

Abstract
Shigella flexneri is a human enteropathogen that infects ca. 165 million people and claims more than 1 million lives per year worldwide. Although shigellosis has been considered a disease of the “Third World,” like many other contagious diseases, it does occur in developed countries. The emergence of drug and multi-drug-resistant strains of Shigella emphasize the need for novel antibiotic development. VirF, an AraC-type transcriptional regulator, is responsible for the expression of all downstream virulence factors that control intracellular invasion and cell-to-cell spread of Shigella. Gene knockout studies have validated that inhibition of VirF expression is sufficient to block the normal life cycle of Shigella in the host and thereby increase susceptibility to the host immune system. The authors have developed a high-throughput, cell-based assay to monitor inhibition of VirF using β-galactosidase as a reporter protein. Using an avirulent strain of Shigella, they have screened libraries containing ~42,000 small molecules. Following confirmation and dose-response analysis, they have identified 25 compounds that demonstrate VirF inhibition in vivo ≥55% in comparison to the controls and little general antibacterial activity (measured by cell growth, OD600). The authors are in the process of confirming these “hits” in several secondary assays to assess the mechanism of action.
Keywords: VirF, Shigella flexneri, AraC family, HTS, transcriptional activators
INTRODUCTION
Shigella Flexneri is a Severe Enteropathogen that plagues more than 1 million victims per year worldwide, leading to severe dysentery in humans.1 The complex mechanism by which Shigella infects the host cells of the gastrointestinal tract has been the subject of much research and was recently reviewed.2 VirF is an AraC-type transcriptional activator that directly regulates transcription of the secondary positive regulator of Shigella virulence, VirB, and the actin-polymerizing enzyme, VirG (IcsA) (Fig. 1).3–8 As the master regulator of positive transcriptional activation, VirF is of particular interest as a novel target in the treatment of shigellosis.
FIG. 1.

Model of Shigella pathogenesis. Many virulence proteins work in concert to promote infection and evade the host immune system response (i.e., engulfment/degradation by macrophages). The primary Shigella spp. virulence factor, VirF, activates transcription of 2 critical virulence genes, virB and virG(icsA). VirB is the secondary transcription factor, which activates transcription of many downstream virulence genes, including the secreted invasion plasmid-associated genes (i.e., ipaBCD). VirG(IcsA) is an actin polymerase and is responsible for promoting cell-to-cell spread in the host.
The use of antibiotics in the food industry and everyday household products has exacerbated the emergence of antibiotic-resistant strains of medically relevant bacteria.9 Accordingly, there has been increased interest in the development of novel antibiotics that target virulence factors.10–12 The pharmaceutical industry has historically focused on development of bactericidal antimicrobial agents (i.e., inhibitors of DNA/RNA, protein, or cell wall synthesis). However, this approach provides a selection pressure for bacteria to become resistant to the antibiotics as resistant bacteria will outgrow the sensitive population. Because expression of virulence factors is not required for cell viability, there should be less selective pressure for the pathogens to develop resistance to inhibitors of such targets.
Transcriptional activators that are regulators of bacterial virulence are often found to be of the AraC family (i.e., Rns from Escherichia coli, ExsA in Pseudomonas aeruginosa, TcpN from Vibrio cholerae, and VirF from S. flexneri). Commonly found in enteropathogens, AraC-type activators have also been identified in virulent bacteria that invade the respiratory and urinary tracts.13 AraC family members that function as homodimers are regulated by either small molecules or physical stimuli.14,15 A third class of AraC transcriptional activators are monomeric and typically respond to cellular stress.16 Despite these differences in protein structure and regulation mechanisms, AraC family members share a high degree of sequence homology in a region of approximately 100 amino acids that constitute the DNA binding domain in the C-terminus.
AraC family members have been difficult to study in vitro, partly due to issues with protein solubility and expression of recombinant proteins.17 Much of the structural evidence to probe the DNA binding properties of the AraC family comes from the monomeric transcriptional activators (including Rob and MarA).18 Researchers at Paratek Pharmaceuticals have studied several members of the AraC family related to bacterial virulence in vitro.19,20 Using a coupled approach with in silico screening of small molecules and crystal structures of 3 AraC members from E. coli (MarA, SoxS, and Rob) and an in vitro DNA binding assay, researchers identified a promising class of inhibitors of DNA binding: hydroxybenzimidazole derivatives.20 Although the compounds demonstrated inhibition of DNA binding in vitro with IC50 values in the low micromolar range, their study only addressed one aspect of AraC-type regulator function. Recently, the same group reported activity of a similar compound set (N-hydroxbenzimidazoles) against LcrF in Yersinia spp.19 LcrF is a Mar-like protein that activates transcription of the type III secretion system. Following synthesis of various analogs, compounds were validated in a cell-based cytotoxicity assay as well as DNA binding analysis with an LcrF homologue (ExsA from Salmonella) and an unrelated transcriptional activator (SlyA from Salmonella) to establish specificity. In the case of VirF and other homodimeric AraC-type activators, there are at least 3 steps in the gene activation process that could be targeted by potential inhibitors (although the order is not clearly understood): DNA binding, dimerization, and recruitment of the transcription complex. A DNA binding screen would not detect inhibition of either dimerization or transcription complex recruitment, nor is this assay amenable to high-throughput screening (HTS).
Manipulation of recombinant, dimeric AraC-type proteins has been problematic in vitro (e.g., poor expression, insolubility). A cell-based assay would also have the advantage of screening for cell permeability, an important factor when screening large compound libraries. We therefore elected to study this class of transcriptional activators in a cell-based, high-throughput assay. Using a β-galactosidase reporter gene in avirulent S. flexneri BS103, VirF-specific activation of the virB promoter (controlling transcription of the lacZ reporter gene) was monitored spectrophotometrically with the substrate CPRG (chlorophenol red β-D-galactopyranoside). Small molecules that demonstrated confirmed inhibition of the reporter were analyzed further in a dose-response analysis.
MATERIALS AND METHODS
Reagents
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). Gelase™ Agarose-Gel Digesting Preparation was from Epicentre (Madison, WI). The QIAprep Spin Miniprep Kit was from Qiagen (Valencia, CA). Carbenicillin (disodium salt), Corning microplates, bactotryptone, and yeast extract were from Fisher Scientific (Hampton, NH). Induction Control E BL21(DE3) was from Novagen (Madison, WI). CPRG (monosodium salt) was from Roche (Basel, Switzerland). All restriction enzymes and Vent® DNA polymerase were from New England Biolabs (Ipswich, MA). SeaPlaque agarose was from Cambrex (East Rutherford, NJ). T4 DNA Ligase and all synthetic oligonucleotides were from Invitrogen (Carlsbad, CA). The deoxynucleotide triphosphates (dNTPs, monosodium salts) were from Promega (Madison, WI). S. flexneri BS103, cured of the virulence plasmid, was a generous gift of Professor Anthony Maurelli (Uniformed Services University of the Health Sciences, Bethesda, MD).
Reporter plasmid construction
The virf gene was subcloned into the maltose binding protein fusion vector, pMAL-c2x, as previously described (named pMALvirF).5 The virB promoter (virB Op) (from pTB6015) and β-galactosidase α-complement (lacZα, from pTZ18U) were amplified by PCR. In a total volume of 50 µL, samples containing 500 ng plasmid, oligonucleotide primers (20 pmol each), dNTP mix (0.24 mM each NTP), and Vent® DNA polymerase (2U) were incubated according to the following temperature sequence: 30 cycles—94°C for 30 s, 50°C for 1 min, and 72°C for 2 min. Both PCR products (20 µL DNA, 40 µL total volume) were treated with AatII (40 U) at 37°C for 1 h. The digested samples were gel purified from Seaplaque agarose gel with Gelase™ according to the vendor’s protocol (Epicentre). The virB Op and lacZα fragments were ligated overnight at 16°C (2:1 volume ratio, 20 µL total volume) with T4 DNA Ligase (2 U). The virB Op-lacZα fragment was subcloned into pMALvirF between the PstI and XbaI restriction sites. Briefly, in a total volume of 50 µL, samples containing 500 ng virB Op-lacZα, 2 mM MgSO4, dNTP mix (2.5 mM each NTP), oligonucleotide primers (20 pmol each), and Vent® DNA polymerase (4 U) were incubated as described above. The amplified virB Op-lacZα sequence and pMALvirF (15 µL DNA) were treated in double-restriction enzyme digestions with PstI and XbaI (40 U each enzyme, 30 µL reaction) at 37°C for 1 h. Both the plasmid and PCR product were gel purified as described above. The purified samples were ligated with T4 DNA ligase (7:1 volume ratio, insert to plasmid) according to the vendor’s protocol (Invitrogen) and transformed into 250 µL of chemically competent TG2 cells. The resultant plasmid, pMALvirF-LacZα, was isolated via miniprep (Qiagen) from 3 mL 2xTY cultures (16 g bactotryptone, 10 g yeast extract, 5 g NaCl/L of water with 100 µg/mL carbenicillin), and samples were confirmed with DNA sequencing (DNA Sequencing Core Facility, University of Michigan).
Full-length lacZ (3 kb) was amplified by PCR from the Novagen Induction Control E (pET28a derivative). An internal AatII restriction site was altered with a silent mutation introduced by site-directed mutagenesis (according to Stratagene Quik-change protocol). The corresponding lacZ open reading frame was amplified by PCR as follows (30 µL volume): 5 µg pET28a–LacZ(ΔAatII), PCR primers (10 pmol each), dNTP mix (0.25 mM each NTP), 2 mM MgSO4, and Vent® DNA polymerase (2 U) were incubated according to the following temperature sequence: 30 cycles—94°C for 1 min, 55°C for 1 min, and 72°C for 2 min. The samples were combined and ethanol precipitated at –20°C overnight. The DNA pellet was resuspended in water (30 µL). The lacZ gene was subcloned into pMALvirF-LacZα in place of the lacZ α-peptide via a double-restriction enzyme digestion. Briefly, samples containing 5 µL DNA (10 µL total volume) were treated with AatII and XbaI (20 U each) at 37°C for 1.5 h. Both the plasmid and PCR product were gel purified, ligated (5:1 volume ratio, insert to plasmid), and transformed into TG2 cells as previously described. The resultant plasmid, pMALvirF-LacZ, was isolated via miniprep from 3 mL 2xTY cultures with carbenicillin, and samples were confirmed with DNA sequencing.
A positive control vector was created for pMALvirF-LacZ as a measure of β-galactosidase activity in the absence of VirF. Each reporter plasmid (10 µL, 20 µL total volume) was treated with the restriction endonuclease BglII (20 U) at 37°C for 90 min. The digested plasmids were gel purified as previously described to remove the resultant 1200-nucleotide fragment of the malE-virF open reading frame. The purified samples were religated at 17°C overnight with T4 DNA ligase (2 U). The ligation products (5 µL) were transformed into 250 µL chemically competent TG2 cells and grown at 37°C on L-carbenicillin plates. Representative plasmid diagrams with key restriction enzyme sites for pMALvirF-LacZ and pMAL(ΔvirF)-LacZ are shown in Figure 2.
FIG. 2.

Construction of the VirF-LacZ reporter plasmid. The OCR products virB Op and LacZ were amplified from pTB601 and Novagen Induction Control E, respectively.
Small-molecule libraries
In the primary screen, approximately 42,000 compounds were tested at the Center for Chemical Genomics (CCG, University of Michigan) with the VirF-driven β-galactosidase reporter assay. Among the libraries tested were the Chemical Diversity set, the Maybridge HitFinder library, MicroSource Spectrum 2000, and the NCC BioFocus library.
High-throughput β-galactosidase reporter assay
Samples containing S. flexneri BS103 harboring pMAL-virF-LacZ (or the positive control for no β-galactosidase activity, pMAL(ΔvirF)-LacZ) were grown to saturation in 2xTY at 37°C with shaking. Small molecules (10 µM, n = 1, 0.2 µL) from the CCG compound library were added to 20 µL 2xTY by Biomek HDR (Beckman, Fullerton, CA). Cells were diluted with 2xTY supplemented with 100 µg/mL carbenicillin to a final OD600 = 0.004, and 10 µL was added to the 384-well microplates with the Multidrop dispenser (Thermo Scientific, Waltham, MA; 30 µL total volume). Plates were incubated for 24 h at 30°C. Cell density (OD600) was measured prior to addition of an equal volume (30 µL) of 0.5 mg/mL CPRG and 0.1% Triton X-100 in Miller’s Z-buffer (60 mM Na2HPO4, pH 7.0; 40 mM NaH2PO4; 10 mM KCl; 1 mM MgSO4).21 The samples were incubated at room temperature for 10 min before measuring chlorophenol red (CPR) absorbance (A570) in the PHERAstar (BMG Labtech, Cary, NC) plate reader with a narrow bandpass filter.
Data analysis
Potential hits were considered active by the following criteria: ≥55% inhibition by plate of the VirF-LacZ assay in comparison to controls (A570 < 1.4), with 100% inhibition defined as the mean A570 reading for the positive control (pMAL(ΔvirF)-LacZ reporter) replicates for each plate (A570 ≅ 0.7–1.0) and inhibition of cell growth (OD600) ≤20%. Compounds meeting these initial criteria (1273 small molecules) were confirmed via rescreening in triplicate according to the above assay protocol. Further data triage criteria for removal of unfavorable properties included “flagged” compounds with reactive or toxic functionalities, molecular weight ≥600 Da, topological polar surface area (TPSA) > 140 Å2, and logP > 7.
Counterscreen with purified β-galactosidase
Triaged compounds with confirmed activity were preincubated with purified β-galactosidase in Z-buffer (25 nM, 30 µL total volume) for 1 h at 30°C in a preliminary in vitro counter-screen of the enzyme (n = 3). The CPRG/Triton X-100 mixture was added to each well, and the absorbance measurements were made following a 2.5-min incubation at room temperature. Compounds that exhibited inhibition of β-galactosidase activity ≥35% were removed from further consideration.
Dose-response analysis
Compounds that passed the triage, confirmation, and counterscreening (238 small molecules) were assayed in a dose-response study (n = 4) according to the screening protocol with the following exception: the concentration of small molecules was varied in 2-fold serial dilutions, ranging from 100 to 1.5 µM. The data were plotted as a function of the concentration of small molecule (µM) versus absorbance (570 nm). For those compounds that exhibited a sigmoidal dose-response curve, the data were fit by nonlinear regression to the following equation using Kaleidagraph (Synergy Software, Essex, VT):
where “lower” is the lower limit of the assay, and “upper” is the upper limit of the assay, [I] is the inhibitor concentration, M1 is the IC50, and M2 is the Hill slope. During the fitting process, the upper limit was manually entered (from the no-inhibitor control) owing to the fact that the data generally did not go to low enough concentrations to sufficiently define the upper portion of these curves. The lower limit was also manually entered as defined by the average absorbance of the positive control for each individual plate.
RESULTS
Construction of β-galactosidase reporter plasmids
The virB Op-lacZα fragment was subcloned directly into pMALvirF, followed by replacement of the β-galactosidase α-peptide (lacZα) with the full-length lacZ gene (pMALvirF + virB – lacZ). This single, high-copy number plasmid was used to express β-galactosidase activity following VirF-specific activation of the virB Op-lacZ open reading frame. A counter-screen plasmid was generated via deletion of a portion of the malE-virF open reading frame, eliminating expression of VirF from these samples (pMAL(ΔvirF) + virB – lacZ). Both VirF-LacZ reporter constructs were assayed in S. flexneri BS103.22
Optimization of high-throughput VirF-LacZ primary assay
The microplate-based assay was optimized at the CCG (University of Michigan). The most reproducible cell-based β-galactosidase assay (Z′ = 0.59, n = 352) was observed when cells (OD600 = 0.004) were incubated at 30°C for approximately 24 hours and assayed with 0.50 mg/mL CPRG + 0.1% Triton X-100 in a 60-µL total volume (data not shown). The primary HTS screen of approximately 42,000 small molecules in the VirF-LacZ cell-based assay was run with several small-molecule libraries, as described in Materials and Methods. Assays were performed with both the negative (pMALvirF + virB – lacZ) and positive (pMAL(ΔvirF) + virB – lacZ) controls for VirF inhibition monitored in the absence of small molecules in replicate (n = 32) on each plate in the assay campaign. To assay β-galactosidase activity, the CPRG/Triton X-100 mixture was added to each well, and the reaction proceeded at room temperature for 10 min prior to measuring CPR accumulation (A570). Representative absorbance values for the negative control (A570 = 2.0–2.5) and the positive control (A570 = 0.7–1.0) were recorded for each individual plate. In the primary assay, each small molecule was tested in a single well in a 384-well microplate, and 1273 compounds were selected as potential hits based on the selection criteria outlined in the Materials and Methods (3% hit rate, Fig. 3A).
FIG. 3.

VirF-β-galactosidase reporter assay confirmation screen and dose-response analysis. (A) Compounds identified as potential hits in the primary screen (1273 small molecules, 10 µM) were reassayed to confirm activity in triplicate. The plot depicts absorbance (A570) versus compound number tested in the assay campaign. The assay was carried out as described in the text. (B) Two representative compounds fit by nonlinear regression for sigmoidal kinetics following dose-response analysis. Each compound was confirmed in replicate (n = 4).
The 1273 initial hits were reassayed for confirmation of inhibition in triplicate. In addition, these compounds were counterscreened in triplicate against purified β-galactosidase, in vitro, to filter out any false positives that directly inhibit the reporter. Four compounds were found to inhibit purified β-galactosidase in vitro (Table 1), 3 with modest inhibition (~40%) and 1 that demonstrated significant inhibition of the purified enzyme (92%, CCG-28697). Three compounds (CCG-28697, 28450, and 28967) share a common tricyclic structure with a bridging piperdine-sulfonyl moiety, derivatized with various aromatic functionalities. The structures are distinct from galactose, and it is unclear how these compounds inhibit β-galactosidase activity.
Table 1.
Confirmed Inhibitors of Purified β-Galactosidase In Vitro
|
Compound Name |
Library |
Percent Inhibition (Average by Plate), % |
Structure |
|---|---|---|---|
| CCG-28697 | ChemDiv | 92 | 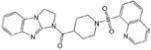 |
| CCG-28450 | ChemDiv | 40 | |
| CCG-39161 | MicroSource | 40 | 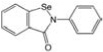 |
| CCG-28967 | ChemDiv | 35 | 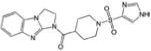 |
Further selection criteria were imposed for identification of optimized hit-to-lead scaffolds. Ultimately, a test set of 238 compounds was chosen for dose-response studies. Each compound was tested in replicate (n = 4) in serial dilutions ranging in concentration from 100 to 1.5 µM according to the screening assay protocol. The data were plotted (concentration vs absorbance). Those compounds that exhibited a sigmoidal-type dose-response curve were fit by nonlinear regression, and the IC50 values were calculated. From this assay, 7 compounds were confirmed by dose-response analysis, with typical IC50 values ranging between 1 and 90 µM (Table 2). Representative data for the dose-response analyses are shown in Figure 3B.
Table 2.
Dose-Response Analysis for VirF-β-galactosidase Reporter Assay
|
Compound Name |
Library | IC50, µM | Structure |
|---|---|---|---|
| CCG-38543 | MicroSource | 1.0 | 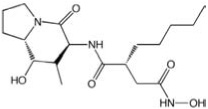 |
| CCG-25354 | ChemDiv | 2.7 | 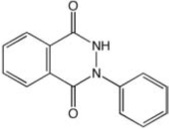 |
| CCG-24904 | ChemDiv | 4.5 | 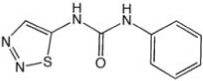 |
| CCG-50009 | Maybridge | 10 | 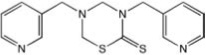 |
| CCG-55215 | Maybridge | 12 | 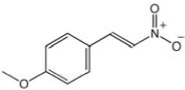 |
| CCG-42010 | Maybridge | 43 | 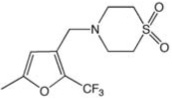 |
| CCG-21496 | ChemDiv | 89 | 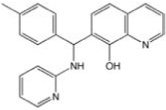 |
Small molecules identified with micromolar IC50 values. Note that these initial IC50 values, using samples from the screening collection libraries, are preliminary and will be reassessed using fresh, neat samples purchased from the sample sources.
DISCUSSION
Many pathogens use transcriptional activators of the AraC family as regulators of bacterial virulence (i.e., Rns from E. coli, ExsA in P. aeruginosa, TcpN from V. cholerae, and VirF from S. flexneri). Previous work on monomeric AraC-type virulence regulators that used an inhibition of a DNA binding assay was not amenable to HTS and would not probe for inhibition of either dimerization or recruitment of the transcription complex for the dimeric AraC-type activators. Although the dimeric AraC-type activators have been difficult to study in recombinant form, we find that transcriptional activation can be sensitively monitored in a cell-based, high-throughput assay using a β-galactosidase reporter gene fused to the appropriate target promoter sequence.
In considering a robust, cell-based assay to screen for inhibitors against VirF activity, β-galactosidase was initially chosen as a reporter for its history as a molecular biology tool and its enzymatic stability.21 With a wide variety of substrates described in the literature, there are several potential options to suit a range of β-galactosidase assay protocols. The most common substrate for measuring LacZ expression on solid media is X-gal (5-bromo-4-chloro-3-indolyl β-D-galactopyranoside), but detection of the insoluble blue product is less amenable to liquid culture. The classical substrate for β-galactosidase enzyme kinetics is o-nitrophenyl β-D-galactopyranoside (ONPG).21 The ONP product of hydrolysis is yellow in color and detected by absorbance (A420). This spectrophotometrically active substrate is best suited to in vitro reactions because the signal-to-noise ratio is high in colorless buffer. For use in cell-based assays, however, detection of ONP is masked by the inherent absorbance in the liquid media. The fluorescent substrate 4-methylumbelliferyl β-D-galactopyranoside (MUG) was recently described for use in a whole-cell assay for β-galactosidase activity.23 Without significant dilution of the starting cell culture, however, there is interference from background fluorescence of components in the media.
CPRG has been described for cell-based applications, and this substrate is particularly sensitive to low concentrations of β-galactosidase.24 In addition, the yellow substrate turns a deep red upon hydrolysis by β-galactosidase, and accumulation of the chlorophenol red product is easily detected by absorbance. One potential concern with the use of CPRG was achieving the necessary signal-to-background ratio in the absorbance measurements. The whole-cell assay required long growth periods (24 h) to produce enough VirF protein that could in turn activate expression of a reasonable amount of β-galactosidase. An increased cell density (A600) could complicate the measurement of chlorophenol red accumulation at A570. This sensitivity was achieved by monitoring absorbance in the PHERAstar spectrophotometer with a narrow bandpass filter at A570.
Previous studies have shown that in E. coli, Rns will recognize VirF promoters; therefore, one would expect interfering background expression if the screening were performed in E. coli.25 In fact, we did observe this (data not shown). There is no other known transcriptional activator in Shigella that can activate VirF promoters, and therefore the VirF reporter assay was optimized in an avirulent strain of S. flexneri, BS103.22 BS103 is a derivative of S. flexneri 2a that has been cured of the 220-kb virulence plasmid and hence lacks endogenous virulence gene expression (including virF, virB, and virR (hns)). A deletion in the malE-virF open reading frame was constructed as a control for background LacZ expression not related to VirF-specific activation. This construct was in every other aspect identical to the reporter discussed previously. We confirmed that LacZ expression in BS103 is the direct result of VirF activation of the virB promoter, with the ΔvirF reporter responding similarly to the Δlac BS103 cell control (data not shown).
Conditions for optimal cell growth and β-galactosidase activity were identified in a 384-well microplate format in collaboration with the Center for Chemical Genomics (University of Michigan). Although enzymatic activity was relatively consistent across the plate, we observed a small amount of evaporation around the perimeter of each microplate. These “edge effects” were also observed in other cell-based assays performed at the CCG. In these wells, although the OD600 reading was not altered significantly, the total volume was reduced by 5 to 10 µL. For completeness, these wells were considered in the overall calculation of Z′ for each plate in the assay campaign. Although there were discrepancies in some cases between wells and between individual plates, the internal controls for each plate were used to normalize the data. In general, more consistent data can often be obtained through use of in vitro assays with purified protein. However, 2 major advantages of a cell-based assay in the primary screen are the selection of compounds that are cell permeable and the relative ease of preparation of an overnight cell culture versus target enzyme purification (which in this case would also include the reporter construct and an in vitro transcription/translation cocktail). Overall, the variability in the high-throughput data in the cell-based assay was reflected in a lower Z′ factor ≅ 0.45 for the overall assay campaign.
We observed a 3% hit rate (1273 small molecules of 42,000 tested) for the compounds tested in the primary screen, with molecules demonstrating modest to significant inhibition of the VirF-LacZ assay. These compounds were confirmed in triplicate according to the primary assay protocol. As an initial counterscreen, purified β-galactosidase was assayed with the same small molecules in vitro. Most compounds tested in the confirmation screen did not inhibit the reporter directly (Table 1). Although this counterscreen does not address the possibility of “off-target” inhibition, most off-target activities that would result in reduced β-galactosidase expression are likely to be toxic (e.g., RNA polymerase inhibition, ribosome inhibition) and would be factored out when the inhibition of the cell growth screen was applied. A potential counterscreen to address off-target activity could involve a parallel cell-based system where LacZ expression was activated by an endogenous Shigella transcription complex from a housekeeping promoter (i.e., trp promoter). The construction of a pMAL(ΔvirF) vector with subcloned trp Op-lacZ fusion is in progress.
Following data triage to identify molecules with favorable medicinal chemistry properties (i.e., molecular weight ≤600, no reactive or toxic functionalities, logP < 7, etc.), a set of 238 compounds was selected for initial dose-response analysis. Although most compounds did not demonstrate dose-dependent inhibition of the VirF-LacZ assay, 7 small molecules were identified with IC50 values in the low to mid micromolar range (Table 2). The data were fit by nonlinear regression for sigmoidal kinetics, and the reported pIC50 values were converted to IC50 values. Although structurally distinct, most compounds were characterized by aromatic or heterocyclic rings that could conceivably interact directly with DNA or with aromatic amino acids that typically associate with nucleic acids (i.e., histidine, phenylalanine).26 Note that these initial IC50 values, using samples from the screening collection libraries, are preliminary and will be reassessed using fresh, neat samples purchased from the sample sources. Secondary in vitro assays (i.e., in vitro DNA binding assay and in vitro transcription assay) will also be performed from these fresh samples to confirm the mechanism of action for each small molecule.
Interestingly, several compounds identified were also characterized as “active” in other bacterial cell-based assays targeting transcription factors at the CCG. The 7 confirmed compounds identified in the VirF-β-galactosidase assay have each been tested in ≥35 individual assays, including screens against prokaryotic and eukaryotic targets in vitro and in vivo. In general, the compounds have demonstrated little activity against eukaryotic targets. The similarity in effects against assays with structurally distinct proteins performing similar functions (i.e., DNA binding, transcriptional activation) suggests that these small molecules may target a conserved function of bacterial transcription factors. Further studies are pending to confirm the specificity and mechanism of action for these small molecules.
Monitoring VirF activity in a cell-based assay addresses all 3 functions of dimeric AraC-type transcriptional activators: DNA binding, dimerization, and recruitment of RNA polymerase. Although researchers at Paratek Pharmaceuticals have successfully studied inhibition of DNA binding of several monomeric AraC-type regulators in vitro, this assay is limited to selection of compounds that address only one aspect of AraC-type functionality. In addition, generating the purified reagents for the DNA binding assay is more expensive and less amenable to HTS of large compound libraries. This approach is more suited for use in screening potential inhibitors as a secondary assay, to assess mechanism of action on a smaller subset of molecules. We report a cell-based assay for S. flexneri VirF that circumvents both of these issues and presents a new approach to monitoring transcriptional activation by oligomeric AraC-type activators for high-throughput applications.
CONCLUSIONS
In collaboration with the CCG (University of Michigan) we have developed a cell-based bacterial transcription assay amenable for HTS of large compound libraries against VirF from S. flexneri. Initial HTS results screening ~42,000 small molecules in the VirF-LacZ reporter assay identified 238 potential hits with favorable medicinal chemistry properties. In addition, very few compounds in these small-molecule libraries directly inhibited β-galactosidase, suggesting the usefulness of this reporter gene in cell-based bacterial transcription assays. Dose-response data for 7 compounds with confirmed inhibition in the VirF-LacZ assay revealed IC50 values in the micromolar range (1–90 µM). Future work will be conducted with several secondary assays to probe the mechanism of inhibition for these “hits.” This report demonstrates proof of principle that monitoring activity of the AraC family of transcriptional activators in vivo is a successful alternative to overcoming insoluble expression of recombinant protein in vitro.
ACKNOWLEDGMENTS
We gratefully acknowledge Dr. Paul Kirchhoff with the University of Michigan Vahlteich Medicinal Chemistry Core for his assistance with the VirF high-throughput assay data triage. We also thank Prof. Anthony Maurelli (Uniformed Services University of the Health Sciences, Bethesda, MD) for the avirulent Shigella strain, BS103. This work was supported by the University of Michigan, College of Pharmacy Vahlteich and Upjohn Research funds (GAG) and NIH-NIGMS GM007767 (JKH, trainee).
REFERENCES
- 1.World Health Organization. Geneva, Switzerland: Initiative for Vaccine Research, World Health Organization; State of the Art of New Vaccines: Research & Development. 2003
- 2.Dorman CJ. The virulence plasmids of Shigella flexneri. In: Schwartz E, editor. Microbial Megaplasmids. Berlin: Springer; 2009. pp. 151–170. [Google Scholar]
- 3.Adler B, Sasakawa C, Tobe T, Makino S, Komarsu K, Yoshikawa M. A dual transcriptional activation system for the 230 kb plasmid genes coding for virulence-associated antigens of Shigella flexneri. Mol Microbiol. 1989;3:627–635. doi: 10.1111/j.1365-2958.1989.tb00210.x. [DOI] [PubMed] [Google Scholar]
- 4.Jost BH, Adler B. Site of transcriptional activation of virB on the large plasmid of Shigella flexneri 2a by VirF, a member of the AraC family of transcriptional activators. Microbiol Pathogenesis. 1993;14:481–488. doi: 10.1006/mpat.1993.1047. [DOI] [PubMed] [Google Scholar]
- 5.Tobe T, Yoshikawa M, Mizuno T, Sasakawa C. Transcriptional control of the invasion regulatory gene virB of Shigella flexneri: activation by virF and repression by H-NS. J Bacteriol. 1993;175:6142–6149. doi: 10.1128/jb.175.19.6142-6149.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lett MC, Sasakawa C, Okada N, Sakai T, Makino S, Yamada M, et al. VirG, a plasmid-coded virulence gene of Shigella flexneri: identification of the VirG protein and determination of the complete coding sequence. J Bacteriol. 1989;171:353–359. doi: 10.1128/jb.171.1.353-359.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T, Saga S, Sasakawa C. Functional analysis of Shigella VirG domains essential for interaction with vinculin and actin-based motility. J Biol Chem. 1996;271:21878–21885. doi: 10.1074/jbc.271.36.21878. [DOI] [PubMed] [Google Scholar]
- 8.Wing HJ, Yan AW, Goldman SR, Goldberg MB. Regulation of IcsP, the outer membrane protease of the Shigella actin tail assembly protein IcsA, by virulence plasmid regulators VirF and VirB. J Bacteriol. 2004;186:699–705. doi: 10.1128/JB.186.3.699-705.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DuPont HL. The growing threat of foodborne bacterial enteropathogens of animal origin. Clin Infect Dis. 2007;45:1353–1361. doi: 10.1086/522662. [DOI] [PubMed] [Google Scholar]
- 10.Alekshun MN, Levy SB. Targeting virulence to prevent infection: to kill or not to kill? Drug Discov Today. 2004;1:483–489. [Google Scholar]
- 11.Barczak AK, Hung DT. Productive steps toward an antimicrobial targeting virulence. Curr Opin Microbiol. 2009;12:490–496. doi: 10.1016/j.mib.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 13.Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. Arac/XylS family of transcriptional regulators. Microbiol Mol Biol Rev. 1997;61:393–410. doi: 10.1128/mmbr.61.4.393-410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soisson SM, MacDougall-Shackleton B, Schleif R, Wolberger C. Structural basis for ligand-regulated oligomerization of AraC. Science. 1997;276:421–425. doi: 10.1126/science.276.5311.421. [DOI] [PubMed] [Google Scholar]
- 15.Falconi M, Colonna B, Prosseda G, Micheli G, Gualerzi CO. Thermoregulation of Shigella and Escherichia coli EIEC pathogenicity: a temperature-dependent structural transition of DNA modulates accessibility of virF promoter to transcriptional repressor H-NS. EMBO J. 1998;17:7033–7043. doi: 10.1093/emboj/17.23.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin RG, Rosner JL. The AraC transcriptional activators. Curr Opin Microbiol. 2001;4:132–137. doi: 10.1016/s1369-5274(00)00178-8. [DOI] [PubMed] [Google Scholar]
- 17.Schleif R. AraC protein: a love-hate relationship. Bioessays. 2003;25:274–282. doi: 10.1002/bies.10237. [DOI] [PubMed] [Google Scholar]
- 18.Kwon HJ, Bennik MH, Demple B, Ellenberger T. Crystal structure of the Escherichia coli Rob transcription factor in complex with DNA. Nat Struct Biol. 2000;7:424–430. doi: 10.1038/75213. [DOI] [PubMed] [Google Scholar]
- 19.Kim OK, Garrity-Ryan LK, Bartlett VG, Grier MC, Verma AK, Medjanis G. N-hydroxybenzimidazole inhibitors of the transcription factor LcrF in Yersinia: novel antivirulence agents. J Med Chem. 2009;52:5626–5634. doi: 10.1021/jm9006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowser TE, Bartlett VJ, Grier MC, Verma AK, Warchol T, Levy SB, et al. Novel anti-infection agents: small-molecule inhibitors of bacterial transcription factors. Bioorganic Med Chem Lett. 2007;17:5652–5655. doi: 10.1016/j.bmcl.2007.07.072. [DOI] [PubMed] [Google Scholar]
- 21.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, NY: CSH Laboratory Press; 1972. [Google Scholar]
- 22.Maurelli AT, Blackmon B, Curtiss RIII. Loss of pigmentation in Shigella flexneri 2a is correlated with loss of virulence and virulence associated plasmid. Infect Immun. 1984;43:397–401. doi: 10.1128/iai.43.1.397-401.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vidal-Aroca F, Giannattasio M, Brunelli E, Vezzoli A, Plevani P, Muzi-Falconi M, et al. One-step high-throughput assay for quantitative detection of beta-galactosidase activity in intact gram-negative bacteria, yeast, and mammalian cells. Biotechniques. 2006;40:433–434. doi: 10.2144/000112145. [DOI] [PubMed] [Google Scholar]
- 24.Eustice DC, Feldman PA, Colberg-Poley AM, Buckery RM, Neubauer RH. A sensitive method for the detection of beta-galactosidase in transfected mammalian-cells. Biotechniques. 1991;11:739. [PubMed] [Google Scholar]
- 25.Porter ME, Smith SG, Dorman CJ. Two highly related regulatory proteins, Shigella flexneri VirF and enterotoxigenic Escherichia coli Rns, have common and distinct regulatory properties. FEMS Microbiol Lett. 1998;162:303–309. doi: 10.1111/j.1574-6968.1998.tb13013.x. [DOI] [PubMed] [Google Scholar]
- 26.Baker CM, Grant GH. Role of aromatic amino acids in protein-nucleic acid recognition. Biopolymers. 2007;85:456–470. doi: 10.1002/bip.20682. [DOI] [PubMed] [Google Scholar]