

Abstract
Background
Polymorphisms in carcinogen detoxification enzymes, NAT2 and GSTM1, have been suggested as susceptibility factors for DNA damage and lung cancer. However, little information is available on DNA adduct burden in the lung tissue and polymorphisms in NAT2 and GST genes. We investigated the independent and combined effects of the metabolic gene polymorphisms of NAT2 and GSTs on DNA adduct formation in different tissues (lung and blood) in lung cancer patients.
Methods
DNA adducts were measured in lung and blood by the 32P-postlabeling assay. Multiple regression models were used to assess adjusted percent change in DNA adduct levels associated with GST and NAT2 genotypes.
Results
After adjusting for potential confounders, as well as for other GST gene variants, lung adduct levels significantly increased by 150.3% (95% CI, 35.4% to 362.6%) for the GSTM1 null and by 73.9% (95% CI, -3.2% to 212.4%) for NAT2 slow acetylator genotype, respectively. No association was seen with polymorphisms of other GST genes such as GSTT1 and GSTP1. The high-risk group, the combined GSTM1 null plus NAT2 slow, had significantly enhanced levels of lung adducts by 295% (95% CI, 72.7% to 803.5%) over those associated with single genes, suggesting a synergistic effect on DNA damage in the target lung tissue.
Conclusions
Increased DNA adduct levels in lung associated with the GSTM1 null and NAT2 slow genotypes alone or in combination.
Impact
These results suggest that GSTM1 and NAT2 genotypes play an independent and interactive role in the formation of carcinogen DNA adduct in the lung.
Keywords: DNA adducts, genetic polymorphisms, molecular epidemiology, lung cancer
Introduction
DNA damage is considered an important step in the carcinogenic process (1-3). Exposure to mutagenic carcinogens such as tobacco-derived aromatic amines (AA), polycyclic aromatic hydrocarbons (PAHs) and their derivatives, nitro-PAHs, can cause DNA adduct formation after metabolic activation (4-8). These carcinogen DNA adducts occur in the early stage of mutation and, when the damage is excessive and left unrepaired, may ultimately lead to induction of lung cancer (9, 10).
Inherited differences in the metabolic capacity of carcinogens can play a role in DNA damage, and therefore increase the risk of lung cancer. Polymorphisms in two important carcinogen detoxification genes, NAT2 and GSTM1, have been suggested as susceptibility factors for lung cancer (11, 12). Epidemiologic evidence shows the associations of GSTM1 null genotype with increased carcinogen DNA adduct burden (13, 14) and risk of lung cancer (15). However, no information is available linking the polymorphisms in NAT2 and GST genes with the formation of DNA adducts in the lung tissue, nor is there information on the potential interaction of these factors. Furthermore, data suggesting the influence of NAT2 acetylator status in the lung cancer population has been conflicting (11, 16-18). Notwithstanding, we previously reported a correlation between DNA adduct levels in different tissues (10). Tissue-specific differences in DNA adduct patterns can be caused by the metabolic activation of tobacco-derived carcinogens in different tissues (19, 20).
Therefore, the present study was designed to examine the independent and combined effects of genetic variation in NAT2 and GSTM1 on carcinogen-induced DNA damage measured in two different tissues: lung and blood mononuclear cells (MNC).
Materials and Methods
Study population and collection of specimens
The study population was described previously (21). This study was approved by the Committees on the Use of Human Subjects in Research at the Massachusetts General Hospital and the Harvard School of Public Health. Lung cancer patients at Massachusetts General Hospital were recruited between December 1992 and December 2000. Surgically resected noninvolved lung tissue was sampled from 142 patients who were undergoing surgical operation for histologically confirmed, newly diagnosed lung cancer. Among the participants, non-smokers (n = 7) were excluded. Lung tissue specimens were frozen immediately on dry ice and stored deep-frozen at -70°C until DNA adduct analysis. Blood samples were collected into heparin-treated tubes from each subject and applied to Ficoll-Hypaque density gradients to separate mononuclear cells (MNCs) from erythrocytes and granulocytes. Among the cases, blood volumes ≥ 20mL considered sufficient for DNA adduct analysis were available for 53 patients. Information on demographic factors, smoking history and occupation were obtained by trained interviewers using a modified standardized American Thoracic Society respiratory questionnaire.
Genotyping of GST polymorphisms and NAT2
The details of the methods for GST polymorphisms and NAT2 genotyping have been previously described (15, 18). All of the genetic polymorphisms were analyzed using PCR-RFLP techniques. Individuals carrying wildtype alleles (i.e., two “rapid” alleles, NAT2 *4/*4) were defined as the “rapid acetylator” genotype and those with only one copy of a rapid allele (heterozygous variant genotype, NAT2 *4) or those with two slow alleles (homozygous variant genotypes; NAT2 *5A, *5B, *5C, *6, or *7) were labeled “slow acetylators”.
Analysis of DNA adducts
We analyzed our previously reported the data on DNA adducts in lung and blood MNCs samples using the 32P-postlabeling assay (10, 21). In these prior studies, total relative DNA adducts were measured in the diagonal reactive zone plus discrete adducts (in nine regions) and were considered primarily to represent aromatic hydrophobic adducts, mainly PAH-DNA adducts (10). The DNA adducts level was expressed as relative adduct level (RAL) as reported previously (10). As a validation analysis, each sample was repeated at least twice and average adduct levels were obtained from the combination of all experiments of the RALs. The coefficient of variation for the repeated measurements was 14% for the positive control.
Statistical analysis
Dependent variables, DNA adduct levels per 1010 nucleotides, were transformed using natural logarithms to improve normality. For computational purposes, samples below the limit of detection (LOD), which is 1 adduct per 1010 nucleotides, were assigned a value of half the LOD as in prior reports (12). All the results for adduct levels are expressed as geometric mean (GM) with the corresponding 95% confidence intervals (CIs). Additionally, multivariate-adjusted geometric mean adduct levels were estimated by using linear regression models. Potential confounders such as age at diagnosis, sex, smoking status (ex-smoker and current smoker) and pack years of smoking were included in the multivariate analysis. The reference group included patients with GSTM1 present, GSTT1 present, GSTP1 common (AA) or NAT2 rapid type. Using this approach, we examined the independent effect of even a single at-risk genotype, compared with those who carried common alleles for the GSTs or NAT2. We estimated the percent changes in DNA adduct levels for the risk genotype compared with the common allele as [eβ – 1] × 100%, with 95% CI [e(β ± 1.96 × SE) – 1] × 100%, where β and SE are the estimated regression coefficient and its standard error. To examine the combined effects of the GSTM1 and NAT2, we constructed a model that included all possible combinations among GSTM1 and NAT2 polymorphisms, with the low-risk combination of GSTM1 wildtype plus NAT2 rapid acetylation, as a reference category. All statistical analyses were performed using SAS version 9.1 (SAS Institute Inc., Carry, NC).
Results
Baseline characteristics
To determine associations between GST and NAT2 polymorphisms and DNA adduct levels, we assessed a population of 135 lung cancer patients who completed genotyping and DNA adducts analysis. Demographic and clinical characteristics and DNA adduct levels in lung and blood MNC on the patient group are shown in Table 1. Overall, 59% of the subjects were former smokers who had not smoked for at least 1 year, leaving 41% as current smokers. Geometric mean (GM) DNA adduct levels were 86.2 adducts per 1010 nucleotides in lung tissues and 36.4 adducts per 1010 nucleotides in MNC samples. As expected, the GM of DNA adduct levels was higher in current smokers than those in ex-smokers, 179.4 versus 51.3 adduct per 1010 nucleotides in lung (P < 0.001) and 60.0 versus 24.0 adduct per 1010 nucleotides in blood MNC (P = 0.005), respectively. Additional details of histological and clinical stage data were published in another report (10).
Table 1.
Characteristics of patients with lung cancer and DNA adduct levels in the lung and blood MNC (N = 135)
| Characteristic | n (%) or mean ± SD |
|---|---|
| Sex (male) | 78 (57.8) |
| Race (Caucasian) | 131 (97.0) |
| Smoking status | |
| Current smokers | 56 (41.5) |
| Ex-smokers | 79 (58.5) |
| Histologic analysis | |
| Adenocarcinoma | 65 (48.1) |
| Squamous | 44 (32.6) |
| Others† | 26 (19.3) |
| Age at diagnosis (years) | 66.3 ± 10.6 |
| DNA adduct levels, adducts per 1010 nucleotides‡ | |
| Lung DNA adduct level (n = 135) | 86.2 ± 4.7 |
| Blood MNC DNA adduct level (n = 53) | 36.4 ± 3.2 |
| Pack-years (years)§ | 63.5 ± 40.6 |
Others include small-cell, large-cell, and mixed cell types.
GM ± GSD
Pack-years = number of packs per day × years of smoking.
Association between individual GSTs, NAT2 and DNA adduct in lung and MNC
The genotype distribution of the polymorphisms and their association with DNA adduct levels are given in Table 2. The DNA adduct levels are expressed as covariate-adjusted geometric mean (GM) with the corresponding 95% CIs estimated by linear regression models. Regarding the difference in adduct levels in lung tissue according to genotype, the adjusted-GM of DNA adduct levels with the variant genotype of GSTM1 null and the slow NAT2 acetylator genotype were higher than those with presence of the wild type GSTM1 genotype or rapid NAT2 acetylator, 173.9 versus 69.5 (P = 0.005) and 144.9 versus 83.3 (P = 0.068), respectively. We examined multiple regression models for the adjusted percent change in DNA adduct levels, associated with each GSTs genotype and NAT2 activity. The DNA adduct levels in lung tissue significantly increased by 150.3% (95% CI, 35.4% to 362.6%) for GSTM1 null and by 73.9% (95% CI, -3.2% to 212.4%) for NAT2 slow acetylator after controlling for potential confounders and other GSTs genes. We also observed an increase in DNA adduct levels in blood MNC with variant homozygous deleted GSTM1 null by 267.4% (95% CI, 28.2% to 952.9%), but none of GSTP1 AG/GG, GSTT1 null, and NAT2 slow types were statistically significant.
Table 2.
Estimated percent changes and 95% CIs in DNA adducts in lung and blood MNC per 1010 nucleotides by GSTs polymorphisms and NAT2 activity
| DNA adduct in lung |
DNA adduct in blood MNC |
|||||
|---|---|---|---|---|---|---|
| n (%) | Adjusted GM (95% CI)† | %change (95% CI) | n (%) | Adjusted GM(95% CI)† | %change (95% CI) | |
| GSTM1 | ||||||
| Null | 56 (43.8) | 173.9 (100.9 to 299.7) | 150.3 (35.4 to 362.6)** | 24 (46.1) | 88.2 (33.5 to 231.9) | 267.4 (28.2 to 952.9)** |
| Wild-type | 72 (56.2) | 69.5 (41.1 to 117.3) | Reference | 28 (53.9) | 24.0 (8.9 to 64.4) | Reference |
| GSTP1 | ||||||
| AG or GG | 57 (48.3) | 115.1 (65.9 to 201.0) | 9.7 (-40.7 to 103.0) | 27 (54.0) | 67.5 (22.5 to 202.7) | 115.4 (-30.1 to 564.1) |
| AA | 62 (51.7) | 104.9 (63.0 to 174.8) | Reference | 23 (46.0) | 31.3 (12.9 to 76.1) | Reference |
| GSTT1 | ||||||
| Null | 17 (14.0) | 119.7 (53.0 to 270.5) | 18.6 (-50.5 to 184.0) | 5 (9.4) | 57.2 (12.1 to 269.8) | 54.6 (-68.2 to 652.1) |
| Wild-type | 104 (86.0) | 100.9 (73.0 to 139.5) | Reference | 48 (90.6) | 37.0 (21.8 to 62.7) | Reference |
| NAT2 activity | ||||||
| Slow | 48 (58.5) | 144.9 (87.1 to 241.2) | 73.9 (-3.2 to 212.4)* | 23 (67.7) | 39.0 (16.6 to 91.8) | -28.2 (-70.5 to 74.8) |
| Rapid | 34 (41.5) | 83.3 (48.5 to 143.2) | Reference | 11 (32.3) | 54.3 (20.0 to 147.4) | Reference |
P < 0.1
P < 0.05
***P < 0.01
Note: Coefficients are expressed as percent changes in aromatic DNA adducts-associated GST polymorphisms and NAT2 activity adjusted for age at diagnosis, sex, smoking status, pack years, and GSTM1, GSTT1, GSTP1, and NAT2 gene.
Adjusted for age at diagnosis, sex, smoking status, pack years, and GSTM1, GSTT1, GSTP1, and NAT2 gene.
Combined effects of GSTM1 and NAT2 on DNA adduct levels
Figure 1 illustrates the combined effects of GSTM1 genotype with NAT2, since they are thought to act as potential concurrent risk factors of individual genetic susceptibility to DNA damage. We constructed a model that included all of the possible combinations among GSTM1 and NAT2, with the low-risk combination of GSTM1 present and NAT2 rapid acetylator, as a reference group. The high-risk group, defined as the combination of GSTM1 null plus NAT2 slow, was highly significantly associated with an increased level of DNA adducts in the lung of 295% (95% CI, 72.7% to 803.5%) compared to the low-risk combination. In the case of blood analysis, the sample number was small; thus, our combined analysis suffers from a lack of statistical power to find such associations.
Figure 1.
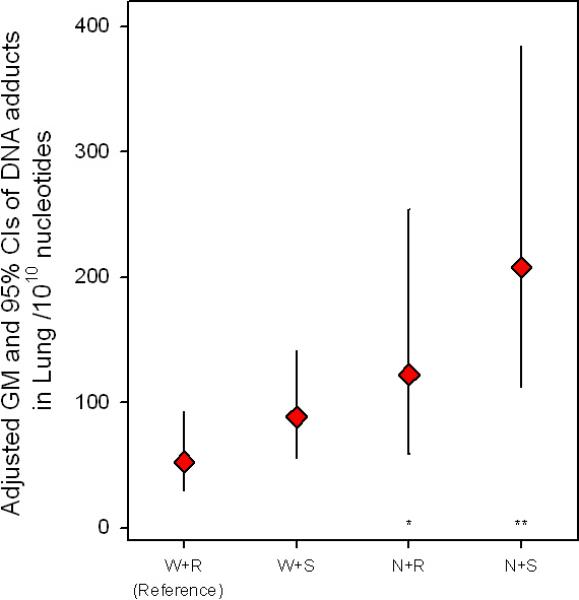
The adjusted geometric mean (GM) and 95% CIs of the DNA adducts levels in lung by all two-way combination between GSTM1 and NAT2. W, GSTM1 wild-type; N; GSTM1 null; S, NAT2 slow acetylator; R, NAT2 rapid acetylator. *P < 0.1, **P < 0.05, *** P < 0.01.
Discussion
To date, there have been no studies examining the associations of independent and combined carcinogen-metabolizing gene polymorphisms with DNA adduct levels in the target organ, lung. This study provides evidence for increased DNA adduct levels associated with the GSTM1 null and NAT2 slow genotypes alone or in combination. Moderate to strong associations for the GSMT1 null and NAT2 slow alleles were found, such that these polymorphisms could account for 150% and 74% increases in DNA adduct levels in the lung, respectively. The combination of both ‘at risk’ genotypes, GSTM1 null/NAT2 slow, which are related to reduced phase II enzyme activity, could lead to a 295% (95% CI, 72.7% to 803.5%) increase in DNA adducts in the lung. Compared with the increases associated with single alleles alone, this indicates an estimated 71% synergism [295% - (150% + 74%)]. Despite the biological plausibility, none of other GSTs polymorphisms evaluated in this study was associated with DNA adduct levels.
The evidence for the independent role of metabolic genotypes and their combinations on carcinogen-DNA adducts in different tissues is insufficient. A few studies have reported a relationship between NAT2 and/or GSTM1 polymorphisms and DNA adducts. As we found in this study, individuals with the GSTM1 null genotype have a greater level of lung DNA adducts formed by Benzo[a]pyrene diol epoxide (BPDE) in currently smoking lung cancer patients (13, 14). With regard to blood adducts, GSTM1 null was associated with increased BPDE-DNA adduct levels in leukocytes of smokers (22). Elevated DNA adduct levels in placental tissue were associated with GSTM1 null genotype among subjects living in a polluted area in the Czech Republic (23). Conversely, no association was seen between DNA adducts in total WBC with the GSTM1 null genotype among 89 cases of primary lung cancer (12) and 296 healthy adults (31). However, we measured DNA adducts in MNC, a longer-lived fraction of the WBC (10). Therefore, the DNA adducts in MNC is a better representation of long term DNA damage, with relatively less variation over time, which may explain the difference between our results and those of earlier studies. NAT2 gene polymorphisms have been proposed as a cancer risk factor (24). The slow NAT genotype has been considered as the recessive trait and linked to a 2-fold increase in lung cancer (17) and an increased risk of bladder cancer (25). Although the evidence for the NAT2 acetylator status as a risk factor in lung cancer has been conflicting (11, 16-18), its effect on DNA damage in lung tissue has not been studied. We found that lung adducts with the NAT2 slow acetylation genotype increased by 74% (95% CI, -3.2% to 212.4%, P = 0.068), as compared with the NAT2 fast genotype, while no such association was seen in blood adducts. Our study was based upon lung cancer patients who are former and current smokers. Thus, they might be exposed to tobacco-carcinogens at relatively high levels, overwhelming the relatively modest effects of common genetic variants (26). Other studies of smoking lung cancer populations support our findings on blood adducts (17, 27, 28).
Very few studies have reported associations for the combined NAT2 slow acetylator and GSTM1 null genotype and increased susceptibility to DNA damage in peripheral blood among current smoking lung cancer cases (16), and elevated risk of bladder cancer (29). We found that genetic factors influence individual susceptibility to DNA damage, and that the combination of NAT2 slow and GSTM1 null genotype is significantly associated with increased lung adduct levels compared to those in low-risk genotypes (NAT2 rapid-GSTM1 null). This result indicates that carrying more than one of the risk polymorphisms may have synergistic effects on DNA damage in the target tissue, and could potentially lead to an increase in carcinogenic potency.
Blood MNC, accounting for about 30% of total WBC, has been suggested as a surrogate tissue for estimating the burden of lung adducts (10). Positive correlations between blood and lung adduct levels (r = 0.77, P < 0.001) were reported in a lung cancer population (10, 21). However, we found independent effects of GSTM1 null on DNA adducts in both tissues, but the effect of NAT2 polymorphisms differed by tissue. Tissue-specific differences were also observed for combinations of these factors. Possibly, metabolic activation of PAHs by various cell types in various tissues leads to tissue-specific differences (19). The metabolism of Benzo[a]pyrene differed among rat lung, liver and peripheral blood lymphocytes (20) and among human samples from bronchus, esophagus, colon, and duodenum (30). Differences in individual tissue repair capabilities and in cell division may also influence tissue-specific variations in expression of carcinogen-metabolizing enzymes and in the lifetime of adducts (20). These could explain the differential results of lung and blood MNC. Alternatively, our blood data were limited due to small sample sizes, thus we acknowledge that our combined analyses suffer from lack of statistical power to detect some associations.
We measured DNA adducts by the nuclease P1 version of the 32P-postlabeling technique. This assay is generally applied to analyze a mixture of adducts, often referred to as aromatic or bulky adducts (2), which are induced mainly from the ubiquitous PAHs. However, the technique is also effective for the detection of DNA adducts from other aromatic compounds such as aromatic amines (AA) or heterocyclic amines (HA) (31). Further study is needed to address chemical characterization of tobacco carcinogen-induced DNA damage in smoking populations if the chemical nature of DNA adducts differs by smoking status (21).
In conclusion, this study suggests that GSTM1 null and NAT2 slow genotypes lead to enhanced DNA damage from mutagens in tobacco smoke. In particular, a synergistic deleterious effect was noted when these high risk genotypes were considered together with the target lung tissue. Therefore, the assessment of a single polymorphic genotype may not be sufficient for evaluating individual susceptibility to DNA damage (32). These results provide a better understanding for the identification of vulnerable populations to the DNA damage linked to lung cancer.
Acknowledgements
This study was supported by NIH grants # CA074386, CA092824, CA090578. The authors acknowledge the technical assistance of Drs. John Wiencke and Kofi Asomaning.
Abbreviations
- AA
aromatic amine
- CI
95% confidence interval
- GM
geometric mean
- GSD
geometric standard deviation
- GST
glutathione S-transferase
- HA
heterocyclic amine
- NAT2
N-acetyltransferase-2
- MNC
mononuclear cell
- PAHs
polycyclic aromatic hydrocarbons
- RAL
relative adduct level
References
- 1.Peluso M, Munnia A, Hoek G, et al. DNA adducts and lung cancer risk: a prospective study. Cancer Res. 2005;65:8042–8. doi: 10.1158/0008-5472.CAN-04-3488. [DOI] [PubMed] [Google Scholar]
- 2.Poirier MC. Chemical-induced DNA damage and human cancer risk. Nat Rev Cancer. 2004;4:630–7. doi: 10.1038/nrc1410. [DOI] [PubMed] [Google Scholar]
- 3.Veglia F, Loft S, Matullo G, et al. DNA adducts and cancer risk in prospective studies: a pooled analysis and a meta-analysis. Carcinogenesis. 2008;29:932–6. doi: 10.1093/carcin/bgm286. [DOI] [PubMed] [Google Scholar]
- 4.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 5.Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer. 2003;3:733–44. doi: 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]
- 6.Vineis P, Bartsch H, Caporaso N, et al. Genetically based N-acetyltransferase metabolic polymorphism and low-level environmental exposure to carcinogens. Nature. 1994;369:154–6. doi: 10.1038/369154a0. [DOI] [PubMed] [Google Scholar]
- 7.Fu PP, Herreno-Saenz D, Von Tungeln LS, et al. DNA adducts and carcinogenicity of nitro-polycyclic aromatic hydrocarbons. Environ Health Perspect. 1994;102(Suppl 6):177–83. doi: 10.1289/ehp.94102s6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talaska G, Underwood P, Maier A, Lewtas J, Rothman N, Jaeger M. Polycyclic aromatic hydrocarbons (PAHs), nitro-PAHs and related environmental compounds: biological markers of exposure and effects. Environ Health Perspect. 1996;104(Suppl 5):901–6. doi: 10.1289/ehp.96104s5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denissenko MF, Pao A, Tang M, Pfeifer GP. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science. 1996;274:430–2. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 10.Wiencke JK, Kelsey KT, Varkonyi A, et al. Correlation of DNA adducts in blood mononuclear cells with tobacco carcinogen-induced damage in human lung. Cancer Res. 1995;55:4910–4. [PubMed] [Google Scholar]
- 11.Cascorbi I, Brockmoller J, Mrozikiewicz PM, Bauer S, Loddenkemper R, Roots I. Homozygous rapid arylamine N-acetyltransferase (NAT2) genotype as a susceptibility factor for lung cancer. Cancer Res. 1996;56:3961–6. [PubMed] [Google Scholar]
- 12.Perera FP, Mooney LA, Stampfer M, et al. Associations between carcinogen-DNA damage, glutathione S-transferase genotypes, and risk of lung cancer in the prospective Physicians’ Health Cohort Study. Carcinogenesis. 2002;23:1641–6. doi: 10.1093/carcin/23.10.1641. [DOI] [PubMed] [Google Scholar]
- 13.Kato S, Bowman ED, Harrington AM, Blomeke B, Shields PG. Human lung carcinogen-DNA adduct levels mediated by genetic polymorphisms in vivo. J Natl Cancer Inst. 1995;87:902–7. doi: 10.1093/jnci/87.12.902. [DOI] [PubMed] [Google Scholar]
- 14.Shields PG, Bowman ED, Harrington AM, Doan VT, Weston A. Polycyclic aromatic hydrocarbon-DNA adducts in human lung and cancer susceptibility genes. Cancer Res. 1993;53:3486–92. [PubMed] [Google Scholar]
- 15.Liu G, Miller DP, Zhou W, et al. Differential association of the codon 72 p53 and GSTM1 polymorphisms on histological subtype of non-small cell lung carcinoma. Cancer Res. 2001;61:8718–22. [PubMed] [Google Scholar]
- 16.Hou SM, Falt S, Yang K, et al. Differential interactions between GSTM1 and NAT2 genotypes on aromatic DNA adduct level and HPRT mutant frequency in lung cancer patients and population controls. Cancer Epidemiol Biomarkers Prev. 2001;10:133–40. [PubMed] [Google Scholar]
- 17.Seow A, Zhao B, Poh WT, et al. NAT2 slow acetylator genotype is associated with increased risk of lung cancer among non-smoking Chinese women in Singapore. Carcinogenesis. 1999;20:1877–81. doi: 10.1093/carcin/20.9.1877. [DOI] [PubMed] [Google Scholar]
- 18.Zhou W, Liu G, Thurston SW, et al. Genetic polymorphisms in N-acetyltransferase-2 and microsomal epoxide hydrolase, cumulative cigarette smoking, and lung cancer. Cancer Epidemiol Biomarkers Prev. 2002;11:15–21. [PubMed] [Google Scholar]
- 19.Ross J, Nelson G, Erexson G, et al. DNA adducts in rat lung, liver and peripheral blood lymphocytes produced by i.p. administration of benzo[a]pyrene metabolites and derivatives. Carcinogenesis. 1991;12:1953–5. doi: 10.1093/carcin/12.10.1953. [DOI] [PubMed] [Google Scholar]
- 20.Ross J, Nelson G, Kligerman A, et al. Formation and persistence of novel benzo(a)pyrene adducts in rat lung, liver, and peripheral blood lymphocyte DNA. Cancer Res. 1990;50:5088–94. [PubMed] [Google Scholar]
- 21.Wiencke JK, Thurston SW, Kelsey KT, et al. Early age at smoking initiation and tobacco carcinogen DNA damage in the lung. J Natl Cancer Inst. 1999;91:614–9. doi: 10.1093/jnci/91.7.614. [DOI] [PubMed] [Google Scholar]
- 22.Lodovici M, Luceri C, Guglielmi F, et al. Benzo(a)pyrene diolepoxide (BPDE)-DNA adduct levels in leukocytes of smokers in relation to polymorphism of CYP1A1, GSTM1, GSTP1, GSTT1, and mEH. Cancer Epidemiol Biomarkers Prev. 2004;13:1342–8. [PubMed] [Google Scholar]
- 23.Binkova B, Chvatalova I, Lnenickova Z, et al. PAH-DNA adducts in environmentally exposed population in relation to metabolic and DNA repair gene polymorphisms. Mutat Res. 2007;620:49–61. doi: 10.1016/j.mrfmmm.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 24.Harris CC. Interindividual variation among humans in carcinogen metabolism, DNA adduct formation and DNA repair. Carcinogenesis. 1989;10:1563–6. doi: 10.1093/carcin/10.9.1563. [DOI] [PubMed] [Google Scholar]
- 25.Risch A, Wallace DM, Bathers S, Sim E. Slow N-acetylation genotype is a susceptibility factor in occupational and smoking related bladder cancer. Hum Mol Genet. 1995;4:231–6. doi: 10.1093/hmg/4.2.231. [DOI] [PubMed] [Google Scholar]
- 26.Kuljukka-Rabb T, Nylund L, Vaaranrinta R, et al. The effect of relevant genotypes on PAH exposure-related biomarkers. J Expo Anal Environ Epidemiol. 2002;12:81–91. doi: 10.1038/sj.jea.7500204. [DOI] [PubMed] [Google Scholar]
- 27.Martinez C, Agundez JA, Olivera M, Martin R, Ladero JM, Benitez J. Lung cancer and mutations at the polymorphic NAT2 gene locus. Pharmacogenetics. 1995;5:207–14. [PubMed] [Google Scholar]
- 28.Bouchardy C, Mitrunen K, Wikman H, et al. N-acetyltransferase NAT1 and NAT2 genotypes and lung cancer risk. Pharmacogenetics. 1998;8:291–8. doi: 10.1097/00008571-199808000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Closas M, Malats N, Silverman D, et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005;366:649–59. doi: 10.1016/S0140-6736(05)67137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Autrup H, Grafstrom RC, Brugh M, et al. Comparison of benzo(a)pyrene metabolism in bronchus, esophagus, colon, and duodenum from the same individual. Cancer Res. 1982;42:934–8. [PubMed] [Google Scholar]
- 31.Agudo A, Peluso M, Sala N, et al. Aromatic DNA adducts and polymorphisms in metabolic genes in healthy adults: findings from the EPIC-Spain cohort. Carcinogenesis. 2009;30:968–76. doi: 10.1093/carcin/bgp062. [DOI] [PubMed] [Google Scholar]
- 32.Hirvonen A. Combinations of susceptible genotypes and individual responses to toxicants. Environ Health Perspect. 1997;105(Suppl 4):755–8. doi: 10.1289/ehp.97105s4755. [DOI] [PMC free article] [PubMed] [Google Scholar]