

Abstract
Background
Hookworm infection is one of the most important neglected diseases in developing countries, with approximately 1 billion people infected worldwide. To better understand hookworm biology and nematode parasitism, the present study generated a near complete transcriptome of the canine hookworm Ancylostoma caninum to a very high coverage using high throughput technology, and compared it to those of the free-living nematode Caenorhabditis elegans and the parasite Brugia malayi.
Results
The generated transcripts from four developmental stages, infective L3, serum stimulated L3, adult male and adult female, covered 93% of the A. caninum transcriptome. The broad diversity among nematode transcriptomes was confirmed, and an impact of parasitic adaptation on transcriptome diversity was inferred. Intra-population analysis showed that A. caninum has higher coding sequence diversity than humans. Examining the developmental expression profiles of A. caninum revealed major transitions in gene expression from larval stages to adult. Adult males expressed the highest number of selectively expressed genes, but adult female expressed the highest number of selective parasitism-related genes. Genes related to parasitism adaptation and A. caninum specific genes exhibited more expression selectivity while those conserved in nematodes tend to be consistently expressed. Parasitism related genes were expressed more selectively in adult male and female worms. The comprehensive analysis of digital expression profiles along with transcriptome comparisons enabled identification of a set of parasitism genes encoding secretory proteins in animal parasitic nematode.
Conclusions
This study validated the usage of deep sequencing for gene expression profiling. Parasitic adaptation of the canine hookworm is related to its diversity and developmental dynamics. This comprehensive comparative genomic and expression study substantially improves our understanding of the basic biology and parasitism of hookworms and, is expected, in the long run, to accelerate research toward development of vaccines and novel anthelmintics.
Background
Genomic data is revolutionizing molecular parasitology and has been used to prioritize drug targets in parasites at a genomic level [1]. Similarly, pan-phylum genomic studies in parasitic nematodes have identified both highly conserved nematode-specific proteins [2], which are attractive as drug candidates as their targeting will not affect the host, and nematode-specific indels in essential proteins [3], which could also be good candidates for "indel-based" drug design in nematodes. Complementing the genomic data, expression data reflects the dynamics of genetic information. Analysis of digital expression data, obtained by sequencing cDNAs, is crucial for studying and understanding organism's development, physiology, and environmental adaptation. Knowledge of these mechanisms in parasites is essential to substantially accelerate research toward the development of both new therapies to prevent parasite infections and vaccines (or novel anthelmintics) needed to control them.
Infection of humans by parasitic nematodes results in substantial human mortality and morbidity, especially in tropical regions of Africa, Asia, and the Americas. Hookworms, probably the most significant public health threat of these nematodes, are the second largest contributor to the 26.7 million annual DALYs (Disability Adjusted Life Years) from iron-deficiency anemia due to blood feeding by adult worms. Chronic anemia from hookworm infection is particularly devastating to children, who suffer from stunted growth and impaired intellectual development, to mothers who are at increased risk for anemia during pregnancy and childbirth, and to the elderly [4-6]. Current hookworm control strategies are limited to deworming of infected people using anthelmintic drugs. However, rapid re-infection in endemic areas and the lack of sterile immunity necessitates repeated treatments, which will in turn result in resistance. The high rates of re-infection after drug therapy mean that vaccines remain the best hope for worm control in humans in the future. No vaccine is yet available, despite substantial support from The Bill and Melinda Gates Foundation specifically for the development of a hookworm vaccine [7]. Until safe and effective vaccines are developed, anthelmintics will continue to be used for treatment and control of nematode infections in humans. Thus, there is a critical need for further research to identify new vaccine and drug targets which requires better understanding of the biology and parasitism of these devastating parasites.
Ancylostoma caninum, a canine hookworm closely related to the human parasites Ancylostoma duodenale and Necator americanus [8], is the most widely used model for human hookworm infections [9]. Similar to other hookworms, adult A. caninum inhabit the small intestine and produce eggs that pass in the feces and hatch in the soil. The first stage larva feeds on bacteria and molts twice to form the non-feeding, infective third stage (iL3). iL3 enters the host by penetrating the skin, molts twice, and matures in to the adult (Ad) stage in the small intestine. A. caninum iL3 can also infect a host, temporarily abort maturation and enter an arrested state (hypobiosis) within the host's somatic tissues [10], reactivating in response to host physiological changes such as pregnancy [11].
A. caninum is a Clade V nematode [12] that also includes the well-studied free-living model nematode Caenorhabditis elegans. C. elegans was the first multicellular genome to be sequenced [13] and it remains the only metazoan for which the sequence of every nucleotide is known to high confidence. Recently, the genome of the human parasite Brugia malayi, has been sequenced and analyzed [14]. Brugia malayi is phylogenetically classified in Clade III [15]. The distant phylogenetic relationship between A. caninum and B. malayi (compared to A. caninum and C. elegans) makes investigation of nematode adaptation to parasitism easier, as similarities shared by A. caninum and B. malayi (but not by C. elegans) are likely to be associated with adaptation to parasitism. Our previous studies based on limited coverage of A. caninum revealed the existence of genes unique to hookworm and the different selective pressures on these genes [16,17]. Another study using microarray technology [18] found several hundred genes in A. caninum changed their expression during the worm's transition from a free living to a parasitic larva. However, because of the limitation of the data and/or methods of these studies, many questions, especially those related to parasitism, remain to be fully explored.
To better understand the biology of parasitism and facilitate prioritization of potential vaccine and drug targets, the present study deeply sequenced the A. caninum transcriptome with a combination of two distinct sequencing technologies, ABI Sanger capillary and 454/Roche massively parallel sequencing platforms. Over 1.5 million cDNAs were generated from different cDNA libraries constructed from pre-parasitic, parasitic larval and adult stages. These reads covered over 90% of the A. caninum transcriptome with an average depth of 10×. This dataset was also used to perform comprehensive comparative analysis among A. caninum, B.malayi and C. elegans, and the unprecedented depth of coverage enabled comparison of digital expression profiles leading to reliable identification of differentially expressed genes during development. This study provides the first nearly complete transcriptome from a parasitic nematode and provides valuable information about nematode adaptations to parasitism, in addition to revealing several candidates for further study as drug target or vaccine components.
Results
Sequence acquisition, organization and transcriptome coverage
Over 1.5 million ESTs were generated from 4 stages, infective L3 larva (iL3), activated L3 larva (ssL3), adult male (M), and female (F), of A. caninum (Table 1). These 1,567,105 reads include 1,483,002 pyrosequencing reads (Roche/454 reads, average length 232 bases) and 84,103 Sanger reads (average length 748 bases). The larval stages were represented by nearly half a million reads, and the adult stages with nearly 300,000 reads (Table 1).
Table 1.
Sequence characteristics
| Reads (#) | ||||||
|---|---|---|---|---|---|---|
| infective L3 | serum stimulated L3 | Female | Male | Total reads | Mean Length (bp) | |
| Roche/454FLX | 474,766 | 458,249 | 277,319 | 272,668 | 1,483,002 | 232 |
| ABI Sanger | 23,429 | 21,722 | 18,960 | 19,992 | 84,103 | 748 |
| Total | 498,195 | 479,971 | 296,279 | 292,660 | 1,567,105 | - |
Assembly, which was performed to reduce data redundancy and improve sequence quality and length, grouped the sequences into 48,326 transcripts longer than 90 bp, for a total of 23 Mb. The transcript consensus sequences are available at http://nematode.net[19]. The average transcript length was 477 bp and average coverage was 10×. Using the core eukaryotic genes as a reference, we estimated that 93% of the A. caninum transcriptome is identified (See additional file 1), making this the first parasitic nematode with a near complete sequenced transcriptome.
Nematode transcriptome diversity and parasitism related genes
Even though A. caninum and C. elegans fall in the same phylogenetic Clade (Clade V)[12], only about 20% of A. caninum transcripts are homologous to C. elegans coding genes, and even lower number (14%) to B. malayi coding genes (Figure 1A). However, when only considering the highly expressed transcripts (those sequenced deeply enough to provide confident stage selectivity in this case) about 43% of A.caninum transcripts are homologous to C. elegans. When all the transcripts were considered, the vast majority (77%) of the A. caninum transcripts were species-specific. This indicates high transcriptome diversity among nematodes. However, this diversity did not correspond to a drastic difference on functional level. The total unique number of KOs associated to the A. caninum and C. elegans genes were very similar (Table 2), with only one (amino acid metabolism pathway; P < 0.001) out of the 33 identified pathways having a statistically significant increased number of unique KOs (359 vs. 315) in A. caninum.
Figure 1.

Venn diagram showing distribution of BLAST matches. Amino acid level homologies with bitscore of 50 or better were considered. (A) A. caninum transcripts homologous to B. malayi and C. elegans. Only 23% of the transcripts (11,025/48,326) shared homology, leaving 37,301 transcripts to be specific to A. caninum. (B) B. malayi genes homologous to A. caninum and C. elegans. About 62% of the 11,609 B. malayi genes shared homology. Higher number of B. malayi genes had homologs to the parasitic A. caninum compared to the free-living C. elegans, and 4,406 B. malayi genes did not share homology.
Table 2.
KEGG pathway mappings for A. caninum and C. elegans orthologs
| Major Category | Pathway | A.caninum KOs | C.elegans KOs | Total KO in the pathway |
|---|---|---|---|---|
| 1. Metabolism | 1467 | 1386 | 2230 | |
| 1.1 Carbohydrate Metabolism | 357 | 327 | 547 | |
| 1.2 Energy Metabolism | 255 | 247 | 406 | |
| 1.3 Lipid Metabolism | 234 | 237 | 332 | |
| 1.4 Nucleotide Metabolism | 118 | 122 | 165 | |
| 1.5 Amino Acid Metabolism | 359 | 315 | 474 | |
| 1.6 Metabolism of Other Amino Acids | 86 | 85 | 125 | |
| 1.7 Glycan Biosynthesis and Metabolism | 113 | 119 | 158 | |
| 1.8 Biosynthesis of Polyketides and Nonribosomal Peptides | 4 | 4 | 4 | |
| 1.9 Metabolism of Cofactors and Vitamins | 180 | 173 | 297 | |
| 1.10 Biosynthesis of Secondary Metabolites | 47 | 46 | 57 | |
| 1.11 Xenobiotics Biodegradation and Metabolism | 122 | 115 | 180 | |
| 2. Genetic Information Processing | 489 | 513 | 793 | |
| 2.1 Transcription | 70 | 77 | 85 | |
| 2.2 Translation | 149 | 145 | 175 | |
| 2.3 Folding, Sorting and Degradation | 191 | 213 | 368 | |
| 2.4 Replication and Repair | 84 | 83 | 174 | |
| 3. Environmental Information Processing | 1049 | 1106 | 1846 | |
| 3.1 Membrane Transport | 120 | 140 | 391 | |
| 3.2 Signal Transduction | 555 | 571 | 919 | |
| 3.3 Signaling Molecules and Interaction | 462 | 483 | 713 | |
| 4. Cellular Processes | 886 | 912 | 1176 | |
| 4.1 Cell Motility | 162 | 165 | 203 | |
| 4.2 Cell Growth and Death | 172 | 181 | 246 | |
| 4.3 Cell Communication | 231 | 234 | 250 | |
| 4.4 Endocrine System | 239 | 247 | 281 | |
| 4.5 Immune System | 260 | 267 | 393 | |
| 4.6 Nervous System | 62 | 62 | 66 | |
| 4.7 Sensory System | 31 | 34 | 38 | |
| 4.8 Development | 99 | 99 | 102 | |
| 4.9 Behavior | 7 | 8 | 10 | |
There were 1,643 transcripts with B. malayi homologs (1,365 genes) but no C. elegans homologs (Figure 1A) despite A. caninum being more closely related phylogenetically to C. elegans. The majority of these transcripts (1,093 out of 1,643) failed to find any GO annotations. Nevertheless, functions of the 550 transcripts having GO annotation are enriched in 3 GO terms, prolyl oligopeptidase activity (GO:0004287, P = 3.5e-6), nucleic acid binding (GO: 0003676, P = 5.1e-5), and DNA binding (GO: 0003677, P = 1.7e-3), with the most enriched category being prolyl oligopeptidase activity. In addition, malic enzyme activity was enriched (P = 5.2e-3) though it failed our FDR cutoff because of the small number of entries in this activity. As a comparison, no GO term enrichment was detected when considering the B. malayi genes with homology to C. elegans but not A. caninum. Meanwhile, homology comparison among the free-living C. elegans and the parasites A. caninum and B. malayi found that more B. malayi genes share homology with A. caninum (5,991) than with C. elegans (5,532) (Figure 1B). The higher number of homologous genes among parasites was statistically significant (P < 1.0e-4, Chi-square test). Since B. malayi (Clade III) is phylogentically more distant from A. caninum than C. elegans (both are in Clade V) [12], B. malayi would share a similar level of homology with both C. elegans and A. caninum if parasitism had no effect on gene evolution. Therefore, we hypothesize that the 1,643 transcripts represent putative parasitism related genes.
Comparative genomics of gene expression during development of A. caninum
The deep sequencing of this study allowed us to examine differential expression of inferred transcripts and shed light on their functions due to the association of the gene expression with molecular function. Figure 2 summarizes the expression selectivity of the 16,359 transcripts that have sequencing depth for confident estimation of expression selectivity, and Table 3 contains the ten most abundant transcripts selectively expressed in different stages (The list of the top 20 most abundant transcripts is available as additional file 2). Only half of the transcripts are expressed through all the stages. The most dramatic change observed between the developmental stages studied was the transition from larvae to adult: more than 3,000 transcripts were turned off and nearly 1,500 turned on. This is not unexpected, especially since the developing L4 stage was not examined. The serum stimulation turned off 78 transcripts, and turned on 401 transcripts. More than one third of the turned-on genes were turned off again in M, F, or M and F. Consistent with these expression changes, comparison of the preparasitic to parasitic (iL3 vs. ssL3) and parasitic larval to adult stages (ssL3 vs. M + F) revealed that nearly twice the number of transcripts have significant expression changes in the latter transition than the former (See additional file 3).
Figure 2.

Distribution of A. caninum transcripts based on stage or origin of each read. iL3: infective L3; ssL3: serum stimulated L3.
Table 3.
The most abundantly represented transcripts in the A. caninum cDNA library expressed genes
| Stage | Contig id | Female | Male | iL3 | ssL3 | Total | GO id | Go descriptor |
|---|---|---|---|---|---|---|---|---|
| Female specific | ||||||||
| contig00905 | 168 | 0 | 0 | 0 | 168 | - | ||
| contig03165 | 118 | 0 | 0 | 0 | 118 | GO:0004252 | aserine-type endopeptidase activity | |
| contig05541 | 103 | 0 | 0 | 0 | 103 | GO:0005529 | asugar binding | |
| contig02012 | 94 | 0 | 0 | 0 | 94 | GO:0005529 | asugar binding | |
| contig05749 | 93 | 0 | 0 | 0 | 93 | - | ||
| contig05507 | 87 | 0 | 0 | 0 | 87 | - | ||
| contig05999 | 86 | 0 | 0 | 0 | 86 | - | ||
| contig06009 | 84 | 0 | 0 | 0 | 84 | - | ||
| contig02190 | 84 | 0 | 0 | 0 | 84 | - | ||
| contig06122 | 84 | 0 | 0 | 0 | 84 | - | ||
| Male specific | ||||||||
| contig43465 | 0 | 408 | 0 | 0 | 408 | GO:0003676 | anucleic acid binding | |
| contig01761 | 0 | 301 | 0 | 0 | 301 | - | ||
| contig53299 | 0 | 294 | 0 | 0 | 294 | - | ||
| contig39375 | 0 | 247 | 0 | 0 | 247 | GO:0003723 | aRNA binding | |
| contig50336 | 0 | 227 | 0 | 0 | 227 | - | ||
| contig54027 | 0 | 213 | 0 | 0 | 213 | GO:0006879 | bcellular iron ion homeostasis | |
| contig53611 | 0 | 191 | 0 | 0 | 191 | - | ||
| contig40384 | 0 | 190 | 0 | 0 | 190 | - | ||
| contig45190 | 0 | 190 | 0 | 0 | 190 | - | ||
| contig51482 | 0 | 182 | 0 | 0 | 182 | - | ||
| Adult specific (M and F) | ||||||||
| contig49608 | 71 | 189 | 0 | 0 | 260 | GO:0004531 | adeoxyribonuclease II activity | |
| contig47068 | 101 | 144 | 0 | 0 | 245 | GO:0004531 | adeoxyribonuclease II activity | |
| contig51608 | 96 | 133 | 0 | 0 | 229 | GO:0006508 | bproteolysis | |
| contig49469 | 76 | 141 | 0 | 0 | 217 | GO:0006508 | bproteolysis | |
| contig41687 | 76 | 124 | 0 | 0 | 200 | GO:0006508 | bproteolysis | |
| contig52673 | 129 | 69 | 0 | 0 | 198 | - | ||
| contig45734 | 176 | 22 | 0 | 0 | 198 | GO:0008289 | alipid binding | |
| contig41787 | 105 | 92 | 0 | 0 | 197 | GO:0005576 | cextracellular region | |
| contig45747 | 39 | 157 | 0 | 0 | 196 | GO:0005576 | cextracellular region | |
| contig09753 | 68 | 120 | 0 | 0 | 188 | GO:0006508 | bproteolysis | |
| IL3 specific | ||||||||
| contig20982 | 0 | 0 | 88 | 0 | 88 | - | ||
| contig53045 | 0 | 0 | 84 | 0 | 84 | GO:0004129 | acytochrome-c oxidase activity | |
| contig46922 | 0 | 0 | 82 | 0 | 82 | - | ||
| contig48104 | 0 | 0 | 76 | 0 | 76 | - | ||
| contig54731 | 0 | 0 | 75 | 0 | 75 | GO:0004129 | acytochrome-c oxidase activity | |
| contig20212 | 0 | 0 | 73 | 0 | 73 | - | ||
| contig52851 | 0 | 0 | 60 | 0 | 60 | GO:0004129 | acytochrome-c oxidase activity | |
| contig01666 | 0 | 0 | 54 | 0 | 54 | GO:0005506 | airon ion binding | |
| contig22515 | 0 | 0 | 46 | 0 | 46 | - | ||
| contig41986 | 0 | 0 | 43 | 0 | 43 | - | ||
| ssL3 specific | ||||||||
| contig43707 | 0 | 0 | 0 | 59 | 59 | - | ||
| contig54118 | 0 | 0 | 0 | 57 | 57 | - | ||
| contig21715 | 0 | 0 | 0 | 55 | 55 | - | ||
| contig42564 | 0 | 0 | 0 | 53 | 53 | - | ||
| contig47041 | 0 | 0 | 0 | 29 | 29 | GO:0008152 | bmetabolic process | |
| contig29443 | 0 | 0 | 0 | 25 | 25 | GO:0005525 | aGTP binding | |
| contig33913 | 0 | 0 | 0 | 24 | 24 | GO:0005576 | cextracellular region | |
| contig43357 | 0 | 0 | 0 | 24 | 24 | GO:0005576 | cextracellular region | |
| contig43120 | 0 | 0 | 0 | 23 | 23 | GO:0005576 | cextracellular region | |
| contig17582 | 0 | 0 | 0 | 23 | 23 | - | ||
| Larval specific (iL3 and ssL3) | ||||||||
| contig03786 | 0 | 0 | 465 | 200 | 665 | - | ||
| contig22000 | 0 | 0 | 331 | 119 | 450 | GO:0004289 | asubtilase activity | |
| contig00904 | 0 | 0 | 211 | 237 | 448 | - | ||
| contig20229 | 0 | 0 | 207 | 224 | 431 | GO:0003723 | aRNA binding | |
| contig46936 | 0 | 0 | 232 | 175 | 407 | GO:0003779 | aactin binding | |
| contig43418 | 0 | 0 | 285 | 104 | 389 | - | ||
| contig03955 | 0 | 0 | 274 | 111 | 385 | GO:0003735 | astructural constituent of ribosome | |
| contig52848 | 0 | 0 | 234 | 148 | 382 | - | ||
| contig02679 | 0 | 0 | 138 | 232 | 370 | GO:0004190 | aaspartic-type endopeptidase activity | |
| contig50665 | 0 | 0 | 221 | 146 | 367 | - | ||
| Constant expression | ||||||||
| contig54178 | 3 | 1148 | 13 | 11 | 1175 | - | ||
| contig53913 | 22 | 837 | 35 | 66 | 960 | GO:0008137 | aNADH dehydrogenase (ubiquinone) activity | |
| contig50938 | 362 | 7 | 382 | 191 | 942 | GO:0005215 | atransporter activity | |
| contig26411 | 105 | 359 | 166 | 200 | 830 | GO:0003824 | acatalytic activity | |
| contig51127 | 174 | 385 | 154 | 94 | 807 | GO:0005622 | cintracellular | |
| contig44592 | 170 | 36 | 264 | 286 | 756 | GO:0005515 | aprotein binding | |
| contig49300 | 138 | 222 | 241 | 154 | 755 | GO:0004365 | aglyceraldehyde-3-phosphate dehydrogenase (phosphorylating) activity | |
| contig43896 | 257 | 64 | 199 | 229 | 749 | GO:0004013 | aadenosylhomocysteinase activity | |
| contig40294 | 195 | 197 | 171 | 139 | 702 | GO:0004611 | aphosphoenolpyruvate carboxykinase activity | |
| contig40430 | 135 | 144 | 225 | 190 | 694 | GO:0031419 | acobalamin binding | |
a Molecular Function; b Biological Processes; c Cellular Component
Among the 401 transcripts turned on by serum stimulation, 13 of them are parasitism related (Figure 2). Given that there are a total of 562 parasitism related transcripts among all the 16,359 transcripts (whose expression selectivity could be confidently ascertained), there is no evidence to support that serum stimulation triggers an extensive expression of parasitism related genes. However, parasitism related genes are more selective expressed in adults. Nearly 18% of these genes (99 out of 562) exhibit M, F, or M and F selectivity, which is significant (P s < 1.0e-4, Chi-square test) when compared to the overall of 9% (1,505 out of 16,359; Figure 2). Interesting, compared to the small fraction of male specific transcripts related to parasitism (17/604), a large fraction of female specific transcripts (35 out of 213) are parasitism related.
The majority of nematode conserved transcripts (2,789 of 4,056) exhibited constant expression over all stages, while less than 40% (3,446 of 8,783) of the A. caninum specific transcripts exhibited the same expression pattern (Figure 2). This difference is highly significant statistically (P < 1.0E-10, Chi-square test). More than 80% of iL3 selective transcripts (66 out of 78) are A. caninum specific. The different expression pattern of conserved transcripts and A. caninum specific transcripts suggests caution in using cDNA data to estimate transcriptome diversity. Using limited number of cDNA reads can underestimate the diversity. For example, the homolog rate between A. caninum and C. elegans would be 54% ((8074-3446-304)/8074) if only the transcripts expressed constantly across the life cycle were considered while that is 20% when all transcripts are included.
The expression profiles defined by our sequencing were compared to the data published by Datu et al., [18]. Datu et al. studied transcriptional changes in the hookworm, A. caninum, during the transition from a free-living to a parasitic larva using suppression subtractive hybridization (SSH) and custom oligonucleotide microarray printed with the SSH expressed sequence tags. Comparison of the two expression profiles of the most highly up-regulated mRNAs associated with serum stimulation obtained by different orthogonal approaches confirmed consistency in expression of 9 out of the 10 up-regulated mRNA associated with serum stimulation. The only one not consistent is cDNA that has been broken into several transcripts in our assembly, therefore giving rise to this discrepancy (additional file 3).
Functional profile of transcripts with different expression selectivity
Examining the function of transcripts with different expression patterns using GO terms revealed that enriched and depleted GO terms correlate to the biology of the corresponding stages. The heatmaps of molecular function GO terms (Figure 3 and Figure 4; See additional file 4) shows that transcripts with different expression selectivity exhibit different functional profiles, although that of the transcripts selectively expressed in both iL3 and ssL3 is closer to that of those constantly expressed, and those of the transcripts selectively expressed in F, M, and both F and M look more similar. Significantly enriched GO terms in the category of molecular functions are shown in Table 4. The top three enriched GO terms of transcripts expressed constantly over all stages are zinc ion binding (GO:0008270, P = 7.4e-12), protein binding (GO:0005515, P = 1.8e-11), and nucleic acid binding (GO:0003676, P = 9.3e-11). The top three transcripts selectively expressed in both F and M are astacin activity (GO:0008533, P = 8.8e-17), a structural constituent of cuticle (GO:0042302, P = 3.5e-8), and cysteine-type endopeptidase activity (GO:0004197, P = 4.0e-11). These terms have all been previously associated with genes involved in parasitism [20-22]. The three most significant enriched GO terms of the selectively expressed transcripts in both iL3 and ssL3 are rhodopsin-like receptor activity (GO:0001584, P = 3.1e-9), N-acetyltransferase activity (GO:0008080, P = 6.6e-6), and sugar hydrogen symporter activity (GO:0005351, P = 4.5e-5). We were only able to detect 2 significant terms from the transcripts exclusively expressed in F: serine-type endopeptidase activity (GO:0004252, P = 3.8e-4) and glucosylceramidase activity (GO:0004348, P = 6.6e-11). Interesting, the most significant enriched GO term of the transcripts exclusively expressed in M is serine-type endopeptidase inhibitor activity (GO:0004867, P = 2.6e-8).
Figure 3.

Enriched A. caninum GO terms of differentially expressed transcripts. F: female; M: male; iL3: infective L3; ssL3: serum stimulated L3.
Figure 4.

Depleted A. caninum GO terms of differentially expressed transcripts. F: female; M: male; iL3: infective L3; ssL3: serum stimulated L3.
Table 4.
Enriched GO terms associated with transcripts with differential expression patterns
| Stage | GO id* | GO descriptor | A. caninum trancripts |
|---|---|---|---|
| Female & Male | |||
| GO:0003674 | Molecular Function | 198 | |
| GO:0004194 | pepsin A activity | 7 | |
| GO:0004197 | cysteine-type endopeptidase activity | 16 | |
| GO:0004245 | neprilysin activity | 6 | |
| GO:0004365 | glyceraldehyde-3-phosphate dehydrogenase (phosphorylating) activity | 4 | |
| GO:0004415 | hyalurononglucosaminidase activity | 2 | |
| GO:0004531 | deoxyribonuclease II activity | 4 | |
| GO:0004623 | phospholipase A2 activity | 3 | |
| GO:0004806 | triacylglycerol lipase activity | 8 | |
| GO:0005529 | sugar binding | 7 | |
| GO:0008270 | zinc ion binding | 22 | |
| GO:0008533 | astacin activity | 19 | |
| GO:0042302 | structural constituent of cuticle | 7 | |
| GO:0051287 | NAD binding | 6 | |
| Female | |||
| GO:0003674 | Molecular Function | 52 | |
| GO:0004252 | serine-type endopeptidase activity | 4 | |
| GO:0004348 | glucosylceramidase activity | 5 | |
| Male | |||
| GO:0003674 | Molecular Function | 168 | |
| GO:0003735 | structural constituent of ribosome | 27 | |
| GO:0004396 | hexokinase activity | 4 | |
| GO:0004672 | protein kinase activity | 23 | |
| GO:0004674 | protein serine/threonine kinase activity | 15 | |
| GO:0004867 | serine-type endopeptidase inhibitor activity | 13 | |
| GO:0005198 | structural molecule activity | 35 | |
| Infective L3 & serum stimulated L3 | |||
| GO:0003674 | Molecular Function | 1388 | |
| GO:0001584 | rhodopsin-like receptor activity | 43 | |
| GO:0003735 | structural constituent of ribosome | 89 | |
| GO:0003849 | 3-deoxy-7-phosphoheptulonate synthase activity | 4 | |
| GO:0003924 | GTPase activity | 23 | |
| GO:0004377 | glycolipid 2-alpha-mannosyltransferase activity | 4 | |
| GO:0004673 | protein histidine kinase activity | 5 | |
| GO:0005249 | voltage-gated potassium channel activity | 8 | |
| GO:0005351 | sugar:hydrogen symporter activity | 14 | |
| GO:0005525 | GTP binding | 57 | |
| GO:0008080 | N-acetyltransferase activity | 14 | |
| GO:0015035 | protein disulfide oxidoreductase activity | 7 | |
| GO:0016491 | oxidoreductase activity | 206 | |
| GO:0016564 | transcription repressor activity | 4 | |
| GO:0016616 | oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor | 27 | |
| GO:0016829 | lyase activity | 43 | |
| GO:0020037 | heme binding | 35 | |
| GO:0050662 | coenzyme binding | 44 | |
| Infective L3 | |||
| GO:0003674 | Molecular Function | 20 | |
| GO:0004129 | cytochrome-c oxidase activity | 3 | |
| GO:0005507 | copper ion binding | 3 | |
| GO:0020037 | heme binding | 3 | |
| Serum stimulated L3 | |||
| GO:0003674 | Molecular Function | 8 | |
| GO:0004806 | triacylglycerol lipase activity | 2 | |
| All stages | |||
| GO:0003674 | Molecular Function | 3052 | |
| GO:0003676 | nucleic acid binding | 558 | |
| GO:0003700 | transcription factor activity | 85 | |
| GO:0003707 | steroid hormone receptor activity | 30 | |
| GO:0003723 | RNA binding | 96 | |
| GO:0003924 | GTPase activity | 44 | |
| GO:0004000 | adenosine deaminase activity | 6 | |
| GO:0004674 | protein serine/threonine kinase activity | 95 | |
| GO:0004713 | protein tyrosine kinase activity | 77 | |
| GO:0005515 | protein binding | 381 | |
| GO:0008026 | ATP-dependent helicase activity | 33 | |
| GO:0008138 | protein tyrosine/serine/threonine phosphatase activity | 12 | |
| GO:0008270 | zinc ion binding | 258 | |
| GO:0008508 | bile acid:sodium symporter activity | 4 | |
| GO:0008705 | methionine synthase activity | 4 | |
| GO:0017111 | nucleoside-triphosphatase activity | 188 | |
| GO:0019787 | small conjugating protein ligase activity | 39 | |
| GO:0031072 | heat shock protein binding | 23 | |
| GO:0046983 | protein dimerization activity | 19 | |
* Only Go terms with more than 50 hits to Mol. Function are presented.
Cut-offs: FRD < 0.1; hypogeometrics test P < 0.05.
Secretory parasitism related genes in A. caninum
Next we attempted to identify transcripts encoding secreted proteins associated with parasitism. Of the 562 transcripts whose differential expression we were able to define (the top 60 differentially expressed A. caninum transcript with homology to B. malayi but not C. elegans are available as additional file 5), 112 were not expressed in the preparasitic iL3 stage but expressed in other stages (Figure 2), suggesting a potential role in parasitism. However, we were only able to detect a secretory signal peptide in 9 transcripts (See additional file 6). This low number might underestimate the true number due to the fragmented nature of our data. Another possibility is that some secretory proteins are released by alternate, poorly characterized secretory pathways, and would therefore be missed by searching for secretory peptide sequences, as was shown recently for B. malayi secreted proteins [23]. In addition, we found more than 700 transcripts with signal peptides from the transcripts that have neither B. malai nor C. elegans homologs (data not shown).
Intra-population polymorphism, Synonymous/non-synonymous Single Nucleotide Polymorphism and positive selection
Due to their high evolutionary rates [24,25], nematodes are believed to have a significant number of single nucleotide polymorphisms (SNPs). A large number of sites with SNP (76,568) were detected over the total 23,038,913 assembled bases (total length of the 48,326 transcripts). Since the average coverage of our transcripts is about 10×, we estimated θ (= 4Nμ) for A. caninum as 1.2 × 10-3. Among the SNP sites only 345 sites have more than two alleles. We obtained confident translations for 6,502 cDNAs containing 20,715 of the 76,568 SNPs. Of these, 10,848 were non-synonymous, and 9,867 were synonymous, with an average dN/dS ratio of 0.3. Among the 518 transcripts possessing more than 9 polymorphic sites, 39 were under positive selection (dN/dS > 1.0), and there were two functional categories identified by the associated GO terms: GO:0004298 (threonine endopeptidase activity and GO:0006511 (ubiquitin-dependent protein catabolic activity). Of these, one parasitism related transcript (contig43771 encoding a protein histidine kinase) exhibited more than 9 polymorphic sites and was under positive selection.
Discussion
The A. caninum transcriptome was sequenced with unprecedented coverage in the present study. While fragmentation is still an issue, the non-biased cloning-free transcript sampling using the Roche/454 technology combined with the conventional Sanger technology in this study enabled an in depth sampling of over 93% of the A. caninum transcriptome. Comparing the A. caninum transcriptome with the coding sequences of C. elegans and B. malayi confirmed the high diversity of nematode transcriptomes. Intra-species studies revealed high expression dynamics of the nematode transcriptome, and suggested an impact of the adaptation to parasitism on A. caninum genes and gene expression.
Nematodes have higher evolutionary rate than most other eukaryotes [24,25]. Only 20% of our A. caninum transcripts shared homology to C. elegans genes. Since A. caninum and C. elegans are from the same phylogenetic clade [12], this lowly shared homology clearly illustrates a high evolutionary rate. The high rate can lead to high polymorphism within species. Our estimation of θ for A. caninum is 1.2 × 10-3, which is about 2 times higher than that of human coding regions [26]. We may have significantly underestimated θ because each final transcript is probably derived from multiple individuals rather than single worms. This high DNA polymorphism is in agreement with the high evolutionary rate of A. caninum. Most SNPs are di-allelic just as detected in human, which suggest a similar mechanism shaping SNPs in both human and nematodes. High mutation rate and diversity are features of the phylum Nematoda. A previous survey of more than 30 nematode species distributed over four nematode clades found that only about 15% of sequenced ESTs could be found in all four nematode clades [27]. The same study suggested that about 30-50% of nematode genes are species specific. Interestingly, our previous studies [16,17], based on limited number of A. caninum genes (9,000 and 4,000 genes respectively), found that about 50% of the A. caninum genes had homologs in Caenorhabditis species. This discrepancy is likely because the conserved genes tend to be expressed at higher levels and therefore are sequenced more deeply. The previous transcriptome studies analyzed transcripts generated using conventional cDNA libraries (cloning based with capillary sequencing), therefore only the most abundant transcripts were represented in those studies. In fact, when only considering the highly expressed transcripts (at least 10 reads sequenced), the homologous rate between A.caninum and C. elegans is 43%.
The high evolutionary rate and diversity of nematodes may contribute to their ability to adapt to nearly every habitat on earth [28]. In addition, parasitism has evolved independently at least nine times in nematodes [29], and the evolution of parasitism plays a role in shaping the nematode transcriptome. The comparative genomic analysis showed that significantly more B. malayi coding genes share homology with A. caninum than with C. elegans. B. malayi is in clade III and both A. caninum and C. elegans are in clade V, thus it is expected that B. malayi share similar homologs with A. caninum and C. elegans without parasitism adaptation. Parasitic nematodes originated from non-parasites, with subsequent adaptation to the host environment. One major difference between free-living and host environments is the availability and abundance of oxygen. Intestinal parasites like hookworms must adapt to the low oxygen levels in their host by using alternative energy and metabolism pathways. They also need to develop systems to evade the host defense mechanisms. The enriched GO terms of the A. caninum transcripts that have homology with B. malayi genes but not with C. elegans reveal the effect of these adaptations. The most significantly enriched GO term is prolyl oligopeptidase activity. Prolyl oligopeptidase is a family of serine-type endopeptidases [30]. One of its members in the parasitic kinetoplastid, Trypanosoma cruzi, is critical for the parasite to invade mammalian host cells [31]. Another highly represented transcript is malic enzyme. Malic enzyme converts malate to pyruvate in the mitochondrion, and is important for adaptation to low oxygen environment in the host [32]. In addition, transcripts encoding nucleic acid binding and DNA binding proteins were also significantly enriched. It is possible that parasitic nematodes interfere with the host's transcription and translation system during invasion, or these transcripts encode endogenous enzymes required for further development and morphological changes that occur in the host. In contrast to the GO term enrichment of these A. caninum transcripts, the B. malayi genes sharing homology with C. elegans only did not exhibit any GO term enrichment.
Despite the strong adaptive capability of nematodes, we failed to find evidence of strong positive selection in A. caninum. Only 7.5% of transcripts are under possible positive selection by the dN/dS test. We also failed to detect an extensive positive selection for the parasitism related genes. The lack of extensive positive selection in nematodes could suggest that these organisms have a high mutation rate. We expect our on-going nematode genomic projects to provide additional information about nematode evolution http://www.genome.gov/10002154.
Gene expression in A. caninum is highly dynamic, with only half of the genes being constantly expressed over all four stages. This 50% is actually a conservative estimation because we excluded the transcripts that lacked sequencing depth. The most drastic difference is found among expression profiles of larvae and adults, indicating the strong effect of adaptation to the host environment. This is confirmed by the functional GO term enrichments. Genes selectively expressed in parasitic adults (both in F and M) are enriched in functions important for parasitism. In fact, 10 out of the 14 GO terms enriched in parasitic adults are related to parasitism. Cysteine endopeptidase is indispensable for parasites, and its numerous functions enabling parasites to defend against their hosts are well-documented [20,21]. Cysteine proteases are also important for digestion of protein and hemoglobin in the blood meal [33]. Astacin plays a crucial role in A. caninum tissue migration [22]. The cuticle is a protective external layer of nematodes that provides the primary defense for parasitic nematodes. Several cuticlar changes associated with parasitism have been documented [34]. The glyceraldehyde-3-phosphate dehydrogenase enzyme is necessary for parasites to utilize host glucose as an energy source. In contrast, genes selectively expressed in larvae tend to be enriched in functions related to basic cellular functions such as oxidoreductase activity, signal transduction, and ribosome structure. The most significant term (based on P-value of GO enrichment test) is rhodopsin-like receptor activity, which are chemosensory receptors in C. elegans and may be required for larval nematodes to interact with their free-living environment.
Surprisingly, only a small number of genes were turned on by serum stimulation, which indicate that exposure to host-like conditions in vitro does not trigger immediate gene expression changes. Also, serum stimulation did not turn on expression of an increased number of parasitism related genes in our study. This suggests that hookworms do not drastically change gene expression during the transition to parasitism upon entering their hosts, i.e. many of the molecules that are released during infections are already synthesized in the iL3 and stored for rapid release during infection [35-37]. However, using differential subtractive hybrization, Datu et al [18] found that the genes expressed in serum stimulated A. caninum L3 did not overlap with genes associated with developmental changes during recovery of C. elegans dauers, and suggested that genes expressed in response to activation with serum were involved in parasitism rather than development [18]. One transcript selectively expressed in ssL3 stage in our study encoded triacylglycerol lipase, which is known to play a role during dauer recovery in C. elegans [38]. Invading hookworm L3 have been compared to recovering dauer larvae [39]. Another group of transcripts expressed selectively in ssL3 versus iL3 are the allergen V5/Tpx-1 related proteins or Ancylostoma secreted proteins (ASPs), originally isolated from excretory/secretory products of A. caninum L3 [35], and subsequently from numerous other nematodes. While its function is unknown, a second, related set of ASPs have been described from the adult stages [40], and were among the most abundant transcripts in adult M and F in this study (Table 3). Overall, expression dynamics correlated with progression through the stages of the life cycle.
Genes with different evolutionary conservation exhibit different expression pattern. Genes conserved among nematodes tend to be expressed consistently in all stages, and A. caninum specific genes tend to be expressed more selectively. These expression patterns can be associated with their corresponding functions. Conserved genes are involved in basic cellular activities and thus are required for all stages. Species-specific genes are functionally more closely related to the specific life cycle stage and niche. For example, the iL3 selective transcripts are mainly A. caninum specific and are depleted of primary metabolic process (GO:0044238) and macromolecule metabolic process (GO:0043170) (data not shown). In addition, the parasitism related genes are more like to be selectively expressed in adult stages. One would expect this since the adults are parasitic whereas the infective larvae are usually free-living. We found that the male adult differentially expressed transcripts are enriched in serine-type endopeptidase inhibitor activity. The male reproductive tract of mammals is enriched in peptidase inhibitors that function in protection and regulation of fertilization [41], suggesting a similar mechanism may be at work in nematodes.
Conclusion
In summary, this study allowed generation and cataloging of all genes expressed in four transcriptomes of A. caninum. Our analysis segregated those genes in multiple dimensions including functional, developmental and phylogenetic categories. The observations agreed with, but also extended, information on previously described genes, suggesting that the newly discovered genes will provide additional unique value. This information identified sets of stage-specific genes, as well as pre-parasitic and parasitic genes that defined differences in metabolic and cellular processes between stages. Furthermore, parasitic adaptation has been shown to be related to transcriptome diversity and developmental dynamics. This dataset is a resource for more complete microarrays, RT-PCR, RNA interference and proteomics. The latter can identify parasite proteins that occur in specific developmental stages, parasite excretory-secretory products, and the external cuticular surface. In addition, the cDNAs generated will enable better annotation of the upcoming genome sequence http://www.genome.gov/10002154. Such extended genomic studies will aid in the identification of genes involved in host recognition, infection, migration and immune invasion as well as the categorization of targets for vaccine and anthelmintic drugs. Finally, the methodology developed in this study illustrates the effectiveness of deep sequencing as a means for analyzing differential gene expression.
Methods
Nematode extraction
The Baltimore strain of A. caninum (U.S. National Parasite Collection accession 100655.00) was maintained in beagles as described [42]. Animals were housed and treated in accordance with George Washington University Institutional Care and Use Committee guidelines. Infective L3 (iL3) were recovered from 7-10 day old coprocultures using a modified Baermann technique, washed clean of debris with BU buffer (50 mM Na2PO4/22 mM KH2PO4/70 mM NaCl, pH 6.8; Hawdon et al., 1991), and treated with 1% HCl in BU for 30 min at 22°C. The larvae were washed twice with sterile BU and snap-frozen by immersion in liquid N2. Frozen larvae were stored at -80°C until used for library construction. Activated (serum stimulated) larvae were generated as described previously (Brand et al, 2004). Briefly, approximately 5,000 A. caninum L3 were incubated in a 500 μl volume of RPMI1640 medium supplemented with 25 mM HEPES (pH 7.0) and antibiotics (RPMI-complete) containing 10% canine serum filtrate (<10 kDa ultrafiltrate) and 15 mm S-methyl-glutathione (Sigma Chemical). Negative-control (non-activated) L3 were incubated in RPMI-c alone. L3 were incubated in 10% CO2 at 37°C for 24 hours. Following incubation, the medium containing the L3 was collected, transferred to microcentrifuge tubes and centrifuged for 5 minutes at 14,000 rpm. The supernatant was removed, and an aliquot of L3 was tested for activation as described previously [43]. L3 pellets showing greater than 85% feeding were pooled and used to isolate nucleic acids. Adult male and female were collected from intestines of infected dogs following euthanasia, and frozen at -80 C until nucleic acids were isolated.
Preparation of A. caninum staged RNA and cDNA libraries
Frozen worm pellets were pulverized using an Alloy Tool Steel Set (Fisher Scientific International). Total RNA from adult and larval parasites was prepared using TRIzol Reagent (GibcoBRL, Life Technologies or Invitrogen, Carlsbad, CA). cDNA libraries from four stages, infective L3 larva (iL3), activated L3 larva (ssL3), adult male (M), and female (F), were generated as previously described [17,44].
Capillary and high throughput sequencing
The cDNA libraries from the four stages were sequenced using the Roche/454 FLX platform [44] and capillary based Sanger sequencing using ABI 3730 and 3700 platforms [17]. All sequences were deposited to GenBank: accession numbers of the Sanger sequences are BM077300 - BM077991; EW741128 - EW744730; EX534506 - EX567272; EX827505 - EX828593; EY458148 - EY473938; FC539038 - FC555743; the Roche/454 SFF files can be found in SRA, libraries SRX000115-SRX000118. The sequences from different platforms underwent different methods for base calling and detection of high quality regions, trimming of linkers, screening for low complexity regions and contaminants, and returning high-quality sequences. Raw cDNA sequences were processed i.e. quality trimmed and screened for vector sequences using SeqClean http://compbio.dfci.harvard.edu/tgi/. The hybrid assembly was dominated by FLX reads (sff format files), therefore we used the 454 Life Sciences' Newbler assembler v1.1.03.21 followed by addition of the ABI reads. All subsequent analyses were based on these contigs, hereafter referred as transcripts, and their constituent reads.
Comparative analysis and functional assignments
The core eukaryotic genes (CEGs)[45] were used to estimate the completeness of the A. caninum transcriptome. A hidden Markov model profile search of the 48,326 transcripts against the 248 CEG profiles of C. elegans genes was carried out using the HMMER [46]. Significant hits were identified according to the suggested cutoffs [45].
C. elegans and B. malayi coding genes were downloaded from Biomart [47]. The A. caninum transcripts were compared against these coding genes using WU-BLAST to identify homologs and matches with a raw BLAST bitscore larger than 50 were considered significant homologs [27]. By this way, A. caninum transcripts could be classified in 4 groups: those sharing homology with both C.elegans and B. malayi genes, those sharing homology with only C. elegans genes or only B. malayi genes, and those sharing no homology with any other species. We defined the first group as nematode conserved, the last group as A. caninum specific, and those sharing homology only with B. malayi genes as parasitism related. KEGG orthology (KO)[48] of the transcripts were identified through BLAST searching against the KEGG database. As with the homologs, bitscores larger than 50 were used as a cut-off. The recorded KOs were mapped to the cellular and biochemical pathways using the KEGG reference maps [48]. The number of shared and unique KOs for each pathway was compared and statistically evaluated by Chi-square test with Bonferroni correction.
Gene Ontology (GO) associations of the parasitism related transcripts were performed using Interproscan [49], and significant enrichment of GO terms were computed based on the hypergeometric distribution using FUNC [50]. A probability refinement was done to remove the GO terms identified as significant due to their children terms. GO term enrichment analysis is vulnerable to false discovery. We used the false discovery rate (FDR) computed by FUNC to reduce false discovery. Therefore, unless specified otherwise, the GO term enrichment was selected based on both p-value < 0.05 (after refinement) and FDR <0.1.
Expression pattern examination
Expression patterns were defined by stage specificity and stage bias of the transcript's constituent reads. After assembly, transcripts with length greater than 90 base pairs (bp) were subjected to expression pattern examination as follows: the numbers of reads originating from the different cDNA libraries for each transcript was recorded, and transcripts with reads originating from only one stage (or a set of stages) were designated to be specific to that stage (or that set of stages). To increase the confidence with which specificity was assigned, we required the transcripts to be sequenced deeply enough to ensure that an observed stage absence (i.e. 0 reads from a specific stage) have a confidence interval above 95%. It turned out that this required the transcript be sequenced at least 10 times (i.e. transcripts have at least 10 constituent reads). Specifically, for each transcript, its observed absence from a stage (if any) was compared to its expected number of stage-specific reads (calculated by multiplying the marginal read frequency of the four stages by the total number of reads in that transcript). If the probability of the observed absence (to a Poisson distribution with the mean as the expected number of reads) was higher than 0.05, the transcript was considered to lack depth of coverage and was excluded from the analysis (including the function-related analyses). This requirement excluded 32,099 transcripts, leaving 16,359 transcripts with defined stage-specificity (with high confidence).
Stage-specific expression implies that a specific gene is expressed during one developmental stage (or one set of development stages) but not in the other. However, gene expression does not always follow the on/off model. Its expression dynamics also includes the expression fluctuation in different stages. In some stages one gene may have enhanced expression while it may have depleted expression in other stages. Such transcripts were designated as stage-biased in our analysis. These transcripts that were biased in expression towards a certain stage were selected by comparing the numbers of reads in different stages (iL3, ssL3, and Adult) using a statistical approach defined by Audic et al. [51] with a significance of P < 1e-05. We focused on the comparisons of iL3 vs ssL3 and ssL3 vs Adult (male and females were treated together as adult stage).
Functional examination of transcripts with different expression pattern
The predicted functions of the transcripts with different expression patterns were examined with GO association as described previously. In consideration of the amount of data and parasitism, we focused on 5 groups of transcripts: those constantly expressed in all four stages; those selectively expressed in both iL3 and ssL3; those exclusively expressed in F, those exclusively expressed in M; and those selectively expressed in both F and M. To visualize the functional profile of these groups, we used heatmap to illustrate the enrichment and depletion of GO terms related to molecular function. To increase the visibility, we included only the GO terms that are enriched or depleted in at least one of the 5 groups. Meanwhile, significantly enriched GO terms in molecular function of these groups were recorded and manually examined to explore the functional association of selective expression.
Identification of secretory parasitism related genes
Upon the identification of parasitism related genes, several criteria were required to further identify parasitism genes encoding secretory proteins [52] that the parasite might use to interfere with the host cellular functions and enable successful parasitism. These criteria require the genes i) to share homology to B. malayi genes but not C. elegans (are in the subset of parasitism related genes identified above), ii) are expressed in the parasitic stages (ssL3, M, F) but should not be expressed in the preparasitic iL3 stage; iii) to have signal peptide for secretion (signal peptides for secretions and trans-membrane domains were identified using PHOBIUS [53], with an additional requirement that the SP is within the first 70 amino acids). Hence, the transcripts expressed in parasitic stages, without homologs in C. elegans but with a significant homology to B. malayi genes and with a signal peptide for secretion but without membrane containing domains were classified as putative secretory parasitism genes. The functions of these transcripts were assigned based on homology found by InterProScan [49].
Intra-population polymorphism and synonymous and non-synonymous Single Nucleotide Polymorphisms (SNPs)
The cDNAs generated in this study originate from RNA isolated from a population of individuals, enabling us to estimate the rate of population polymorphism of the transcriptomic reads. We used Polybayes [54] to detect the SNPs by using the transcript (i.e. contigs) as reference. Based on the detected SNPs, we estimated the θ, a representation of DNA sequence polymorphism which is related to the mutation rate and effective population size, by 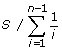 following Watterson [55], where n = 10 (the estimated coverage depth) and S is the fraction of sites with polymorphism. By treating the estimated coverage depth as n, we have likely underestimated θ. To identify if the SNPs contribute to synonymous or non-synonymous changes the transcripts were translated using Prot4Est [56]. The dN and dS were calculated using the method of Nei and Gojobori [57]. To improve reliability of the estimation, only transcripts with more than 9 polymorphic sites were analyzed. Functions of transcripts under positive selection (dN/dS > 1.0) were further investigated for GO term enrichment (as stated above).
following Watterson [55], where n = 10 (the estimated coverage depth) and S is the fraction of sites with polymorphism. By treating the estimated coverage depth as n, we have likely underestimated θ. To identify if the SNPs contribute to synonymous or non-synonymous changes the transcripts were translated using Prot4Est [56]. The dN and dS were calculated using the method of Nei and Gojobori [57]. To improve reliability of the estimation, only transcripts with more than 9 polymorphic sites were analyzed. Functions of transcripts under positive selection (dN/dS > 1.0) were further investigated for GO term enrichment (as stated above).
Abbreviations
L3: third stage larvae; iL3: infective L3; ssL3: serum stimulated L3; F: female; M: male; Ad: adult; cDNA: complementary DNA; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; KO: KEGG Ontology; SNP: Single Nucleotide Polymorphism; dN: non-synonymous polymorphism; dS: synonymous polymorphism.
Authors' contributions
ZW and MM conceived and designed the experiments. JH provided the worms/RNA. JM, SA and ZW carried out experiments and analyses. ZW, RKW, JH and MM interpreted results and prepared the manuscript. All authors have read and approved the final manuscript.
Supplementary Material
Coverage of the A. caninum transcriptome based on low copy number conserved eukaryotic genes.
The top 20 most abundant stage-specific and constantly expressed A. caninum genes.
Stage-biased differential expression of A. caninum genes. A). Comparison among preparasitic and parasitic larval stages, and parasitic larval and adult stage. B) Comparison of expression profiles generated by suppression subtractive hybridization and cDNA sequencing of stage-specific libraries.
Functional enrichment and depletion score derived from the enrichment and depletion probabilities calculated by hypogeometrics test of molecular functional GO terms.
The top 10 most abundant stage-specific and constantly expressed A. caninum genes having homology to B. malayi but not C. elegans.
Putative A.caninum parasitic genes and their functions.
Contributor Information
Zhengyuan Wang, Email: wang@watson.wustl.edu.
Sahar Abubucker, Email: sabubuck@wason.wustl.edu.
John Martin, Email: jmartin@watson.wustl.edu.
Richard K Wilson, Email: rwilson@watson.wustl.edu.
John Hawdon, Email: mtmjmh@gwumc.edu.
Makedonka Mitreva, Email: mmitreva@watson.wustl.edu.
Acknowledgements
We would like to thank the dedicated members of the cDNA production group at the Washington University's Genome Center and to all authors of the numerous algorithms used to perform the analysis. The cDNA analysis and the Nematode.net at the Genome Center is supported by the US National Institute for Allergy and Infectious Disease grant to M.M and the National Human Genome Research Institute grant to RKW.
References
- Aguero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, Campbell RK, Carmona S, Carruthers IM, Chan AWE, Chen F, Crowther GJ, Doyle MA, Hertz-Fowler C, Hopkins AL, McAllister G, Nwaka S, Overington JP, Pain A, Paolini GV, Pieper U, Ralph SA, Riechers A, Roos DS, Sali A, Shanmugam D, Suzuki T, Van Voorhis WC, Verlinde C. Genomic-scale prioritization of drug targets: the TDR Targets database. Nature Reviews Drug Discovery. 2008;7(11):900–907. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Martin J, Abubucker S, Wang Z, Wyrwicz L, Rychlewski L, McCarter J, Wilson R, Mitreva M. Molecular determinants archetypical to the phylum Nematoda. BMC Genomics. 2009;10(1):114. doi: 10.1186/1471-2164-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Martin J, Abubucker S, Yin Y, Gasser R, Mitreva M. Systematic analysis of insertions and deletions specific to nematode proteins and their proposed functional and evolutionary relevance. BMC Evol Biol. 2009;9(1):23. doi: 10.1186/1471-2148-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, Diemert D, Hotez PJ. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. The Lancet. 2006;367(9521):1521–1532. doi: 10.1016/S0140-6736(06)68653-4. [DOI] [PubMed] [Google Scholar]
- Liu C, Zhang X, Qiu D, Xiao S, Hotez PJ, Zhen D, Zhen H, Li M, Ren H, Zhan B, Xue H, Hawdon J, Feng Z. Epidemiology of human hookworm infections among adult villagers in Hejiang and Santai Counties, Sichuan Province, China. Acta Trop. 1999;73(3):243–249. doi: 10.1016/S0001-706X(99)00035-2. [DOI] [PubMed] [Google Scholar]
- de Silva NR, Brooker S, Hotez PJ, Montresor A, Engels D, Savioli L. Soil-transmitted helminth infections: updating the global picture. Trends in Parasitology. 2003;19(12):547–551. doi: 10.1016/j.pt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Diemert DJ, Bethony JM, Hotez PJ. Hookworm vaccines. Clin Infect Dis. 2008;46(2):282–288. doi: 10.1086/524070. [DOI] [PubMed] [Google Scholar]
- Blaxter M. Genes and genomes of Necator americanus and related hookworms. Int J Parasitol. 2000;30(4):347–355. doi: 10.1016/S0020-7519(99)00198-8. [DOI] [PubMed] [Google Scholar]
- Hotez PJ, Hawdon JM, Cappello M, Jones BF, Ghosh K, Volvovitz F, ShuHua X. Molecular approaches to vaccinating against hookworm disease. Pediatric Research. 1996;40(4):515–521. doi: 10.1203/00006450-199610000-00001. [DOI] [PubMed] [Google Scholar]
- Schad GA. In: Hookworm Disease Current Status and New directions. Schad GA, Warren KS, editor. London: Taylor and Francis; 1990. Hypobiosis and related phenomena in hookworm infection; pp. 71–88. [Google Scholar]
- Stone WM, Girardeau M. Transmammary passage of Ancylostoma caninum larvae in dogs. J Parasitol. 1968;54(3):426–429. doi: 10.2307/3277058. [DOI] [PubMed] [Google Scholar]
- Blaxter ML, De Ley P, Garey JR, Liu LX, Scheldeman P, Vierstraete A, Vanfleteren JR, Mackey LY, Dorris M, Frisse LM, Vida JT, Thomas WK. A molecular evolutionary framework for the phylum Nematoda. Nature. 1998;392(6671):71–75. doi: 10.1038/32160. [DOI] [PubMed] [Google Scholar]
- Consortium TCeS. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282(5396):2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, Allen JE, Delcher AL, Guiliano DB, Miranda-Saavedra D, Angiuoli SV, Creasy T, Amedeo P, Haas B, El-Sayed NM, Wortman JR, Feldblyum T, Tallon L, Schatz M, Shumway M, Koo H, Salzberg SL, Schobel S, Pertea M, Pop M, White O, Barton GJ, Carlow CKS, Crawford MJ, Daub J, Dimmic MW, Estes CF, Foster JM, Ganatra M, Gregory WF, Johnson NM, Jin J, Komuniecki R, Korf I, Kumar S, Laney S, Li BW, Li W, Lindblom TH, Lustigman S, Ma D, Maina CV, Martin DMA, McCarter JP, McReynolds L, Mitreva M, Nutman TB, Parkinson J, Peregrin-Alvarez JM, Poole C, Ren Q, Saunders L, Sluder AE, Smith K, Stanke M, Unnasch TR, Ware J, Wei AD, Weil G, Williams DJ, Zhang Y, Williams SA, Fraser-Liggett C, Slatko B, Blaxter ML, Scott AL. Draft genome of the filarial nematode parasite brugia malayi. Science. 2007;317(5845):1756–1760. doi: 10.1126/science.1145406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaxter M. Caenorhabditis elegans is a nematode. Science. 1998;282(5396):2041–2046. doi: 10.1126/science.282.5396.2041. [DOI] [PubMed] [Google Scholar]
- Abubucker S, Martin J, Yin Y, Fulton L, Yang SP, Hallsworth-Pepin K, Johnston JS, Hawdon J, McCarter JP, Wilson RK, Mitreva M. The canine hookworm genome: Analysis and classification of Ancylostoma caninum survey sequences. Molecular and Biochemical Parasitology. 2008;157(2):187–192. doi: 10.1016/j.molbiopara.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitreva M, McCarter JP, Arasu P, Hawdon J, Martin J, Dante M, Wylie T, Xu J, Stajich JE, Kapulkin W, Clifton SW, Waterston RH, Wilson RK. Investigating hookworm genomes by comparative analysis of two Ancylostoma species. BMC Genomics. 2005;6(58) doi: 10.1186/1471-2164-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datu BJD, Gasser RB, Nagaraj SH, Ong EK, O'Donoghue P, McInnes R, Ranganathan S, Loukas A. Transcriptional Changes in the Hookworm, Ancylostoma caninum, during the Transition from a Free-Living to a Parasitic Larva. PLoS Neglected Tropical Diseases. 2008;2(1) doi: 10.1371/journal.pntd.0000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J, Abubucker S, Wylie T, Yin Y, Wang Z, Mitreva M. Nematode.net update 2008: improvements enabling more efficient data mining and comparative nematode genomics. Nucl Acids Res. 2009;37(suppl_1):D571–578. doi: 10.1093/nar/gkn744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CD, Coombs GH, North MJ, Mottram JC. Parasite cysteine proteinases. Perspect Drug Discov Des. 1996;6:99–118. doi: 10.1007/BF02174048. [DOI] [Google Scholar]
- Sajid M, McKerrow JH. Cysteine proteases of parasitic organisms. Molecular and Biochemical Parasitology. 2002;120(1):1–21. doi: 10.1016/S0166-6851(01)00438-8. [DOI] [PubMed] [Google Scholar]
- Williamson AL, Lustigman S, Oksov Y, Deumic V, Plieskatt J, Mendez S, Zhan B, Bottazzi ME, Hotez PJ, Loukas A. Ancylostoma caninum MTP-1, an Astacin-Like Metalloprotease Secreted by Infective Hookworm Larvae, Is Involved in Tissue Migration. Infect Immun. 2006;74(2):961–967. doi: 10.1128/IAI.74.2.961-967.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno Y, Geary TG. Stage- and gender-specific proteomic analysis of Brugia malayi excretory-secretory products. PLoS Neglected Tropical Diseases. 2008;2(10):e326. doi: 10.1371/journal.pntd.0000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushegian AR, Garey JR, Martin J, Liu LX. Large-scale taxonomic profiling of eukaryotic model organisms: A comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res. 1998;8(6):590–598. doi: 10.1101/gr.8.6.590. [DOI] [PubMed] [Google Scholar]
- Aguinaldo AMA, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature. 1997;387(6632):489–493. doi: 10.1038/387489a0. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22(3):231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Parkinson J, Mitreva M, Whitton C, Thomson M, Daub J, Martin J, Schmid R, Hall N, Barrell B, Waterston RH, McCarter JP, Blaxter ML. A transcriptomic analysis of the phylum Nematoda. Nat Genet. 2004;36(12):1259–1267. doi: 10.1038/ng1472. [DOI] [PubMed] [Google Scholar]
- Andrassy I. Evolution as a basis for the systematization of nematodes. London: Pitman Publishing Ltd; 1976. [Google Scholar]
- Dorris M, De Ley P, Blaxter ML. Molecular analysis of nematode diversity and the evolution of parasitism. Parasitology Today. 1999;15(5):188–193. doi: 10.1016/S0169-4758(99)01439-8. [DOI] [PubMed] [Google Scholar]
- Polgar L. The prolyl oligopeptidase family. Cell Mol Life Sci. 2002;59(2):349–362. doi: 10.1007/s00018-002-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana JM, Grellier P, Schrevel J, Teixeira ARL. A Trypanosoma cruzi-secreted 80 kDa proteinase with specificity for human collagen types I and IV. Biochem J. 1997;325:129–137. doi: 10.1042/bj3250129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saz HJ. Energy Metabolisms of Parasitic Helminths: Adaptations to Parasitism. Annu Rev Physiol. 1981;43(1):323–341. doi: 10.1146/annurev.ph.43.030181.001543. [DOI] [PubMed] [Google Scholar]
- Williamson AL, Brindley PJ, Abbenante G, Prociv P, Berry C, Girdwood K, Pritchard DI, Fairlie DP, Hotez PJ, Dalton JP, Loukas A. Cleavage of hemoglobin by hookworm cathepsin D aspartic proteases and its potential contribution to host specificity. FASEB J. 2002. 02-0181fje. [DOI] [PubMed]
- Giblin-Davis RM, Davies KA, Williams DS, Center TD. Cuticular changes in fergusobiid nematodes associated with parasitism of fergusoninid flies. Comp Parasitol. 2001;68(2):242–248. [Google Scholar]
- Hawdon JM, Jones BF, Hoffman DR, Hotez PJ. Cloning and characterization of Ancylostoma-secreted protein - A novel protein associated with the transition to parasitism by infective hookworm larvae. J Biol Chem. 1996;271(12):6672–6678. doi: 10.1074/jbc.271.12.6672. [DOI] [PubMed] [Google Scholar]
- Hawdon JM, Narasimhan S, Hotez PJ. Ancylostoma secreted protein 2. cloning and characterization of a second member of a family of nematode secreted proteins from Ancylostoma caninum. Molecular and Biochemical Parasitology. 1999;99(2):149–165. doi: 10.1016/S0166-6851(99)00011-0. [DOI] [PubMed] [Google Scholar]
- Zhan B, Hotez PJ, Wang Y, Hawdon JM. A developmentally regulated metalloprotease secreted by host-stimulated Ancylostoma caninum third-stage infective larvae is a member of the astacin family of proteases. Molecular and Biochemical Parasitology. 2002;120(2):291–296. doi: 10.1016/S0166-6851(01)00453-4. [DOI] [PubMed] [Google Scholar]
- Selch F, Higashibata A, Imamizo-Sato M, Higashitani A, Ishioka N, Szewczyk NJ, Conley CA. Genomic response of the nematode Caenorhabditis elegans to spaceflight. Advances in Space Research. 2008;41(5):807–815. doi: 10.1016/j.asr.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawdon JM, Schad GA. Albumin and a dialyzable serum factor stimulate feeding invitro by 3rd-stage larvae of the canine hookworm Ancylostoma-caninum. J Parasitol. 1991;77(4):587–591. doi: 10.2307/3283164. [DOI] [PubMed] [Google Scholar]
- Zhan B, Liu Y, Badamchian M, Williamson A, Feng J, Loukas A, Hawdon JM, Hotez PJ. Molecular characterisation of the Ancylostoma-secreted protein family from the adult stage of Ancylostoma caninum. Int J Parasitol. 2003;33(9):897–907. doi: 10.1016/S0020-7519(03)00111-5. [DOI] [PubMed] [Google Scholar]
- Murer V, Spetz JF, Hengst U, Altrogge LM, de Agostini A, Monard D. Male fertility defects in mice lacking the serine protease inhibitor protease nexin-1. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(6):3029–3033. doi: 10.1073/pnas.051630698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawdon JM, Datu B, Crowell M. Molecular cloning of a novel multidomain Kunitz-type proteinase inhibitor from the hookworm Ancylostoma caninum. J Parasitol. 2003;89(2):402–407. doi: 10.1645/0022-3395(2003)089[0402:MCOANM]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Brand A, Hawdon JM. Phosphoinositide-3-OH-kinase inhibitor LY294002 prevents activation of Ancylostoma caninum and Ancylostoma ceylanicum third-stage infective larvae. Int J Parasitol. 2004;34(8):909–914. doi: 10.1016/j.ijpara.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Mitreva M, Mardis ER. Large-scale sequencing and analytical processing of ESTs. Methods in Molecular Biology. 2009;533:1–35. doi: 10.1007/978-1-60327-136-3_8. full_text. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Ning Z, Keane T, Korf I. Assessing the gene space in draft genomes. Nucl Acids Res. 2009;37(1):289–297. doi: 10.1093/nar/gkn916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14(9):755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A. BioMart - biological queries made easy. BMC Genomics. 2009;10(1):22. doi: 10.1186/1471-2164-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Research. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Prufer K, Muetzel B, Do HH, Weiss G, Khaitovich P, Rahm E, Paabo S, Lachmann M, Enard W. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics. 2007;8(41) doi: 10.1186/1471-2105-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Davis EL, Hussey RS, Baum TJ, Bakker J, Schots A, Rosso MN, Abad P. Nematode parasitism genes. Annu Rev Phytopathol. 2000;38(1):365. doi: 10.1146/annurev.phyto.38.1.365. [DOI] [PubMed] [Google Scholar]
- Kall L, Krogh A, Sonnhammer ELL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338(5):1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Marth GT, Korf I, Yandell MD, Yeh RT, Gu ZJ, Zakeri H, Stitziel NO, Hillier L, Kwok PY, Gish WR. A general approach to single-nucleotide polymorphism discovery. Nat Genet. 1999;23(4):452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7(2):256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Wasmuth JD, Blaxter ML. Prot4EST: Translating Expressed Sequence Tags from neglected genomes. BMC Bioinformatics. 2004;5 doi: 10.1186/1471-2105-5-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3(5):418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Coverage of the A. caninum transcriptome based on low copy number conserved eukaryotic genes.
The top 20 most abundant stage-specific and constantly expressed A. caninum genes.
Stage-biased differential expression of A. caninum genes. A). Comparison among preparasitic and parasitic larval stages, and parasitic larval and adult stage. B) Comparison of expression profiles generated by suppression subtractive hybridization and cDNA sequencing of stage-specific libraries.
Functional enrichment and depletion score derived from the enrichment and depletion probabilities calculated by hypogeometrics test of molecular functional GO terms.
The top 10 most abundant stage-specific and constantly expressed A. caninum genes having homology to B. malayi but not C. elegans.
Putative A.caninum parasitic genes and their functions.