the brain suggests that the high concentration of PS favors the kinetics of coagulation reactions in vivo. It has also been reported that fibrin, monocytes, and platelets accumulate in the brain of patients who died from CM*abstract 39 indicating that these vessels display a pro-inflammatory/ -coagulant environment. Actually, platelets may contribute to malaria pathogenesis through exposure of PS and release of pro-inflammatory mediators among other mechanisms 20; besides, one study concluded that trombocytopenia have predictive value for mortality in P. falciparum-infected patients 12.
-
The coagulation-inflammation cycle. The TFM classifies items 1 and 2 above as crucial steps in mounting and sustaining a coagulation-inflammation cycle (Figure I of Box 1) that, when uncontrolled, escalates to severe disease 27–31, 40. In fact, TF is reportedly in the interface of blood coagulation and inflammation 27–32, and TF blockade prevents coagulopathy, reduces inflammation, and attenuates lethality in an experimental model of sepsis 27, 41. The inflammatory effect of coagulation factors occurs through protease-activated receptor (PAR) activation, which mediates expression of adhesion molecules (e.g. ICAM-1; E-selectin) and production of inflammatory cytokines (e.g. TNF-α, IL-1β) found in the plasma of malaria patients 1–3. Of note, cytokines induce TF expression in EC and in monocytes and therefore could potentially perpetuate the coagulation-inflammation cycle 27–32.
The TFM also takes into account events potentially associated with sequestration such as hypoxia 1–3, fibrin formation* 20 , apoptosis 42, and cytokine production 1–3, 11 which may contribute to the procoagulant/inflammatory tonus observed in the disease. Likewise, molecules from pRBC (e.g. hemozoin, glycosylphosphatidylinositol anchor, and oxidized heme) are potentially proinflammatory and may participate in disease pathogenesis 43–45. Microparticle production 21 which reportedly display TF and PS 46, and host responses such as activation of the contact pathway, complement system, and neutrophils—known to produce radical oxygen species (ROS) and to release procoagulant elastase (reviewed in ref 20)—may also play an important pro-inflammatory role in P. falciparum infection 15, 18, 19. Finally, the plasma or local levels of anticoagulants (e.g.: tissue factor pathway inhibitor, thrombomodulin/ protein C, and antithrombin), and antiplatelet substances produced by the endothelium (e.g., prostacyclin) may determine the procoagulant-inflammatory tonus in vessels of patients with malaria. Likewise, bioavailability of vasodilator nitric oxide (NO), which may be impaired in human and in experimental malaria, could potentially aggravate inflammation-related events through enhanced expression of adhesion molecules and the exarcebation of cytoadherence and sequestration 47 48. * Chandrak, P., Carr, RA, Seed, PT, Lucas, SB, Liomba, NG, Whitten, RO, Grau, GE, Mackenzie, CD, Molyneux, ME, Taylor, TE. (1999) Fibrin thrombi in the brain in fatal pediatric malaria correlate with malarial pigment globules (abstract#297). 48th Meeting of the Am. Soc Trop. Med. Hyg. 61.
Acute and ‘compensated’ DIC in malaria. There are compensated and decompensated states of blood coagulation activation in malaria. Mild malaria is usually accompanied by close to normal (or prolonged) PT and PTT, but increased levels of TAT and D-dimers in the absence of bleeding 12–20, which thus characterize ‘compensated’ DIC 23, 27. On the other hand, severe malaria (e.g. CM, 1% of all cases of malaria) is associated with a higher degree of coagulation activation and with bleeding in 10% of cases 13–16. In support of a decompensated state in severe malaria are findings of EC apoptosis 42, identification of circulating microparticles 21, 46, and accumulation of platelets and monocytes in the vessels of the brain 39 from CM cases but not from uncomplicated malaria. It is important to recognize that compensated states occur in baboons infected with escalating doses of Escherichia coli 27, 40, an experimental model for sepsis. Of note, the activation of the coagulation cascade also appears to play an important role in sepsis pathogenesis 3, 20.
-
Orderly steps for disease progression. The TFM also introduces orderly steps to explain the disease didactically, i.e. initiation, amplification, and coagulation-inflammation steps. Figure 1 depicts these events diagrammatically.
Notably, TF expression by endothelium appears not to be an epiphenomenon but a major and differential component of the disease. In fact: (i) high levels of procoagulant cytokines such as TNF-α and monocyte procoagulant activity 14 are found in both P vivax 49 and P falciparum infection 1–3, but a coagulation disorder is consistently detectable only in falciparum 12–16, 19 but not in vivax malaria 14; and (ii) thrombocytopenia is found in vivax and falciparum malaria, but is mediated primarily by a immune mechanism in vivax malaria 50 whereas in falciparum malaria, it appears to be dependent on activation of the coagulation cascade, and splenic clearance 12–20. In other words, identification of TF expression in the endothelium of malaria patients—and the procoagulant role of pRBC—plausibly explains why P. falciparum-infected individuals develop a coagulation disorder that may contribute to organ dysfunction and coma.
Box 1, Figure I. The coagulation-inflammation cycle and the syndrome of multiorgan dysfunction.
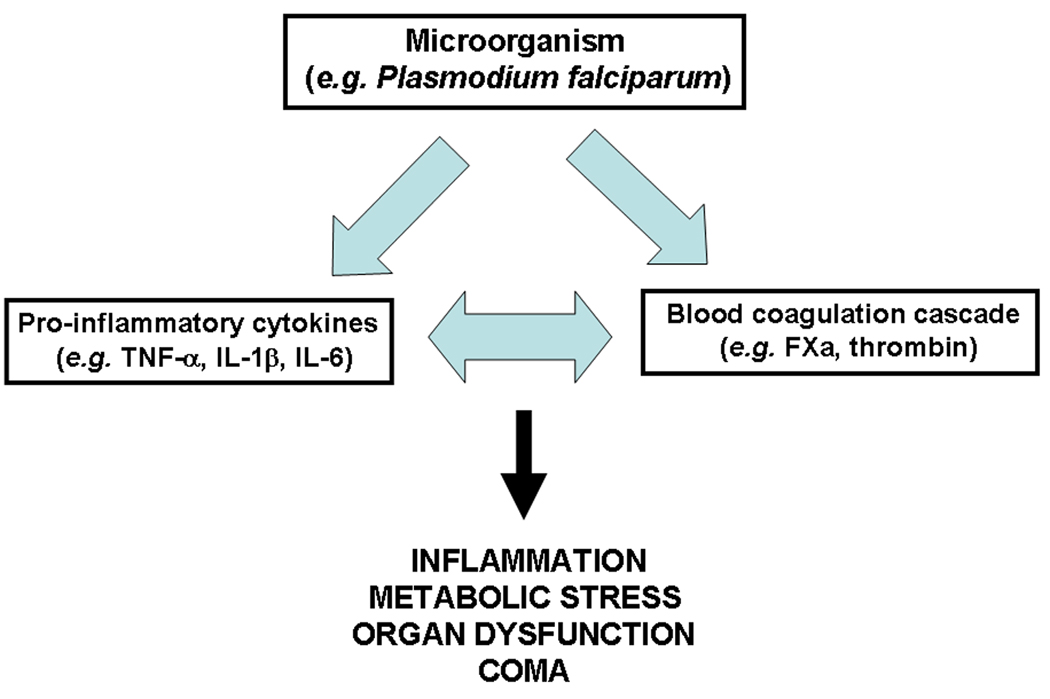
Figure 1. Tissue factor, blood coagulation cascade, inflammation and the Tissue Factor Model for human malaria pathogenesis.
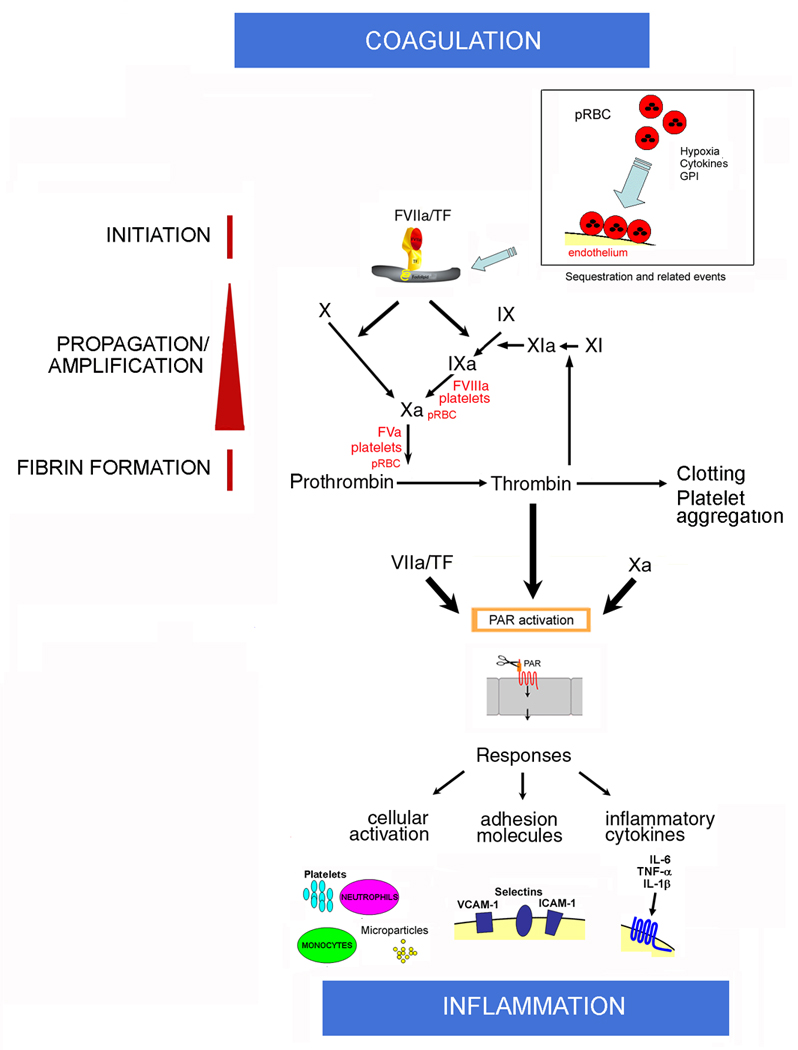
Initiation: Normal, quiescent endothelium does not express functional TF. According to this model, sequestration and sequestration-associated events is associated with EC activation and TF expression in the microvessels of the brain and in other vascular beds. Cytokines, fibrin, hypoxia, apoptosis, proinflammatory molecules released by pRBC (e.g. GPI, hemozoin, oxidized heme) or platelets, and monocytes might also contribute to TF expression (not shown) at sequestration sites and/or paracrinally. Mechanistically, TF initiates the coagulation cascade through binding to coagulation FVIIa and the substrates FIX and FX (extrinsic Xase) in the presence of Ca2+. In this ternary initiation complex, FIXa and FXa are generated.
Amplification: FXa, FVa, and prothrombin assemble in the pRBC surface and/or activated platelets in the presence of Ca2+, with formation of the prothrombinase complex leading to explosive thrombin formation and amplification of the coagulation cascade. Thrombin thus formed promotes fibrin deposition and induces platelet aggregation. Thrombin also activates FXI to FXIa, which activates FIX to FIXa. Factors IXa, FVIIIa, and FX assemble in the membrane of activated platelets and/or pRBC with formation of the intrinsic Xnase complex required for production of additional FXa owing to feedback inhibition of the FVIIa/TF complex by TFPI (not shown). Therefore, the Tissue Factor Model (TFM) for human malaria pathogenesis proposes that both initiation of blood coagulation by TF expression and amplification phase supported by pRBC and/or activated platelets—particularly at sequestration sites—are critical steps for pathogenesis of the disease.
Coagulation–Inflammation: Activated coagulation factors FVIIa/TF, FXa, and thrombin activate PAR in different cell types including mononuclear cells and EC at sequestration sites or paracrinally. PAR activation is accompanied by upregulation of molecules (e.g., ICAM-1, VCAM-1, E-selectin, COX-2, NO synthase) and production of proinflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) that have reportedly been found in CM. Cytokines bind to specific receptors and, together with coagulation factors (and vice versa), perpetuate the inflammatory response, which promotes increased interaction of activated monocytes, elastase- and ROS-producing neutrophils, activated platelets, and/or GPI-containing pRBC with activated EC. The result is a convergence of signals leading to exacerbated TF expression that sustains the coagulation-inflammation cycle in different vascular beds. The cycle is potentially modulated by physiological inhibitors such as NO, prostacyclin, and anticoagulants tissue factor pathway inhibitor, thrombomodulin/protein C pathway, and antithrombin (not shown). As a result of pro- and anti-inflammatory events, a potentially systemic inflammation ensues, leading to metabolic stress, apoptosis, and production of TF-containing microparticles. In extreme cases, organ dysfunction and coma occur.
Concluding remarks
A picture emerges where a local inflammatory reaction initiated by sequestration-associated endothelium TF expression and amplification by pRBC and activated platelets results in production of proinflammatory cytokines and coagulation factors that might act locally and systemically in a synergistic manner, fueling the inflammation–coagulation cycle 27–32. This cycle might be over amplified in the vessels of the brain, a tissue where the anticoagulant thrombomodulin is absent 24. As a result, neutrophil, lymphocyte, and platelet interactions with the capillary endothelium promotes EC injury, increased vascular permeability, vesiculation, and cellular apoptosis 27–32. Accordingly, an apoptosis–coagulation–inflammation imbalance could occur in falciparum malaria, and it is plausible that the vicious cycle of coagulation and inflammation 27–32, 51 operate and contribute to organ dysfunction, particularly in severe P. falciparum infections. It is important to recognize that nitric oxide (NO) availability may be impaired by hemolysis and hypoargininemia in both human 47 and mice 48 infected with Plasmodium sp.; it has also been reported that carbon monoxide (CO) suppresses the pathogenesis of experimental cerebral malaria 45. While pleiotropic mechanisms may explain the action of these gazes 45 47 48, it is documented that NO negatively modulates endothelium activation and platelet function 52 whereas CO inhibits the expression of pro-inflammatory genes (e.g., TF and TNF-α) and prevents inflammatory events in vivo 53. Finally, the finding that TF is potentially a crucial mediator of malaria pathogenesis suggests that therapeutics targeting TF and/or EC 54 could be useful in the treatment of malaria and/or its complications 4, 55.
Box 1. coagulation-inflammation cycle and relevance for multiorgan dysfunction and coma
Severe malaria is associated to a certain degree, with uncontrolled infection, systemic inflammation syndrome and cytokinemia, increased capillary permeability, hypoxia, acidosis, EC activation, microvascular coagulopathy, and multiorgan dysfunction. While much remains to be understood in malaria pathogenesis, the multiorgan dysfunction syndrome often associated with morbidity in malaria appears to be the result, at least in part, of the coagulation-inflammation cycle 1–4, 27–32.
The molecular mechanisms by which activation of coagulation-inflammation cycle is associated with metabolic stress and organ dysfunction have been comprehensively discussed, particularly in the context of sepsis pathogenesis 27–32. Of note, in both conditions activation of blood coagulation takes place3, 10. Usually, an underlying disorder (e.g. P. falciparum infection) could be accompanied by coagulation activation and increase in inflammatory cytokines. Coagulation factors (e.g. FVIIa, FXa, thrombin) are highly proinflammatory molecules through activation of PAR and through induction of cytokine expression. Cytokines, in turn, induce TF expression, leading to the perpetuation of the coagulation responses 27–32. Injection of activated coagulation factors (e.g., FVIIa) in healthy volunteers is accompanied by cytokine production (e.g., IL-6), while administration of cytokines (e.g., TNF-α) is associated with a procoagulant state 27–32. Further, coagulation activation and DIC has been well documented in patients with severe, sometimes untreatable bleeding, and the clinical/pathologic relevance is undisputable; however, major bleeding occurs in a minority of patients with DIC, as it is the case in malaria (reviewed in Ref. 20). More common is the occurrence of organ failure, but there is considerable debate about the extent that (micro)vascular fibrin deposition, as a consequence of DIC, contributes to disease pathogenesis. In other words, does coagulation activation have any functional/pathologic significance in malaria? Several lines of evidence support an important role of coagulation activation/DIC in the development of organ failure. Actually, extensive data have been reported on post-mortem findings of patients with DIC. These autopsy findings include diffuse bleeding at various sites, hemorrhagic necrosis of tissue and, in some cases, fibrin-platelet microthrombi in small blood vessels and sometimes associated with endothelial damage 27–32. Most important, experimental animal studies of DIC and clinical trials in humans show amelioration of hemostatic defects by various interventions targeting the coagulation cascade—it appears to improve organ failure, and, in most cases, to reduce mortality 27, 31, 40, 41. Clinical studies also support the notion of coagulation as an important denominator of clinical outcome, and DIC has been shown to be an independent predictor of mortality in patients with sepsis and severe trauma 27–32. Taking malaria pathology studies into account, intravascular fibrin formation is known to occur in vivo as estimated by increased levels of D-dimers in many patients infected with P. falciparum 12–20. Pathology studies have also detected fibrin (or thrombus) deposition in organs of some patients who died from falciparum malaria 20.abstract
It should be noted that development of EC injury and organ damage in the experimental model for sepsis seems to depend largely on a combination of factors and not in only one component (e.g., fibrin deposition only) associated with the coagulation-inflammation cycle 27–32. For example, reduced fibrinolysis and the effects of inflammatory mediators on the microvascular endothelium seem to be particularly important. Indeed, suppressed fibrinolysis is believed to be one of the most important components of organ dysfunction during DIC. Low levels of α2-antiplasmin-plasmin complex and high levels of plasminogen-activator inhibitor 1 (PAI-1) could therefore function as markers for aggravating disease 27–32. Notably, this coagulation profile has been reported in falciparum malaria in a study involving 76 patients 14. While much remains to be learned about the mechanism of organ dysfunction in sepsis and malaria, there appears to be sufficient evidence to conclude that the coagulation-inflammation cycle and DIC contribute, at least in part, to organ failure under distinct pathologic conditions.
Acknowledgments
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. We thank Drs. José Marcos C. Ribeiro, Thomas E. Wellems, Robert W. Gwadz, and Kathyrin Zoon (NIAID/NIH) for continuous encouragement and support. We are grateful to Karl B. Seydel, Robson Q. Monteiro, Richard O. Whitten, Jerrold M. Ward, and Terrie E. Taylor for helpful discussions and immunohistochemistry assays. We thank NIAID intramural editor Brenda Rae Marshall for assistance.
The author was not able to include many references due to space constraints imposed by the Journal.
References
- 1.Miller LH, et al. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 2.White NJ, Breman JG. Malaria and babesiosis: diseases caused by red blood cell parasites. In: Kasper DL, Brawnwald E, Fauci AS, Hauser SL, Longo DL, Jameson DL, editors. Harrison’s Principles of Internal Medicine. Vol. 1. McGraw Hill: Nwe York, NY; 2005. pp. 1218–1233. [Google Scholar]
- 3.Clark IA, et al. Human malarial disease: a consequence of inflammatory cytokine release. Malar J. 2006;5:85. doi: 10.1186/1475-2875-5-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marsh K, et al. Indicators of life-threatening malaria in African children. N Engl J Med. 1995;332:1399–1404. doi: 10.1056/NEJM199505253322102. [DOI] [PubMed] [Google Scholar]
- 5.Taylor TE, et al. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med. 2004;10:143–145. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- 6.Dondorp AM, et al. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Med. 2005;2:e204. doi: 10.1371/journal.pmed.0020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seydel KB, et al. The distribution and intensity of parasite sequestration in comatose Malawian children. J Infect Dis. 2006;194 doi: 10.1086/505078. 208-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silamut K, et al. A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am J Pathol. 1999;155:395–410. doi: 10.1016/S0002-9440(10)65136-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner GD, et al. Systemic endothelial activation occurs in both mild and severe malaria. Correlating dermal microvascular endothelial cell phenotype and soluble cell adhesion molecules with disease severity. Am J Pathol. 1998;152:1477–1487. [PMC free article] [PubMed] [Google Scholar]
- 10.Francischetti IM, et al. Plasmodium falciparum-infected erythrocytes induce tissue factor expression in endothelial cells and support the assembly of multimolecular coagulation complexes. J Thromb Haemost. 2007;5:155–165. doi: 10.1111/j.1538-7836.2006.02232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown H, et al. Cytokine expression in the brain in human cerebral malaria. J Infect Dis. 1999;180:1742–1746. doi: 10.1086/315078. [DOI] [PubMed] [Google Scholar]
- 12.Gerardin P, et al. Prognostic value of thrombocytopenia in African children with falciparum malaria. Am J Trop Med Hyg. 2002;66:686–691. doi: 10.4269/ajtmh.2002.66.686. [DOI] [PubMed] [Google Scholar]
- 13.Horstmann RD, Dietrich M. Haemostatic alterations in malaria correlate to parasitaemia. Blut. 1985;51:329–335. doi: 10.1007/BF00320043. [DOI] [PubMed] [Google Scholar]
- 14.Mohanty D, et al. Fibrinolysis, inhibitors of blood coagulation, and monocyte derived coagulant activity in acute malaria. Am J Hematol. 1997;54:23–29. doi: 10.1002/(sici)1096-8652(199701)54:1<23::aid-ajh4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 15.Holst FG, et al. Low levels of fibrin-stabilizing factor (factor XIII) in human Plasmodium falciparum malaria: correlation with clinical severity. Am J Trop Med Hyg. 1999;60:99–104. doi: 10.4269/ajtmh.1999.60.99. [DOI] [PubMed] [Google Scholar]
- 16.Hemmer CJ, et al. Activation of the host response in human Plasmodium falciparum malaria: relation of parasitemia to tumor necrosis factor/cachectin, thrombin-antithrombin III, and protein C levels. Am J Med. 1991;91:37–44. doi: 10.1016/0002-9343(91)90071-5. [DOI] [PubMed] [Google Scholar]
- 17.Dennis LH, et al. Depletion of coagulation factors in drug-resistant Plasmodium falciparum malaria. Blood. 1967;29:713–721. [PubMed] [Google Scholar]
- 18.Pukrittayakamee S, et al. Polymorphonuclear leucocyte elastase in Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg. 1992;86:598–601. doi: 10.1016/0035-9203(92)90143-z. [DOI] [PubMed] [Google Scholar]
- 19.Clemens R, et al. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br J Haematol. 1994;87:100–105. doi: 10.1111/j.1365-2141.1994.tb04877.x. [DOI] [PubMed] [Google Scholar]
- 20.Francischetti I, et al. Blood coagulation, inflammation and malaria. Microcirculation. 2007 doi: 10.1080/10739680701451516. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Combes V, et al. Circulating endothelial microparticles in malawian children with severe falciparum malaria complicated with coma. Jama. 2004;291:2542–2544. doi: 10.1001/jama.291.21.2542-b. [DOI] [PubMed] [Google Scholar]
- 22.Toh CH, Hoots WK. The scoring system of the Scientific and Standardisation Committee on Disseminated Intravascular Coagulation of the International Society on Thrombosis and Haemostasis: a 5-year overview. J Thromb Haemost. 2007;5:604–606. doi: 10.1111/j.1538-7836.2007.02313.x. [DOI] [PubMed] [Google Scholar]
- 23.Mammen EF. Disseminated intravascular coagulation (DIC) Clin Lab Sci. 2000;13:239–245. [PubMed] [Google Scholar]
- 24.Broze GJ., Jr Tissue factor pathway inhibitor and the revised theory of coagulation. Annu Rev Med. 1995;46:103–112. doi: 10.1146/annurev.med.46.1.103. [DOI] [PubMed] [Google Scholar]
- 25.Giesen PL, et al. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci U S A. 1999;96:2311–2315. doi: 10.1073/pnas.96.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osterud B, Bjorklid E. Sources of tissue factor. Semin Thromb Hemost. 2006;32:11–23. doi: 10.1055/s-2006-933336. [DOI] [PubMed] [Google Scholar]
- 27.Ruf W. Protease-activated receptor signaling in the regulation of inflammation. Crit Care Med. 2004;32:S287–S292. doi: 10.1097/01.ccm.0000126364.46191.12. [DOI] [PubMed] [Google Scholar]
- 28.Levi M, et al. New treatment strategies for disseminated intravascular coagulation based on current understanding of the pathophysiology. Ann Med. 2004;36:41–49. doi: 10.1080/07853890310017251. [DOI] [PubMed] [Google Scholar]
- 29.Slofstra SH, et al. Disseminated intravascular coagulation. Hematol J. 2003;4:295–302. doi: 10.1038/sj.thj.6200263. [DOI] [PubMed] [Google Scholar]
- 30.Marshall JC. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 2001;29:S99–S106. doi: 10.1097/00003246-200107001-00032. [DOI] [PubMed] [Google Scholar]
- 31.Opal SM, Esmon CT. Bench-to-bedside review: functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Crit Care. 2003;7:23–38. doi: 10.1186/cc1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 33.Berendt AR, et al. Cerebral malaria: the sequestration hypothesis. Parasitol Today. 1994;10:412–414. doi: 10.1016/0169-4758(94)90238-0. [DOI] [PubMed] [Google Scholar]
- 34.Clark IA, Rockett KA. The cytokine theory of human cerebral malaria. Parasitol Today. 1994;10:410–412. doi: 10.1016/0169-4758(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 35.Dondorp A, et al. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet. 2005;366:717–725. doi: 10.1016/S0140-6736(05)67176-0. [DOI] [PubMed] [Google Scholar]
- 36.Fairhurst RM, Wellems TE. Modulation of malaria virulence by determinants of Plasmodium falciparum erythrocyte membrane protein-1 display. Curr Opin Hematol. 2006;13:124–130. doi: 10.1097/01.moh.0000219655.73162.42. [DOI] [PubMed] [Google Scholar]
- 37.Monroe DM, et al. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. 2002;22:1381–1389. doi: 10.1161/01.atv.0000031340.68494.34. [DOI] [PubMed] [Google Scholar]
- 38.Udeinya IJ, Miller LH. Plasmodium falciparum: effect of infected erythrocytes on clotting time of plasma. Am J Trop Med Hyg. 1987;37:246–249. doi: 10.4269/ajtmh.1987.37.246. [DOI] [PubMed] [Google Scholar]
- 39.Grau GE, et al. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J Infect Dis. 2003;187:461–466. doi: 10.1086/367960. [DOI] [PubMed] [Google Scholar]
- 40.Taylor FB., Jr Staging of the pathophysiologic responses of the primate microvasculature to Escherichia coli and endotoxin: examination of the elements of the compensated response and their links to the corresponding uncompensated lethal variants. Crit Care Med. 2001;29:S78–S89. doi: 10.1097/00003246-200107001-00026. [DOI] [PubMed] [Google Scholar]
- 41.Carraway MS, et al. Blockade of tissue factor: treatment for organ injury in established sepsis. Am J Respir Crit Care Med. 2003;167:1200–1209. doi: 10.1164/rccm.200204-287OC. [DOI] [PubMed] [Google Scholar]
- 42.Hemmer CJ, et al. Plasmodium falciparum Malaria: reduction of endothelial cell apoptosis in vitro. Infect Immun. 2005;73:1764–1770. doi: 10.1128/IAI.73.3.1764-1770.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaramillo M, et al. Hemozoin-inducible proinflammatory events in vivo: potential role in malaria infection. J Immunol. 2004;172:3101–3110. doi: 10.4049/jimmunol.172.5.3101. [DOI] [PubMed] [Google Scholar]
- 44.Schofield L, et al. Glycosylphosphatidylinositol toxin of Plasmodium up-regulates intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin expression in vascular endothelial cells and increases leukocyte and parasite cytoadherence via tyrosine kinase-dependent signal transduction. J Immunol. 1996;156:1886–1896. [PubMed] [Google Scholar]
- 45.Pamplona A, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med. 2007;13:703–710. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 46.Furie B, Furie BC. Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol Med. 2004;10:171–178. doi: 10.1016/j.molmed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 47.Yeo TW, et al. Impaired nitric oxide bioavailability and L-arginine reversible endothelial dysfunction in adults with falciparum malaria. J Exp Med. 2007;204:2693–2704. doi: 10.1084/jem.20070819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gramaglia I, et al. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–1422. doi: 10.1038/nm1499. [DOI] [PubMed] [Google Scholar]
- 49.Karunaweera ND, et al. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proc Natl Acad Sci U S A. 1992;89:3200–3203. doi: 10.1073/pnas.89.8.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamaguchi S, et al. Severe thrombocytopenia suggesting immunological mechanisms in two cases of vivax malaria. Am J Hematol. 1997;56:183–186. doi: 10.1002/(sici)1096-8652(199711)56:3<183::aid-ajh9>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 51.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–822. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 52.Villagra J, et al. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110:2166–2172. doi: 10.1182/blood-2006-12-061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mishra S, et al. Carbon monoxide rescues ischemic lungs by interrupting MAPK-driven expression of early growth response 1 gene and its downstream target genes. Proc Natl Acad Sci U S A. 2006;103:5191–5196. doi: 10.1073/pnas.0600241103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kendrick BJ, et al. Drotrecogin alfa (activated) in severe falciparum malaria. Anaesthesia. 2006;61:899–902. doi: 10.1111/j.1365-2044.2006.04752.x. [DOI] [PubMed] [Google Scholar]
- 55.Wellems TE, Miller LH. Two worlds of malaria. N Engl J Med. 2003;349:1496–1498. doi: 10.1056/NEJMp038127. [DOI] [PubMed] [Google Scholar]