

Abstract
Activating mutations in the epidermal growth factor receptor (EGFR) characterize a subset of non–small cell lung cancers (NSCLC) with extraordinary sensitivity to targeted tyrosine kinase inhibitors (TKI). A single secondary EGFR mutation, T790M, arising in cis with the primary activating mutation, confers acquired resistance to these drugs. However, the T790M mutation is also detected in the absence of drug selection, suggesting that it may provide a growth advantage. We show here that although T790M alone has only a modest effect on EGFR function, when combined with the characteristic activating mutations L858R or del746–750, it results in a dramatic enhancement of EGFR activity. The double mutants show potent ligand-independent receptor autophosphorylation associated with altered cellular phenotypes, soft agar colony formation, and tumorigenesis in nude mice. The significant gain-of-function properties of these double mutants may explain their initial presence before drug selection and their rapid selection as the single drug resistance mutation during therapy with gefitinib/erlotinib, and suggests that they may contribute to the adverse clinical course of TKI-resistant NSCLC.
Introduction
Non–small cell lung cancer (NSCLC) is the leading cause of cancer-related deaths in the United States. A subset of NSCLC, primarily adenocarcinomas arising in nonsmokers, is defined by the presence of mutations within the kinase domain of epidermal growth factor (EGF) receptor (EGFR; refs. 1-3). These somatic mutations, which seem to arise more frequently in women and in Asians, are correlated with dramatic clinical responses to treatment with tyrosine kinase inhibitors (TKI), such as gefitinib and erlotinib. These mutations comprise a small number of recurrent, gain-of-function changes clustered around the ATP-binding pocket of the tyrosine kinase domain, of which the most common are in-frame deletions (typically encompassing residues 746–750) and the missense mutation L858R (1-3). The EGFR kinase mutants have increased ligand-activated signaling activity compared with their wild-type counterparts, particularly with respect to downstream effectors involved in cell survival, such as AKT and signal transducers and activators of transcription 5 (STAT5; ref. 4). Treatment of NSCLC cell lines harboring these mutations with either gefitinib/erlotinib or transfection with small interfering RNA targeting EGFR results in massive apoptosis, suggesting that these cells are dependent on mutant EGFR-driven signals for survival. This is consistent with the paradigm of “oncogene addiction” (5), which has been similarly implicated in explaining the unique sensitivity of chronic myeloid leukemia (CML) cells to imatinib dependent on expression of the BCR-ABL fusion protein (6).
Despite a rapid and profound response to TKI therapy, EGFR-mutant NSCLC often recurs rapidly, limiting the effect on overall survival of treated patients (7, 8). A single secondary mutation, T790M, has been observed in approximately half of cases with acquired resistance to gefitinib/erlotinib (9, 10). This mutation arises at a critical “gatekeeper” threonine 790 residue within EGFR (11), analogous to the threonine 315 residue that is mutated (T315I) in Gleevec-resistant BCR-ABL(12). In vitro signaling and modeling studies have suggested that both mutations in these gatekeeper residues restrict binding of the drug within the ATP cleft (9, 11, 12).
Acquired resistance to imatinib in CML may result from a number of different somatic mutations in BCR-ABL in addition to T315I (13-19). This mutation, however, is notorious in that it is equally resistant to dasatinib and nilotinib, second-generation TKIs that are effective against most imatinib-resistant mutations (20, 21), and it has also been shown to mediate increased BCR-ABL tyrosine kinase activity in vitro (22). Moreover, it has recently been shown that T315I and T315A BCR-ABL mutants have altered P-loop phosphorylation signatures compared with wild-type, thereby affecting their kinase function (23). In contrast, the analogous EGFR T790M mutant does not seem to have increased kinase activity in standard in vitro assays (9-11, 24). In addition to being the only secondary acquired EGFR mutation identified in gefitinib/erlotinib–resistant NSCLC, T790M is unique for a number of reasons: First, it has been detected in some NSCLC specimens in the absence of drug selection, including both primary tumors (25-27) and the NSCLC-derived cell line NCI-H1975 (10), where it is present in cis with a characteristic activating EGFR mutation (7). Second, we have recently identified a family with inherited susceptibility to the bronchoalveolar subtype of NSCLC that is associated with transmission of a germline T790M allele (7). In this family, T790M seems to be an initiating genetic event, followed in the tumors by a characteristic somatic-activating EGFR mutation, which is in cis with the T790M mutation. Despite these data suggesting that the T790M mutation might confer growth advantage as well as drug resistance, studies to date have not revealed altered signaling by T790M-EGFR compared with the wild-type receptor.
Here, we analyzed the oncogenic properties of T790M-EGFR, either alone or in cis with characteristic activating mutations, using stable retrovirally directed expression in untransformed NIH3T3 cells and immortalized tracheobronchial epithelial cells. T790M alone confers a modest increase in ligand-dependent EGFR activity; in this context, however, double mutants with T790M in cis with either L858R or del746–750–EGFR exhibit dramatically enhanced ligand-independent signaling and transforming phenotypes. These observations have important implications for our understanding of acquired resistance to TKIs in EGFR mutant NSCLC.
Materials and Methods
Cell culture and ectopic expression
NIH3T3 cells were cultured in DMEM supplemented with 10% bovine serum, glutamine, and penicillin/streptomycin. EGF (Sigma) was used at a final concentration of 100 ng/mL unless indicated. Gefitinib was provided by AstraZeneca, and HKI-272 was provided by Wyeth Pharmaceuticals. Using standard techniques, the entire wild-type human EGFR coding sequence was subcloned into the recombinant retroviral expression vector pRV-HygR (28). An identical strategy was used for PCR-generated mutant constructs (EGFR T790M, EGFR L858R, EGFR del747–753, EGFR T790M/L858R, and EGFR T790M/del746–750). Additionally, EGFR constructs (EGFR wild-type, EGFR T790M, EGFR L858R, EGFR del746–750, EGFR T790M/L858R, and EGFR T790M/del746–750) were subcloned into pBABE puro for infection of human tracheobronchial epithelial cells immortalized with the early region of SV40 and human telomerase reverse transcriptase (hTBE cells; ref. 29). Retroviral infection was used to generate cells with stable ectopic expression of EGFR, whereas lentiviral vectors were used for short hairpin RNA (shRNA) knockdown. For retroviral infection studies, EGFR pRV-HygR constructs were transfected into the 293T-derived BOSC-23 packaging cell lines (30) and EGFR pBABE puro were transfected into the 293T-derived Phoenix packaging cell lines using LipofectAMINE 2000 (Invitrogen). NIH3T3 and hTBE cells were infected with the retroviral supernatant twice in the presence of polybrene. Infected cells were selected using hygromycin (700 μg/mL for NIH3T3; Roche) or puromycin (2.5 μg/mL for NIH3T3 or 1 μg/mL for hTBE; Sigma). hTBE cells were maintained in serum-free defined medium as described (29). For lentiviral production, HEK293 cells were transfected with the Δ8.2 lentiviral construct (encoding gag, pol, rev), VSVG, and either empty pLKO.1puro vector or the pLKO.1puro vector containing the following sequence transcribing shRNAs specific to EGFR: 5′-GCTGGATGATAGACGCAGATA-3′. Virus-containing medium was then used to infect the NIH3T3 T790M/Del stable cell line. For all experiments, uncloned drug selected pools of infected cells were used, so as to avoid potential clonal selection bias. Overall expression of ectopic EGFR constructs was comparable with the level of the endogenous protein in hTBE cells, and infection efficiency was close to 95% (NIH3T3) or 75% (hTBE) of cells.
Immunoblot analysis
Cells were lysed with ice-cold buffer [150 mmol/L NaCl, 1% Triton X-100, 50 mmol/L Tris-HCl (pH 8.0), 1 mmol/L EDTA] containing 1 mmol/L sodium orthovanadate and 1× protease inhibitor cocktail (Roche). Debris was removed by centrifugation. Clarified lysates were boiled in gel loading buffer and separated by 10% or 4% to 15% gradient SDS-PAGE. Proteins were electrotransferred to nitrocellulose and detected with specific antibodies. The phosphorylated EGFR Y845, Y1045, Y1068, and Y1148 antibodies as well as the phosphorylated Src, total Src, phosphorylated mitogen-activated protein kinase (MAPK), total MAPK, total Akt, phosphorylated STAT3 and total STAT3 antibodies were obtained from Cell Signaling. The Ras antibody was obtained from BD Transduction Laboratories, total EGFR antibody was from Santa Cruz Biotechnology, and both phosphorylated Akt and phospho EGFR Y992 antibodies from Biosource. This was followed by a horseradish peroxidase–conjugated secondary antibody. The bands were visualized with enhanced chemiluminescence (Perkin-Elmer) followed by autoradiography.
Transformation assays
For focus formation assays, NIH3T3 cells were transfected with 4 μg of the pRV-HygR/EGFR or 1 μg of the pBABE/H-RasV12 per 6-cm dish. Two days after transfection, cells were seeded in a 10-cm dish and grown for 20 days in DMEM 5% serum. Cells were then fixed with 4% paraformaldehyde and stained with crystal violet. To test for colony formation in soft agar, NIH3T3 cells were infected with pRV-HygR/EGFR constructs or pBABEpuro/H-RasV12, and 1 × 104 selected cells were suspended in DMEM containing 0.5% agarose with or without 100 ng/mL EGF. Cells were then seeded onto a 0.5% agar base. Two to 6 weeks after the initial seed, colony growth was assayed by counting colonies using a grid. Experiments were done in triplicates in six-well plates. DMEM 10% serum (0.5 mL) was supplemented to the wells twice weekly, with 100 ng/mL EGF, 1 μmol/L HKI-272 , or 1 μmol/L gefitinib where indicated. hTBE cells infected with pBABE puro/EGFR constructs were seeded at 1 × 105 per well in 0.4% agarose. For tumorigenicity assays, immunodeficient mice were housed under specific pathogen-free conditions, anesthetized with Avertin (0.017 mL of 2.5% stock solution per gram of body weight), and then injected s.c. with 1 × 106 NIH3T3 cells stably expressing EGFR or H-RasV12. Tumor growth was measured every 2 to 3 days.
Results
Morphologic phenotype induced by T790M/L858R and T790M/del746–750 EGFR double mutants
To assess the effect of expressing mutant EGFRs in nontransformed cells, we infected early-passage NIH3T3 cells with retrovirally driven constructs encoding wild-type EGFR; EGFR with the characteristic activating missense mutations L858R or a frequently detected deletion mutant Del (del747–753); the drug resistance mutation T790M alone; and double mutants with T790M in cis with L858R (T790M/L858R) or with the deletion mutant del746–750 (T790M/Del). To circumvent any potential clonal selection effects, hygromycin drug selection was followed by analysis of uncloned pools of infected cells, which had comparable expression of all constructs (Fig. 1A). The retrovirally driven EGFR was expressed in NIH3T3 cells, which normally express low levels of endogenous EGFR and ErbB2 and have no detectable levels of ErbB3 and ErbB4 (31-34). Unexpectedly, NIH3T3 cells expressing either of the T790M/L858R or T790M/Del double mutants exhibit a striking alteration in appearance, displaying a small, round, and light-refracting morphology that is characteristic of Ras-transformed fibroblasts (Fig. 1B). No such morphologic change was apparent in cells infected with wild-type EGFR or any of the single mutations T790M, L858R, or Del.
Figure 1.

Morphologic transformation of NIH3T3 cells expressing double-mutant EGFR. A, immunoblot of NIH3T3 cells infected with retroviruses encoding the indicated EGFR constructs demonstrating comparable expression of all EGFR constructs (H-RasV12 and actin controls). B, cells expressing EGFR T790M/L858R and EGFR T790M/Del double mutants have striking morphologic differences from parental cells and those expressing EGFR wild-type (wt) or single mutants, as determined by phase-contrast microscopy. Cells expressing H-RasV12 also differ morphologically.
Cells were treated with the selective tyrosine kinase inhibitors gefitinib and HKI-272. Gefitinib is a reversible EGFR inhibitor that inhibits the activity of wild-type as well as most classic EGFR mutants other than T790M, whereas HKI-272 binds EGFR irreversibly and can also inhibit EGFR T790M (9, 10, 24). As expected, gefitinib effectively inactivates the L858R and Del mutant EGFR but is ineffective against the T790M mutation, whereas the irreversible inhibitor HKI-272 inhibits all EGFR mutants, including T790M (Fig. 2A). To verify that the observed morphologic alterations were directly attributable to the expression of EGFR double mutants, cells were treated with 1 μmol/L HKI-272. Indeed, treatment with HKI-272 reverses the transformed morphology in cells expressing the EGFR double mutants (Fig. 2B). As a control, HKI-272 has no effect on the transformed morphology induced by oncogenic H-RasV12. To further test the dependence of morphologic changes on expression of EGFR double mutants, we infected cells with lentiviral constructs encoding shRNA targeting the EGFR mRNA. Effective EGFR knockdown was achieved, and, again, reversion of morphologic transformation was evident in cells expressing the EGFR T790M/Del double mutant (Fig. 2C and D). Thus, expression of EGFR double mutants (T790M/L858R and T790M/Del) in NIH3T3 cells is sufficient to induce a transformed morphology.
Figure 2.
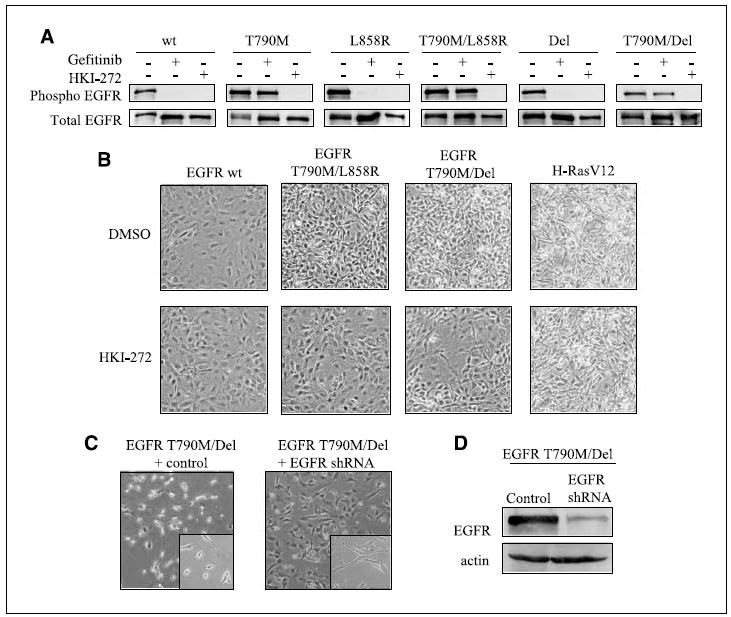
EGFR T790M/Del activity is required for morphologic transformation. A, cells stably expressing various EGFR constructs (shown above) were treated with 10 μmol/L gefitinib, 10 μmol/L HKI-272, or DMSO for 2 h, and were then stimulated with 100 ng/mL EGF for 5 min. Top row, phosphorylated (Phospho) Y992 EGFR; bottom row, total EGFR. B, cells expressing the constructs were grown in 5% serum and treated with 1 μmol/L HKI-272 or DMSO for 6 d to show drug-induced reversion of transformed morphology. C, cells expressing EGFR T790M/Del were infected with lentivirus encoding shRNA against EGFR or a control. D, lysates of infected cells were analyzed by immunoblot to confirm knockdown of EGFR expression. Actin was used as a loading control.
Modulation of EGFR kinase activity by the T790M mutation alone and in cis with characteristic activating mutations
Under normal growth condition, cells infected with EGFR T790M show a significantly higher level of receptor autophosphorylation at tyrosines 845, 992, and 1068, compared with cells infected with wild-type EGFR (Fig. 3A and data not shown). Quantitative analysis normalized for total EGFR levels revealed a 50% increase in receptor autophosphorylation (P = 0.015, t test). Under comparable conditions, the classic EGFR-activating mutation L858R mediates a 2-fold increase in basal EGFR autophosphorylation relative to wild-type (P = 0.009, t test). When present in cis within double mutant constructs, T790M is synergistic with these classic mutations, further increasing EGFR autophosphorylation (Fig. 3B). The effect of both single and double mutants on EGFR autophosphorylation was tested using a panel of phosphospecific antibodies against various tyrosine phosphorylation sites, all showing a consistent pattern of increased activity.
Figure 3.
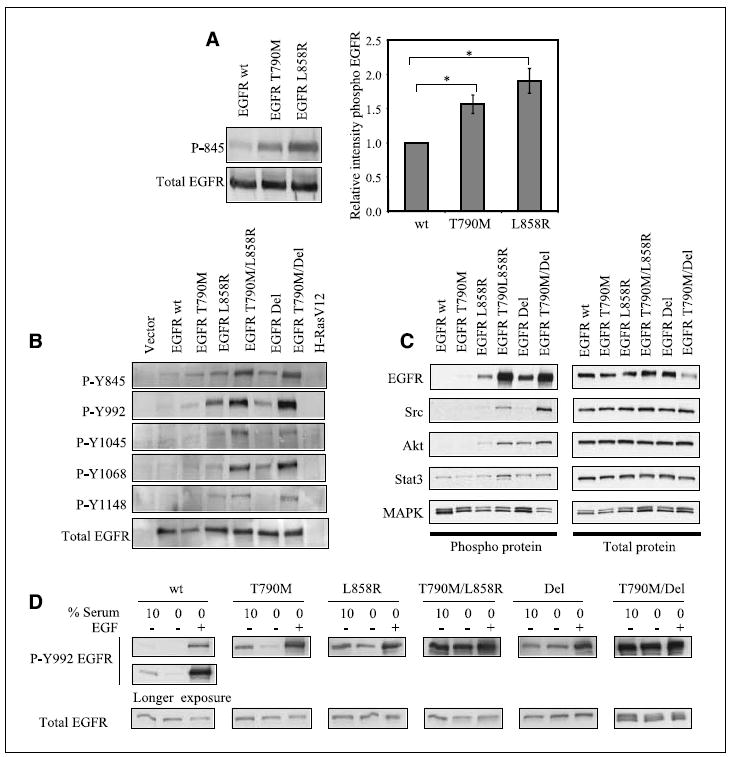
Increased EGFR kinase activity by T790M mutation in cis with classic activating mutations. A, immunoblot of NIH3T3 cells expressing EGFR wild-type, EGFR T790M, and EGFR L858R show increased autophosphorylation of mutant EGFR, as measured by phosphorylated Y845 EGFR (left). Total EGFR was used as a control. The intensity of the phosphorylated Y845 EGFR band in T790M extracts or L858R extracts relative to wild-type extracts was measured in five independent experiments, using total EGFR as an internal control (right). A t test was done to determine the statistical significance (P = 0.015 for T790M versus wild-type and P = 0.009 for L858R versus wild-type). Bars, SE. B, lysates derived from cells expressing EGFR wild-type or mutants (H-RasV12 and vector as controls) were subjected to immunoblot analysis with a panel of phosphorylated EGFR antibodies (Y845, Y992, Y1045, Y1068, Y1148) and total EGFR. C, cells expressing EGFR wild-type or mutants were starved of serum for 24 h. Lysates derived from these cells were subjected to immunoblot analysis against phosphorylated EGFR, Src, Akt, STAT3, and MAPK, and the appropriate total proteins as indicated. D, cells expressing EGFR wild-type or EGFR mutants were grown in 10% serum. Where indicated, cells were serum starved for 24 h, then stimulated with 100 ng/mL EGF for 5 min. Total cell lysate was analyzed by immunoblot against phosphorylated Y992 EGFR and total EGFR. Ligandindependent activity of EGFR double mutants is evident.
To further characterize the activity of the double-mutant EGFR, we examined the ability of these mutants to engage downstream signaling pathways. As previously reported, we observe that Akt and STAT3 activity are enhanced by the classic EGFR mutants, L858R and Del, whereas MAPK activity is not stimulated by these mutants (Fig. 3C; ref. 4). Unexpectedly, however, the T790M double mutants do not show significantly enhanced activation of Akt or STAT3 compared with the single classic activating mutants despite their substantial increase in autophosphorylation. This led us to speculate that the double mutants might have preferences for different substrates. This is consistent with the recently published paper by Skaggs et al. (23), which elegantly showed that the Bcr-Abl T315I–resistant mutant, analogous to EGFR T790M, has a unique phosphosubstrate signature compared with wild-type Bcr-Abl. Indeed, we find that the Src kinase, a known EGFR substrate, exhibits significantly elevated phosphorylation in cells expressing the double mutants compared with the single-mutant alleles, suggesting the involvement of alternative downstream signaling pathways for these kinases harboring gatekeeper mutations.
To test whether T790M-containing EGFR double mutants exhibit altered ligand responsiveness, we repeated these phosphorylation studies in serum-starved cells and after administration of EGF (Fig. 3D). As expected, cells expressing wild-type EGFR have low levels of receptor autophosphorylation at baseline, which was detectable only after a long exposure of the autoradiograms and is abolished after 24 h of serum starvation. Addition of EGF to the culture transiently induces receptor autophosphorylation. Cells expressing the EGFR T790M mutation display higher levels of receptor autophosphorylation under serum-starved conditions, suggesting some degree of ligand-independent activity. Ligand-dependent activation of T790M-EGFR is comparable with that of wild-type EGFR. The classic EGFR mutations, L858R and Del, show a further increase in ligand-independent and ligand-stimulated kinase activity. Remarkably, cells with the T790M/L858R or T790M/Del double mutants exhibit high levels of receptor autophosphorylation despite serum starvation, and this is not further enhanced by treatment with EGF. The ligand-independent autophosphorylation of EGFR double mutants is considerably higher than that of maximally ligand induced wild-type EGFR or the single mutants L858R and Del.
To further characterize the ligand dependency of the various EGFR mutants, we did a dose response assay. As expected, EGFR wild-type is stimulated by low concentrations of EGF and its stimulation is enhanced at increasing concentration of ligand (Supplementary Fig. S1). This pattern is reproduced with EGFR T790M but with a higher level of activation than EGFR wild-type for the same dose of ligand. The classic EGFR Del mutant showed low levels of stimulation in the absence of ligand, and was significantly stimulated by low concentrations of ligand. EGFR Del/T790M mutant, however, retained high levels of autophosphorylation in the absence of ligand and this was not further increased after EGF stimulation, reminiscent of the data shown in Fig. 3D. Hence, T790M confers increased activity to EGFR wild-type and cooperates with the classic activating mutations in both baseline and serum-starved conditions to promote ligand-independent kinase activation.
Induction of oncogenic transformation by EGFR double mutants
The transformed morphology and ligand-independent activity mediated by T790M/L858R and T790M/Del raise the possibility that these EGFR double mutants also have altered transforming properties. We first tested the effect of these mutants on focus formation in culture. NIH3T3 cells maintained at confluence in low serum concentrations for prolonged periods form very few foci, as do those infected with either wild-type EGFR, the T790M mutant, or the L858R mutant (average of 14/plate). Cells expressing EGFR Del give rise to a slightly increased number of foci (29 per plate). Moreover, both the T790M/L858R and T790M/Del EGFR double mutants cause a marked increase in foci (40 and 94 per plate, respectively; Supplementary Fig. S2).
We also tested anchorage-independent growth in soft agar. NIH3T3 cells expressing EGFR T790M/L858R or T790M/Del plated in soft agar in the absence of exogenous EGF give rise to an average of 28 and 69 colonies per well, respectively (Fig. 4A). Cells expressing the single mutants L858R, Del, and T790M form very few colonies in soft agar (<6 per well), whereas those infected with wild-type EGFR or control vector do not grow under these conditions. Of interest, addition of exogenous EGF yields anchorage-independent colonies in NIH3T3 cells infected with any of the EGFR constructs, including wild-type and single-mutant constructs. Therefore, ligand-independent activation of the EGFR double-mutant receptors may be a particularly important distinguishing factor as it relates to oncogenic transformation.
Figure 4.

Synergistic transforming activity of T790M in cis with L858R and Del EGFR mutations. A, soft-agar colony formation of NIH3T3 cells stably expressing the indicated EGFR construct or H-RasV12. Colonies were counted after 5 wk. This experiment was done in duplicate and is representative of three independent experiments (left). Right, similar soft-agar assay in the presence of 100 ng/mL EGF added to the top agar. Colonies were counted after 2 wk. B, soft agar colony formation of immortalized hTBE cells infected with the indicated EGFR constructs (left). Colonies were counted after 1 mo. Immunoblot of hTBE cells stably expressing EGFR constructs showsimilar expression levels of EGFR (right). Tubulin is used as a loading control. C, tumor formation in nude mice injected s.c. with NIH3T3 cells stably expressing the indicated EGFR construct or H-RasV12. Tumor volume was measured every 2 to 3 d. D, number of tumors formed and the total number of inoculations for each construct. Tumor formation was assessed 25 d after inoculation.
To test whether EGFR double mutants are transforming in a more physiologically relevant model, EGFR mutant constructs were stably infected in human tracheobronchial epithelial cells immortalized with the early region of SV40 and human telomerase reverse transcriptase (29). Similar to our results in NIH3T3 cells, hTBE cells expressing EGFR T790M/L858R or T790M/Del plated in soft agar give rise to a significant number of colonies, whereas cells expressing the single activating mutants are only weakly transforming (Fig. 4B). Analysis of EGFR expression level in these stable cells shows low levels of ectopic expression comparable with levels of endogenous EGFR expressed by the parental cell line. Thus, our observations are consistent with the physiologic effect of heterozygous EGFR mutants present in NSCLC.
We have shown that the irreversible tyrosine kinase inhibitor HKI-272 can inhibit EGFR double-mutant activity, as measured by autophosphorylation and cell morphology (Fig. 2). We therefore wanted to confirm that the transforming activity of EGFR double mutants could be reversed by HKI-272. Indeed, HKI-272 treatment of NIH3T3 cells expressing EGFR T790M/L858R or T790M/Del prevents anchorage-independent growth (Supplementary Fig. S3). As expected, gefinitib treatment of these EGFR double mutant–expressing cells does not have any significant effect on soft-agar colony formation because the T790M double mutants are resistant to this drug.
To test whether EGFR double mutants exhibit increased oncogenic activity in vivo, NIH3T3 cells stably expressing the various EGFR constructs were injected s.c. into nude mice. As controls, we established that vector-infected cells are not tumorigenic, whereas cells infected with H-RasV12 form tumors within 1 week after injection. NIH3T3 cells expressing T790M/Del and T790M/L858R give rise to large tumors within 2 weeks (six of six inoculations for each construct), whereas cells expressing the L858R or Del mutations leads to comparable tumors only after 3 weeks (six of six and four of four inoculations for each construct, respectively). Cells expressing the T790M mutation alone form small tumors in only two of six inoculations; cells infected with wild-type EGFR do not give rise to tumors (Fig. 4C and D). Sequencing of EGFR coding regions from tumor tissue showed no acquired mutations during the process of tumor formation (data not shown). Taken together, these results indicate that the EGFR double mutants T790M/L858R and T790M/Del have increased oncogenic activity in vitro and in vivo relative to wild-type EGFR and EGFR single mutants.
Discussion
We have shown potent transforming activity by EGFR harboring two mutations in cis, including the only reported gefitinib/erlotinib resistance mutation, T790M, and either of two classic drug-sensitizing mutations L858R or del746–750. The double EGFR mutants mediate significantly increased ligand-independent activation of the receptor, compared with any of the single mutations. Taken together, these observations suggest that T790M might be particularly advantageous to tumor cells after drug selection as it confers both drug resistance and gain of transforming activity.
The modest increase in EGFR signaling mediated by T790M alone may explain the observation that this drug resistance mutation has been reported as a germ line mutation in a family with inherited susceptibility to lung cancer (7). The relatively weak increase in signaling associated with T790M-EGFR may explain why it is “tolerated” in the germ line. Our tumorigenesis studies in mice further point to a modest oncogenic effect of T790M alone. These results are consistent with T790M mediating susceptibility to NSCLC in humans only at late onset. Of note, previous studies using transient overexpression have not succeeded in demonstrating clear functional distinctions between wild-type EGFR and the T790M mutation (9-11) either in terms of receptor signaling or tumorigenesis, pointing to the potential advantage of using stable retrovirally driven expression in nontumorigenic cells.
The development of tumors in patients with a germ line T790M mutation is typically accompanied by somatic acquisition of a second, classic activating EGFR mutation in cis (7). Similarly, the acquisition of gefitinib/erlotinib resistance in NSCLC patients whose tumor harbors an initial drug-sensitizing EGFR mutation is associated with the T790M mutation arising in cis with the initial mutation. In rare cases, such double mutants in EGFR have also been observed in untreated NSCLC, although use of allele-specific PCR is increasingly revealing the presence of the T790M mutation at low frequency in primary tumors before treatment (10, 25, 26). Thus, genetic evidence strongly points to specific functional properties of these double mutants, in addition to their reported role in gefitinib/erlotinib resistance.
The presence of T790M in cis with either of the classic activating mutations L858R or del746–750 has a profound effect on receptor activity. Most significant is the appearance of potent ligan-dindependent receptor activation. Under conditions tested here, ligand-independent activation is modest with single classic kinase mutations of EGFR. This difference is consistent with the observation that activating EGFR mutations are transforming in the presence of ligand, whereas the double mutants give rise to soft agar colonies in the absence of exogenous EGF. The degree of ligand dependence of the classic activating mutants L858R and Del may vary under experimental conditions (35-38). Indeed, we have found that the classic activating EGFR single mutants show varying degrees of transformation depending on expression levels (data not shown). Our approach appears particularly valuable in distinguishing between EGFR double mutants and EGFR single mutants. Additional work is needed to determine the mechanism by which T790M can confer ligand-independent activity to EGFR. Increased affinity of the mutant EGFR tyrosine kinase domain for the sites of EGFR tyrosine phosphorylation (reduced Km) or increased catalytic activity of the mutant EGFR kinase domain (increased kcat) are possible explanations that should be pursued further. Additionally, further structural studies will be required to determine how the combination of mutations within the ATP pocket may modulate catalytic activity of the receptor. In this respect, it is of interest that the EGFR T790M/Del mutant consistently shows increased activity compared with the T790M/L858R mutant, as measured by morphologic transformation, soft agar colony formation, and tumorigenicity assays. Accumulating clinical findings are pointing to distinct biological properties for the common activating mutations of EGFR, L858R, and in-frame deletion variants, which may well also have different properties when combined with T790M. Of note, EGFR double mutants appear to signal via alternative downstream pathways compared with wild-type and classic mutant EGFR. Similarly, a recent report from Skaggs et al. (23) has shown that Bcr-Abl T315I, the analogous mutation to EGFR T790M, has a unique phosphosubstrate signature compared with wild-type Bcr-Abl.
The potent transforming properties of EGFR double mutants raise an interesting paradox. Their apparent oncogenic effect readily explains why these mutations are detectable in cases that have not been treated with either gefitinib or erlotinib. The presence of some tumor cells expressing these double mutants even before treatment with tyrosine kinase inhibitors may explain the relatively rapid emergence of drug resistance, and also the fact that T790M is the sole mutation associated with acquired drug resistance. This is reminiscent of cases of imatinib-resistant Bcr-Abl mutants present in untreated patients (18), suggesting a global mechanism of resistance by clonal selection of preexisting kinase domain mutations. On the other hand, such a potent proliferative advantage to cells expressing the T790M double mutants would suggest that these cells should have become prevalent in untreated NSCLC, rather than constituting a small subset of the tumor population. It is possible that the frequency of T790M mutations in NSCLC is underestimated by virtue of “allelic dilution.” In the subset of tumors with amplified mutant EGFR, a single doublemutant allele may be below a level of detectability for standard sequencing assays. This hypothesis is strengthened by a recent publication that shows that a T790M mutation present in only a small fraction of amplified EGFR alleles, which goes undetected in sequencing assays, is sufficient to confer resistance to gefitinib in vitro (39). Alternatively, because most EGFR mutant NSCLC do not have amplification of the mutant allele, the frequency of T790M double mutants may be driven by its signaling properties. Considering the weak ligand-independent signaling mediated by the classic EGFR kinase mutations, versus the strong ligandindependent effect of the T790M double mutants, we speculate that the former may be sufficient to drive cellular proliferation in tissues where ligand is abundant, whereas the T790M mutation may provide a selective growth advantage in tissues where such ligands are not present. Tissue context may therefore regulate the relative abundance of single versus double mutants in the absence of drug selection. Following such selection, however, the emergence of T790M double mutants may have important implications not only for drug sensitivity but also for tumor aggressiveness (see model in Fig. 5).
Figure 5.

Model for T790M selection in EGFR mutant-expressing lung cancer. A somatic activating mutation at EGFR (e.g., L858R) is sufficient to initiate the formation of NSCLC (40, 41). A small proportion of cells acquire ligand-independent oncogenic potential by the secondary mutation T790M in cis with the primary mutation. Treatment with the TKIs gefitinib or erlotinib results in killing of cells expressing EGFR L858R, but has no effect on those expressing EGFR T790M/L858R. Drug treatment thus enriches for preexisting NSCLC cell populations expressing the highly transforming EGFR double mutant, resulting in a more aggressive tumor.
Most cases of NSCLC with activating EGFR kinase mutations have a dramatic initial response to tyrosine kinase inhibitors, followed by relatively rapid tumor regrowth. The data presented here suggest that these recurrent, T790M double-mutant, EGFR-driven tumors might be more aggressive than the primary, singlemutant tumors, thereby contributing to the modest survival benefit in these patients (7, 8). This hypothesis suggests that secondgeneration EGFR kinase inhibitors that retain activity against the T790M double-mutant receptors, such as irreversible inhibitors, may prove to have clinical utility.
Supplementary Material
Acknowledgments
Grant support: NIH grants R01 CA115830-02 (J. Settleman and D.A. Haber) and P01 CA95281 (D.A. Haber), the V Foundation Award (J. Settleman), the Doris Duke Foundation Distinguished Clinical Investigator for Cancer Research Award (D.A. Haber), and the Tosteson Postdoctoral Fellowship Award from the Massachusetts Biomedical Research Corporation (N. Godin-Heymann).
We thank Dr. William Hahn and the RNAi Consortium for providing EGFR shRNA constructs and the hTBE cell line; Drs. Deyin Xing, Nicolas Degauque, Sree Sharma, and Sven Diederichs for helpful comments; AstraZeneca for the sample of gefinitib; and Wyeth Pharmaceuticals for the sample of HKI-272 used in this study. D.W. Bell acknowledges the Intramural Program of the National Human Genome Research Institute at NIH.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. see comment. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. see comment. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate antiapoptotic pathways. Science. 2004;305:1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 5.Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 6.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 7.Bell DW, Gore I, Okimoto RA, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. 2005;37:1315–6. doi: 10.1038/ng1671. [DOI] [PubMed] [Google Scholar]
- 8.Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133–44. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. see comment. [DOI] [PubMed] [Google Scholar]
- 10.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blencke S, Ullrich A, Daub H. Mutation of threonine 766 in the epidermal growth factor receptor reveals a hotspot for resistance formation against selective tyrosine kinase inhibitors. J Biol Chem. 2003;278:15435–40. doi: 10.1074/jbc.M211158200. [DOI] [PubMed] [Google Scholar]
- 12.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 13.Barthe C, Gharbi MJ, Lagarde V, et al. Mutation in the ATP-binding site of BCR-ABL in a patient with chronic myeloid leukaemia with increasing resistance to STI571. Br J Haematol. 2002;119:109–11. doi: 10.1046/j.1365-2141.2002.03708.x. [DOI] [PubMed] [Google Scholar]
- 14.Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABLin patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–5. doi: 10.1182/blood.v99.9.3472. [DOI] [PubMed] [Google Scholar]
- 15.Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190–6. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann WK, Jones LC, Lemp NA, et al. Ph(+) acute lymphoblastic leukemia resistant to the tyrosine kinase inhibitor STI571 has a unique BCR-ABL gene mutation. Blood. 2002;99:1860–2. doi: 10.1182/blood.v99.5.1860. [DOI] [PubMed] [Google Scholar]
- 17.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–8. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 18.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 19.von Bubnoff N, Schneller F, Peschel C, Duyster J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002;359:487–91. doi: 10.1016/S0140-6736(02)07679-1. [DOI] [PubMed] [Google Scholar]
- 20.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 21.Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto M, Kurosu T, Kakihana K, Mizuchi D, Miura O. The two major imatinib resistance mutations E255K and T315I enhance the activity of BCR/ABL fusion kinase. Biochem Biophys Res Commun. 2004;319:1272–5. doi: 10.1016/j.bbrc.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 23.Skaggs BJ, Gorre ME, Ryvkin A, et al. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc Natl Acad Sci U S A. 2006;103:19466–71. doi: 10.1073/pnas.0609239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102:7665–70. doi: 10.1073/pnas.0502860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–23. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 26.Tokumo M, Toyooka S, Ichihara S, et al. Double mutation and gene copy number of EGFR in gefitinib refractory non-small-cell lung cancer. Lung Cancer. 2006;53:117–21. doi: 10.1016/j.lungcan.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Inukai M, Toyooka S, Ito S, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66:7854–8. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- 28.Riese DJ, II, DiMaio D. An intact PDGF signaling pathway is required for efficient growth transformation of mouse C127 cells by the bovine papillomavirus E5 protein. Oncogene. 1995;10:1431–9. [PubMed] [Google Scholar]
- 29.Lundberg AS, Randell SH, Stewart SA, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21:4577–86. doi: 10.1038/sj.onc.1205550. [DOI] [PubMed] [Google Scholar]
- 30.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993;90:8392–6. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carraway KL, III, Sliwkowski MX, Akita R, et al. The erbB3 gene product is a receptor for heregulin. J Biol Chem. 1994;269:14303–6. [PubMed] [Google Scholar]
- 32.Cohen BD, Green JM, Foy L, Fell HP. HER4-mediated biological and biochemical properties in NIH 3T3 cells. Evidence for HER1-4 heterodimers. J Biol Chem. 1996;271:4813–8. doi: 10.1074/jbc.271.9.4813. [DOI] [PubMed] [Google Scholar]
- 33.Di Fiore PP, Pierce JH, Fleming TP, et al. Over-expression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell. 1987;51:1063–70. doi: 10.1016/0092-8674(87)90592-7. [DOI] [PubMed] [Google Scholar]
- 34.Hudziak RM, Schlessinger J, Ullrich A. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc Natl Acad Sci U S A. 1987;84:7159–63. doi: 10.1073/pnas.84.20.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagatomo I, Kumagai T, Yamadori T, et al. The gefitinib-sensitizing mutant epidermal growth factor receptor enables transformation of a mouse fibroblast cell line. DNA Cell Biol. 2006;25:246–51. doi: 10.1089/dna.2006.25.246. [DOI] [PubMed] [Google Scholar]
- 36.Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65:8968–74. doi: 10.1158/0008-5472.CAN-05-1829. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi S, Ji H, Yuza Y, et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65:7096–101. doi: 10.1158/0008-5472.CAN-05-1346. [DOI] [PubMed] [Google Scholar]
- 39.Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji H, Li D, Chen L, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–95. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 41.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to downregulation of the receptors. Genes Dev. 2006;20:1496–510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.