

Abstract
Molinate has been widely used as a pre emergent herbicide in the rice fields of California’s Central Valley. In rat studies, the metabolite molinate sulfoxide is suspected of causing testicular toxicity after exposure to molinate. The sulfoxide is generated in the liver and can circulate in the blood, eventually reaching the testis. Man qualitatively produces the same molinate metabolites as the rat. To extrapolate the reproductive risk to man, the present study outlines the development of a preliminary PBPK (Physiologically-based Pharmacokinetic) model, validation in the rat and extrapolation to man.
The preliminary seven-compartment PBPK model for molinate was constructed for the adult, male Sprague-Dawley rat that employed both flow-limited (blood, kidney, liver, rapid-perfused tissues and slowly perfused tissues) and diffusion-limited (fat) rate equations. The systemic circulation connects the various compartments. The simulations predict the molinate blood concentrations of the rat blood and testes compartment favorably with the profiles obtained from 10 and 100 mg/kg po or 1.5 and 15 mg/kg iv doses. Human physiological parameters were substituted into the oral dosed model and the simulations closely predicted the molinate blood concentration obtained from 5.06 mg oral dose. A sensitivity analysis determined for an oral dose that peak blood molinate concentrations were most responsive to the blood flows to kidney and fat compartments while testicular molinate sulfoxide concentrations depended on molinate sulfoxide partition coefficients for the testes compartment and the Km for glutathione conjugation of molinate sulfoxide in the liver compartment.
Keywords: PBPK, Molinate, Rat, Testicular toxicity
INTRODUCTION
s-ethyl hexahydro-1H-azepine-1-1carbothioate (MOLINATE) is pre emergent herbicide used in the rice growing industry around the world. Primary occupational exposure routes include both dermal and inhalation for field workers exposed to the herbicide as well as the oral route from drinking water contaminated from agricultural runoff. Molinate induces numerous biochemical, morphological, and toxicological responses including reproductive toxicities such as a distinctive sperm lesion and delayed release of the late spermatids to the seminiferous tubular lumen. (Cochran et al., 1997, Ellis et al., 1998, Jewell et al., 1998; Berger and Miller, 2000; and Kavlock and Cummings, 2005). Moreover in rat studies, high doses of molinate were shown to cause testicular toxicity and lower dose levels were associated with sperm abnormalities. Further, the testicular toxicity was related to formation of the molinate metabolite, molinate sulfoxide (Figure 1).
Figure 1.

Proposed pathway for molinate metabolism in rat and human.
Current understanding of the mechanism of molinate’s reproductive toxicity implicates a sulfoxide metabolite generated in the liver that can circulate in the blood to reach the testis (see Figure 1). The sulfoxide can be further metabolized to the sulphone, and theoretically both are reactive and capable of covalent protein binding and conjugation with glutathione ultimately appearing after further metabolism (Jewell and Miller, 1998) as mercapturates in the urine. The major urinary metabolites are the mercapturate, the hydroxy – molinate(s) and their respective glucuronides (De Baun et al., 1978). The covalent protein binding can impact both toxicity a) by covalently modifying cysteine residues at the active site of enzymes and altering function (Zimmerman et al., 2002, Jewell and Miller, 1998) as well as b) the kinetics and disposition of the sulfoxide (Campbell et al., 2008).
Presently, understanding of the molecular mechanisms of the developmental reproductive effects of molinate is quite limited. One possible hypothesis suggests that once molinate is bioactivated to molinate sulfoxide, the sulfoxide is free to bind proteins that may be important to the production and activation of the testosterone or other biologically important molecules such as retinoic acid receptor which initiates a cascade of events leading to the effects. In sum, the mechanism of toxicity of molinate sulfoxide remains controversial.
Ellis et al. (1998) describe a variety of toxic effects on the male rat reproductive system induced by molinate. Briefly, they found administration of molinate to rats (40 mg/kg/day for 7 days) caused a distinctive sperm lesion. Higher doses of molinate (140 mg/kg for 7 days) produced morphological changes to the testis that included a delayed release of the late spermatids to the seminiferous tubular lumen. This is a process controlled by the release of testosterone, and by using [3H]molinate, the primary target site appears to be the Leydig cells of the testis. Another important observation in rats dosed with molinate (>/=40 mg/kg) and molinate sulfoxide (>10 mg/kg) was the substantial decrease in both circulating and testicular testosterone concentration.
The morphological changes can be explained by an inhibition of Leydig cell function, including testosterone production, which is required for the maintenance of spermatogenesis. Since molinate sulfoxide inhibits general ester hydrolysis including neutral cholesterol ester hydrolase (nCEH) within the Leydig cells of the rat testis, cholesterol release would be prevented from its storage ester within this cell type. Therefore one explanation for the rodent’s increased susceptibility to molinate testicular toxicity compared to man lies in the difference in major source of cholesterol; rodents utilize high-density lipoproteins (HDLs) in plasma that are hydrolyzed within the cell cytosol by nCEH (Gwynne et al., 1976) while conversely human obtain the majority of their cholesterol from low-density lipoproteins (LDLs) (Payne et al., 1985).
Several studies have examined the pharmacokinetics of molinate in rat, rabbit, monkey and man (Jewell et al., 1998, Dean, 1977, Lythgoe et al., 1992, and Batten et al., 1992). In this study we add to the preliminary pharmacokinetic studies plus several metabolism experiments are utilized to develop a Physiologically-based Pharmacokinetic model (PBPK), a tool that can be used to predict the distribution of molinate, determine target tissue molinate concentrations, and provide simultaneous tissue concentration versus time profiles for the various compartments in the model. Since certain chemicals (arsenic, benomyl, dioxin, dibromochloropropane) can cause toxicity at minute concentrations in experimental animals; linking the temporal relationship between dose, exposure, and response would be an important step towards accurately estimating the potential adverse risk to human health.
A PBPK model is a body composed of compartments, and each compartment contains mathematical descriptions of a chemical’s absorption, distribution, metabolism, and elimination (ADME). Similar to conventional allometery, PBPK models provide a quantitative means of extrapolating ADME properties across species. The difference lies in the PBPK models ability to substitute species-specific physiological and biochemical parameters into the model. Thus by developing a PBPK model that can predict molinate concentrations in adult rat compartments the aim of this work was to extrapolate molinate blood and testes concentration predictions to humans.
In order for a pharmacokinetic model to successfully extrapolate between species the differences in a) metabolism and kinetics and b) physiological changes between species must be incorporated. Since rat and man qualitatively share the same pathway for molinate detoxification and bioactivation, an attempt was made to incorporate the appropriate metabolic and physiologic parameters that would optimize the interspecies prediction. Finally, since documented molinate administration to humans is rare, the opportunity to validate this model in humans is limited; therefore the utility of this model to predict human tissue concentrations is restricted to oral exposures.
MATERIALS AND METHODS
Chemicals
Molinate (s-ethyl hexahydro-1H-azepine-1-1carbothioate) was obtained from ChemService (Westchester, PA). It was 99% pure. Molinate sulfoxide was a gift from Dr William Helke, Syngenta, Greensboro, NC. Metabolite standards for molinate were generously provided by Zeneca Ag Products (Richmond, CA). Glycerol formal was obtained from Sigma-Aldrich (St. Louis, MO). All other chemicals were purchased from Fisher Scientific (Fairlawn, NJ). Molinate and molinate sulfoxide stock solutions (1 mg/mL) in acetonitrile were prepared before each analytical run and diluted to standard concentration in 2:1 acetonitrile: 100 mM PBS, pH 7.4. Purity for molinate, molinate sulfoxide, and the glutathione conjugate were assessed at 99.1%, > 97.1%, and 57.1%, respectively by the manufacturer for molinate or by A218 nm peak area from reverse phase chromatography.
Animals
Double jugular vein catheterized male Sprague-Dawley rats (approximately 300 g obtained from Charles River Laboratories, Wilmington, MA) were used for the iv study whereas both single jugular vein male catheterized or normal Sprague-Dawley rats (approximately 300 g from Charles River Laboratories) were used for the oral dosed study. The animals were housed one per cage in temperature (22.6 ± 1°C) and humidity (50.6 ± 10%) controlled rooms under a 12-h light/dark cycle. Animals were acclimated to housing conditions for at least 1 week before use. Because molinate is a male reproductive toxicant, only males were analyzed. This study was approved by the UC Davis Administrative Panel on Laboratory Animal Care. UC Davis University is fully accredited by the AAALAC and registered with the United States Department of Agriculture. All animals involved in this study received humane care in accordance with “The Guide for the Care and Use of Laboratory Animals” (Institute of Laboratory Animal Resources, National Research Council, Revised 1996) and University of California, Davis Animal Care Guidelines. The Vivarium Service Center (VSC) provided routine housing and husbandry (cage changes, feeding, etc.). A member of the VSC staff observed the animals on study twice daily.
Experimental
Molinate was dissolved in sterile glycerol formal to give the desire dose of 1.5, 10, 15 and 100 mg/kg of body weight in a total volume of 1.5 ml/kg. For the iv study, subjects received 1.5 or 15 mg/kg molinate through the left cannula and serial micro blood samples were taken through the right cannula at predose, 5, 10, 20, 30, 60, 90, 120, and 180 minutes after exposure. For the oral study, rats received 10 or 100 mg/kg molinate through oral gavage and serial micro blood samples were taken through the right cannula at 10, 30, 60, 120, 180 and 240 minutes after exposure. Oral dosing of normal, uncannulated rats followed by serial sacrifices at predose, 10, 30, 60, 120, 180 and 240 minutes after exposure were performed in a separate experiments to obtain brain, fat, kidney, liver, muscle and testes tissue samples. Three to four animals/group were used.
Analytical
Analyte concentrations were measured as described previously by Tjeerdema et al., 1987, and modified by Campbell et al. 2008. Briefly, A C18 column (250 × 4.6mm, i.d. 5 μm, Alltech Associates) is initially equilibrated in dH20: acetonitrile each with 0.1% formic acid (10:90) and then eluted over a linear gradient to final conditions (90:10) over 22 minutes at a flow rate of 1 ml/min. Single Ion Mode liquid chromatography/mass spectral (LC-MS) detection was conducted using an 8:1 split. Blood was extracted by the addition of two volumes of acetonitrile with vortexing, and centrifuged at 1300g for 5 minutes. Tissues were homogenized in 50:50 acetonitrile :100 mM phosphate buffer, pH 7.4 (1:3 ), then 1X volume of 25% NaCl in 100 mM phosphate buffer, pH 7.4 was added and solution was vortexed, followed by a 30 minute room incubation. Finally the solution was extracted by the addition of two volumes of acetonitrile with vortexing followed by centrifugation at 1300g for 5 minutes. Extracted blood and tissue samples were 0.45 μm filtered and stored at −80°C until LC-MS analysis. The extraction method was validated by spike and recovery of both molinate and molinate sulfoxide in each tissue homogenate at three different tissues concentrations (0.1, 0.5 and 1 μg/ml homogenate). All dosing solutions were measured by the LC-MS method.
PBPK Model development
The iv model was developed first since it contains fewer absorption assumptions. The iv PBPK model for molinate consists of seven compartments including blood, fat, liver, testes, kidney, richly and slowly perfused tissue compartments (Figure 2). Next was development of the oral dosed model which used the same metabolic constants as the iv model. Both models assume that each compartment is flow-limited with the exception of fat which best fit a diffusion limited assumption. Validation of this model included several data sets from rats. Extrapolating a model with this many compartments to humans is problematic, due to the limited human data available. While some adult urine concentrations are available from the published literature, rarely is the external molinate concentration documented. Data from other tissues are not readily available. Thus, validating the model predictions in the different compartments in humans would be unlikely.
Figure 2.
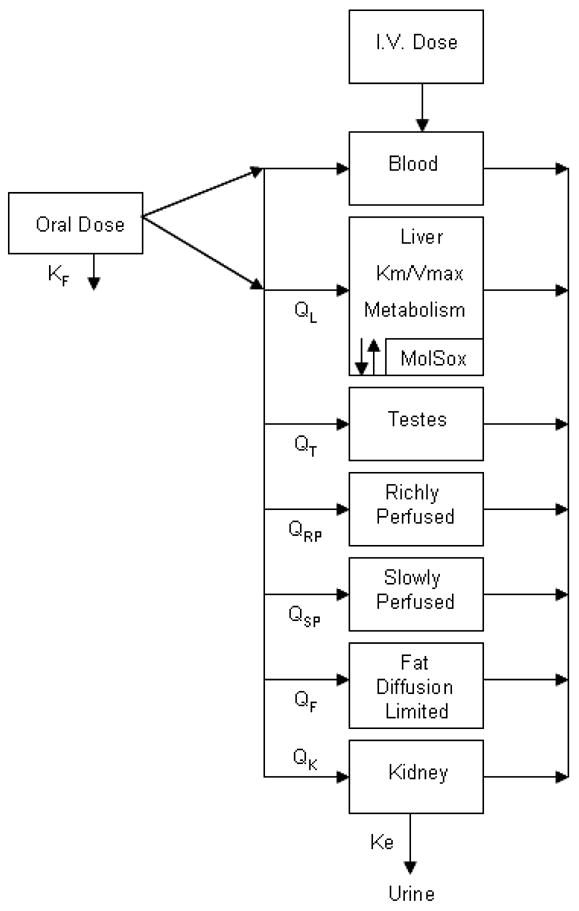
Conceptual representation of PBPK model for rat intravenous and oral exposure to molinate. All compartments flow limited except fat (diffusion limited).
Tissues in this model include those that have important roles in the pharmacokinetics and reproductive toxicity of molinate. A blood compartment was used to describe the systemic circulation, this tissue is readily sampled in rats and humans, and the molinate sulfoxide - hemoglobin adduct formation rate is also included here. Cytochrome P450–mediated metabolism and glutathione conjugation is accounted for in the liver and fat was included in the model because it is a primary storage site of molinate in the first 24 hours of exposure (DeBaun et al., 1978). The kidney was selected because elimination for both parent and metabolite is performed in the kidneys (glomular filtration rate multiplied by blood flow). The testis is the primary target organ and the rest of the body compartment was included as either slowly or richly perfused compartments in order to achieve mass balance. The present description of this simplified PBPK model represents the most important compartments for an adequate description of the pharmacokinetics and metabolism of molinate (Figure 2). A more detailed model description including the equations used for each compartment is available in the Appendix, and the physiological parameters used in the model are in Table 1, the metabolic parameters used in the model are in Table 2, and the chemical specific absorption, injection, and optimized (see Model simulation and fitting procedure section below) parameters are summarized in Table 3.
Table 1.
Physiologic Parameters used in the PBPK model for Rat and Human
| Variable | Symbol | Compartment | Rat | Human |
|---|---|---|---|---|
| Initial Body Weight (Kg) | WtO | Body Weight/Cardiac Output | 0.298A | 60C |
|
| ||||
| Cardiac Output (L/min/Kg0.75) Rat (L/min/Kg) Human | Qcar | Body Weight/Cardiac Output | 0.273C | 0.086C |
|
| ||||
| Fraction of Oral Dose that reaches Blood through Lymphatic System | Fkl | Split | 0.1G | 0.1G |
|
| ||||
| Fecal excretion constant (Fraction of Dose) | Kf | Absorption | 0.1E | 0.1E–F |
|
| ||||
| Tissue Volume Fraction of Bodyweight | Vc | Liver | 0.037 | 0.0257 |
| Testes | 0.013A | 0.00057D | ||
| Kidney | 0.0073 | 0.0044 | ||
| Slowly Perfused | 0.81 | 0.437 | ||
| Richly Perfused | 0.0596 | 0.166 | ||
| Fat | 0.17 | 0.214 | ||
| Blood | 0.07 | 0.08 | ||
| All other valuesC | All other valuesC | |||
|
| ||||
| Fraction of Tissue Volume that is Blood | Vbc | Liver | 0.21 | 0.11 |
| Testes | 0.03A | 0.03D | ||
| Kidney | 0.16 | 0.36 | ||
| Slowly Perfused | 0.04 | 0.04 | ||
| Richly Perfused | 0.21 | 0.21 | ||
| Fat | 0.05 | 0.02 | ||
| All other valuesC | All other valuesC | |||
|
| ||||
| Blood Flow to Tissue (Fraction of Cardiac Output) | Qc | Liver | 0.18 | 0.1854 |
| Testes | 0.01 | 0.0004D | ||
| Kidney | 0.16 | 0.175 | ||
| Slowly Perfused | 0.16 | 0.16 | ||
| Richly Perfused | QTOT-QTOT(ΣQc)F | QTOT-QTOT(ΣQc)F | ||
| Fat | 0.07 | 0.052 | ||
| All other valuesC | All other valuesC | |||
|
| ||||
| Fraction of Tissue Weight that is Protein | Pfl | Liver | 0.348A | 0.348Rat Value |
|
| ||||
| Fraction of Tissue Weight that is Cytosolic Protein | Pfc | Liver | 0.042A | 0.02B |
|
| ||||
| Weight of Tissue (grams) | W | Liver | 12.5C | 1800A |
Source
Experiment
Experiment (Data from SRI)
Estimated
Table 2.
Metabolic Parameters used in the PBPK model for Rat and Human.
| Variable | Symbol | Rat Liver | Rat Optimized | Human Liver |
|---|---|---|---|---|
| Hydroxylation Km (nM) | KmOH | 587,000A | 587,000 | 124,000A |
| Vmax Hydroxylation (nmol/min/g protein) | VmaxOH | 8.33A | 8.33 | 58.6A |
| Km Sulfoxidation (nM) | KmSO | 2,837,000A | 360,000C | 981,000A |
| Vmax Sulfoxidation (nmol/min/g protein) | VmaxSO | 638A | 638 | 510A |
| Km GST conjugation (nM) | KmGST1 | 305,500B | 304,100 | 91,250B |
| Vmax GST Conjugation (nmol/min/g Cytosolic Protein) | VmaxGST1 | 4,208B | 5,383 | 323B |
Table 3.
Chemical Specific and Diffusion-Limited Tissue Parameters used in the PBPK Model for Rat and Human.
| Variable | Symbol | Compartment | Rat | Optimized* | Human |
|---|---|---|---|---|---|
| Absorption Rate Constant (Oral Model) (min−1) | K1 | 0.05 | 0.05 | .05 | |
| K2 | Absorption | 0.004 | 0.004 | .004 | |
| K3 | 0.0001 | 0.0001 | .0001 | ||
| All valuesA | All valuesA | All valuesA | |||
|
| |||||
| Fraction of starting bioavailable dose at absorption site at time zero, ( Dose – Kf* Dose) | |||||
| allocated to rapid phase absorption | DFa | Absorption | 0.2 | 0.2 | 0.2 |
| allocated to slow phase absorbtion | DFb | 0.05 | 0.05 | 0.05 | |
| allocated to slowest phase absorbtion | DFc | 0.75 | 0.75 | 0.75 | |
| All valuesA | All valuesA | All valuesA | |||
|
| |||||
| Injection Rate Constant (IV Model) | la | Bolus Injection | 0.09A | 0.09A | N/A |
|
| |||||
| Glomular Filtration Rate (L filtrate/Kg* min) | GFR | Kidney | 0.011B | 0.074B | |
|
| |||||
| Blood Binding Rate Constant (min−1) | Kbb | Blood | −0.017C | −0.017 (Rat Value) | |
|
| |||||
| Molinate Tissue/Blood Partition Coefficient | P | Liver | 14.1 | 0.9 | 0.9 |
| Testes | 4.48 | 0.25 | 0.25 | ||
| Kidney | 14.1(Liver Value) | 3.75 | 3.75 | ||
| SlowlyPerfused | 4.17 (Muscle Value) | 40 | 40 | ||
| RichlyPerfused | 14.1(Liver Value) | 14.1 | 14.1 | ||
| Fat | 278 | 450 | 450 | ||
| All valuesD | |||||
|
| |||||
| Molinate Sulfoxide Tissue/Blood Partition Coefficient | PS | Liver | 1.08 | 0.163 | 0.163 |
| Testes | 1.03 | 0.116 | 0.116 | ||
| Kidney | 1.08 (Liver Value) | 1.18 | 1.18 | ||
| SlowlyPerfused | 0.95 (Muscle Value) | 0.84 | 0.84 | ||
| RichlyPerfused | 1.08 (Liver Value) | 1.08 | 1.08 | ||
| Fat | 4.85 | 0.3 | 0.3 | ||
| All valuesD | |||||
|
| |||||
| Tissue PA cross-product (L/min) | PA | Fat | 1.15 × 10−3 A | 1.15 × 10−3 A | |
|
| |||||
| Molinate Binding Capacity (nmol/L) | Bmax | 1A | 1A | 1A | |
| Molinate Sulfoxide | Fat | ||||
| Binding Capacity (nmol/L) | BSmax | 1A | 1A | 1A | |
|
| |||||
| Molinate Dissociation Constant (nmol/L) | Kd | 0.714A | 0.714A | 0.714A | |
| Molinate Sulfoxide | Fat | ||||
| Dissociation Constant (nmol/L) | KSd | 0.272A | 0.272A | 0.272A | |
see “Model Simulation and Fitting Procedure” section for optimization procedure details
Source
Optimized
Experiment
PBPK Model parameterization
PBPK models require three different sets of parameters a) species specific physiologic parameters b) physiochemical properties of the compound(s) of interest (estimated experimentally or using a previously published alogorithm, Poulin et al., 1995), and c) Km and Vmax values for metabolism and relevant kinetic constants. All parameters for adult animals were taken from US EPA Physiological parameter values for PBPK models (1994). The algorithm published by Poulin and Krishnan (1995) states that a chemical’s tissue: blood partition coefficient can be calculated by dividing the amount of chemical that partitions into the tissue by the sum of 0.37 X (amount of chemical that partitions into the erythrocyte) + 0.63 X ( amount of chemical that partitions into the plasma). The Poulin paper supplies the algorithm for calculating the amount of chemical in tissue, erythrocyte, and plasma plus the constant fractions of neutral lipid, phospholipid, and water for each tissue. Lastly the calculation requires the Kow or the octanol-water partition coefficient of the chemical (the ratio of the concentration of a chemical in octanol and in water at equilibrium and at a specified temperature): the Kow for molinate was taken from an individual pesticide report, (www.ars.usda.gov) and molinate sulfoxide was estimated with f values (hydrophobicity substituent constant) taken from Hansch and Leo (1979). Both of these figures were also calculated and verified using an online program LogKow, and WSKOWWIN from Syracuse Research Corporation, (www.syrres.com). The formula, the constant fractions, and the Kow of molinate and molinate sulfoxide were then used to estimate the various tissue PC’s for the oral and iv rat model.
The oral absorption bolus is described using a triexponetial equation from a separate absorption compartment with out blood flow. This gave the best fit to the experimental data but does not provide a physiological representation of the process (Craigmill, 2003). The oral absorption constants values were determined by a two step process; first a visual fitting of predicted plasma concentrations of molinate with observed concentrations for the 10 and 100 mg/kg oral dose, then a formal fit optimization was performed using the parameter estimation module, an optimization routine for fitting models to experimentally collected data, from ACSL (Advanced Continuous Simulation Language) Extreme Program (Aegis Corporation. Huntsville, AL). The oral absorption of molinate is described as occurring through both the portal and the lymphatic circulation and fecal elimination is also incorporated into this compartment (Figure 1).
In contrast, the IV administration is directed into the blood compartment using an algorithm for iv dosing provided by ACSL extreme software. Here again the iv injection rate constant value (la, the reciprocal pulse width, so at t=1/la, the pulse is cut off) was determined by visual fitting of predicted plasma concentrations of molinate with observed concentrations for the 1.5 and 15 mg/kg iv dose followed by a formal optimization.
Km and Vmax values for formation of hydroxymolinate and molinate sulfoxide in the liver were taken from Jewell and Miller, 1999 (Table 2). Recent studies have obtained kinetic constants describing the glutathione transferase-catalyzed detoxification of molinate sulfoxide in the liver, the nonenzymatic reaction rate of molinate sulfoxide with glutathione, and the reaction rate for removal of free sulfoxide in whole blood (Campbell et al., 2008). A previous study has demonstrated adduct formation of molinate sulfoxide with blood proteins. (Zimmerman et al., 2004). For the oral model the fecal excretion constant (Kf) (expressed as a fraction of dose and assumed to be unmetabolized parent compound) was taken from Debaun et al., 1978. Finally, cytochrome P450 induction and binding of molinate is not considered in the model, although liver concentrations were substantial. For the human simulation the same model was used including the PC’s optimized for the rat. However, human physiological and the metabolic parameters (as reported in Table 1, 2, 3) were substituted into the model as appropriate.
Model Simulation and Fitting Procedure
The PBPK model was developed with algebraic and differential equations describing the kinetics of molinate and molinate sulfoxide using the commercially available ACSL Extreme software which also contains a maximum likelihood estimation algorithm so that model parameters achieve best possible fit using the default Nelder-Mead algorithm, heteroscedasticity parameters were allowed to vary between 0 and 2, and limits were set on how much parameters can be adjusted to ensure biologically plausibility. In example both the metabolic and kinetic parameters were generally allowed to vary ± 1 standard deviation from the reported value. For this study the parameters that were included in the optimization procedure were all of the absorption constants, the partition coefficients, and the Km of the metabolizing enzyme responsible for molinate sulfoxide production.
Validation of PBPK model
Four experimental rat studies (two iv and two oral doses) were used to validate this model and are described below and in Table 4. The blood profiles from the iv doses of 1.5 and 15 mg/mg molinate were used to evaluate the performance of the iv molinate model. The time course data from the 10 and 100 mg/kg doses by oral gavage were then used to monitor and judge the performance of the oral dosed molinate model. The same chemical specific and physiological parameters that were used in the iv model were also employed in the oral dosed model. The experimental data for muscle concentrations was used as a surrogate for the slowly perfused compartment in the oral dosed model. One experimental human study by Batten et al., 1992 in which volunteers were orally administered 5.06 mg molinate in a gelatine capsule was used to validate the human model.
Table 4.
Experimental data reported on molinate disposition.
| No | Dose Regimen | Dose (mg/kg) | Vehicle | Animal | N per time point | Blood Cmax (ng/ml) | Reference |
|---|---|---|---|---|---|---|---|
| 1 | Single intravenous | 1.5 | Glycerol formal | Rat | 3–4 | 368 | |
| 2 | Single intravenous | 15 | Glycerol formal | Rat | 3–4 | 6,852 | |
| 3 | Single oral | 10 | Glycerol formal | Rat | 3–4 | 152 | |
| 4 | Single oral | 100 | Glycerol formal | Rat | 3–4 | 2,723 | |
| 5 | Single oral | 5.06 (mg) | Gelatin capsule | Human | 2 | 2.3 | Batten et al. (1992) |
Sensitivity analysis
During model simulations, it is important to establish the sensitivity of the parameters to small changes. Each parameter of this model was tested for sensitivity. This evaluation consisted of varying each parameter by a factor of 0.1% (i.e. Δx ÷ x = 0.001) and the dose metrics of interest computed. This study used the central difference formula for calculating sensitivity coefficients using AcslXtreme OptStat module. Finite difference simply varies the parameters by a small delta to calculate the derivatives whereas central difference uses the following equation to calculate finite difference:
where x is the response variable (e.g. molinate blood concentration) and Δx is the change in response variable or the fractional change in the output divided by the fractional change in the input. The two types of normalization are performed as follows. Normalization with respect to response variables is performed by dividing the computed sensitivity coefficient at each time slice by the value of the response variable at that time. Normalization with respect to a parameter is performed by multiplying the computed sensitivity coefficient at each time slice by the value of the parameter. Values were calculated for 49 model parameters for1.5 mg/kg iv and10 mg/kg oral dosed molinate. The iv model was used to calculate the impact on blood molinate and molinate sulfoxide levels whereas the oral dose was used to consider the impact of the various parameters on blood molinate and testicular molinate sulfoxide concentration. These different comparisons provide information about the behavior of the model under different exposure conditions.
RESULTS
IV PBPK Model
Evaluation of the model was conducted by simulating the experimental data obtained from exposing four Sprague-Dawley rats to a single iv dose of either 1.5 or 15 mg molinate/kg (Figure 3A and 3B). Blood concentrations are expressed in ng molinate/ml blood. Lines represent model simulations. Each point represents mean for 4 rats per group per time point. Molinate concentrations were determined in these rats just in blood. Molinate blood concentrations estimated by the model were modestly over predicted for the Cmax (5 min) to 30 min period of the blood molinate concentrations determined experimentally. From 30 minutes to 100 minutes the simulation adequately predicted the decline in HPLC measured blood concentrations. IV molinate dosing solutions measured ±11.2 % of theoretical concentration. The molinate sulfoxide simulations were more accurate in both the Cmax and consequent decline in blood concentration predictions out to100 minutes. The congruence of model prediction with experimentally determined blood concentrations suggests that distribution of molinate between whole blood and serum is equivalent
Figure 3.

Figure 3A and 3B. Blood compartment simulation plus LC-MS data for 1.5 and 15 mg/kg iv molinate dose from iv PBPK model.
Oral Dosed PBPK Model
Figures 4 through 6 show experimental data and the model predictions for molinate and molinate sulfoxide blood and tissue concentrations in rats following oral gavage dosings of molinate at 10 and 100 mg/kg body weight. Symbols (square) represent experimental data for blood or tissues and lines represent model simulations. Each point represents mean for 3 to 4 rats per group per time point. Oral molinate dosing solutions measured ± 17% of theoretical concentration.
Figure 4.

Figure 4A and 4B. Blood compartment simulation plus LC-MS data for 10 and 100 mg/kg oral dosed molinate from oral PBPK model.
Figure 6.

Figure 6A and 6B. Liver and Testes compartment simulation plus LC-MS data for 100 mg/kg oral dosed molinate from oral PBPK model.
In Figure 4A molinate blood predictions are within one standard deviation compared to most of the experimental data, however, the model slightly overestimates the first 60 min molinate blood concentration. Simulations for molinate sulfoxide (Figure 4B) for this exposure result in more accurate predictions of blood concentrations, within factor of 2 for the range of the experimental data for the entire time course.
FIG. 5A and 5B shows experimental data and the model predictions of molinate and molinate sulfoxide fat concentrations in rats. Since flow-limited assumptions in the preliminary model failed to describe the kinetics of molinate in adipose tissue, and both the 10 and 100 mg/kg simulation adequately forecasts the slightly rising molinate concentration levels measured by HPLC from one to four hours, both of these findings further support the diffusion-limited uptake assumption by using a visually fitted PA value of 1.15 × 10−3 L/min or approximately 1.04% of cardiac output. In sum, the molinate predictions are predominately within one standard deviation compared to most of the 100 mg/kg experimental data except the 30 minute HPLC measured time point whereas the model underestimates the molinate concentration for the first hour of the 10 mg/kg dose. The molinate sulfoxide predictions for the 100 mg/kg molinate exposure (Figure 5B) predominately resulted in simulations ± 1 SD from HPLC measured tissue concentrations with the exception of the 30 minute HPLC measured timepoint, whereas the 10 mg/kg simulation mainly fell within a factor of 2 for the range of the experimental data.
Figure 5.

Figure 5A and 5B. Fat compartment simulation plus LC-MS data for 10 and 100 mg/kg oral dosed molinate from oral PBPK model
In Figures 6A and 6B the symbols represent experimental data for molinate (square) and molinate sulfoxide (circle) and the model predictions of molinate and molinate sulfoxide liver and testes concentrations in rats from a 100 mg/kg dose of molinate. In Figure 6A the simulation initially overestimates the molinate concentration for the liver compartment then predominately approximates the rest of the time course data by a factor of ± 2. Further the model accurately estimates both the liver (Figure 6A) and testes (Figure 6B) molinate sulfoxide concentration by approximating the measured concentrations ± 1 SD. Finally the model predominately adheres to the measured testes molinate concentration ± 1 SD with an accurate Cmax prediction at T=30 minutes.
FIG. 7A and 7B shows experimental datum and the model predictions of molinate blood and model predictions for molinate sulfoxide testes concentrations in humans following peroral administration of 5.06mg molinate. The square represents experimental data for blood. The point represents mean for 2 humans (approximately 60 kg each) per group per time point. Lines represent model simulations. Briefly the human mean concentration of 2.3 ng/ml (n= 2) at 0.5 hr was described by the human blood simulation within a factor of 2. The human testes compartment simulation reveals a Cmax and Tmax of approximately 0.041 ng/ml and 51 minutes, respectively.
Figure 7.

Figure 7A. Human blood compartment and Figure 7B. human testes compartment oral PBPK model simulation. HPLC data from human oral dosed with 5.06 mg molinate included in Figure 7A.
Sensitivity Analysis
Sensitivity analysis was performed on all parameters in this PBPK model at one dose level for both iv and oral exposures. Sensitivity coefficients were calculated for two dose metrics for 48 and 55 model parameters for iv and oral exposure, respectively. However, to simplify the presentation of the analysis only parameters that resulted in a normalized sensitivity coefficient |NSC| > 0.1 of are presented and discussed.
For the 1.5 mg molinate/kg iv exposure both blood molinate and molinate sulfoxide concentrations were used. The two sets of coefficients were not correlated with each other (r2 = 0.139 and n = 13 for molinate and n = 21 for molinate sulfoxide). The three parameters which decreased molinate blood concentrations the most when they were increased (listed in descending order) were slowly perfused molinate PC, liver molinate PC and liver weight (w). Conversely, molinate blood concentrations achieved the highest concentrations when the following parameters were increased (listed in descending order); bodyweight, liver Km for sulfoxidation., and liver blood flow rate. Blood molinate sulfoxide concentrations were diminished the most by increasing the slowly perfused tissue volume as % body weight (vc), slowly perfused molinate PC, and liver Km for sulfoxidation. Molinate sulfoxide blood concentrations rose the highest when the following parameters were also increased (listed in descending order); fat molinate sulfoxide binding capacity, fat molinate dissociation constant of binding, and liver Vmax for sulfoxidation.
Given the difference in PC’s between molinate and molinate sulfoxide it is not surprising that their impacts do not vary directly. Blood molinate and molinate sulfoxide levels were the highest when body weight and various liver and fat parameters were increased. Increasing the Km for sulfoxidation appears to have a dual role; decreasing both the consumption of molinate and the production of molinate sulfoxide. As the Km is a direct measure of the enzymes affinity for the substrate, this is not a surprising finding; it is not readily apparent the importance of body weight plays of the molinate blood concentration prediction. While increasing both liver Vmax for sulfoxidation and fat molinate dissociation constant of binding should also increase the molinate sulfoxide blood concentration, it is also unclear by which mechanism increasing the molinate sulfoxide fat binding capacity would also increase the blood molinate sulfoxide concentration.
For the 10 mg molinate/kg oral dose, NSC’s for many parameters for blood molinate and testicular molinate sulfoxide peak concentrations were calculated (Figure 8). The two sets of coefficients n = 12 for molinate and n = 22 for molinate sulfoxide were not correlated with each other (r2 = 0.083). The blood molinate dose metric was most positively increased (listed in descending order of impact) by increasing the blood flows to kidney and fat compartments and also to Km for sulfoxidation in the liver compartment while decreased the most (again listed in descending order of impact) when to the molinate partition coefficient of the liver, slowly perfused compartment and the weight of the liver were increased. Conversely, the testicular molinate sulfoxide concentration was decreased the most by increasing the following three parameters; Km for sulfoxidation in the liver compartment, molinate sulfoxide PC in both the liver and slowly perfused compartment. Finally the testicular molinate sulfoxide dose metric was most sensitive to increases in the molinate sulfoxide PC for the testes compartment, the Km for glutathione conjugation of molinate sulfoxide in the liver compartment and body weight.
Figure 8.

NSC’s for several model parameters using blood molinate and testes molinate sulfoxide peak concentrations as the dose metrics. A 10 mg molinate/kg oral dose was simulated here.
DISCUSSION
The present study is the first attempt to publish a preliminary two species validated PBPK model simulating the concentrations of molinate in various tissues (PubMed Database, 2008), and compares PBPK model predictions of molinate and the toxic metabolite, molinate sulfoxide, to measured concentrations following single oral and iv administrations in the rat and human.
PBPK models are often used in toxicology and risk assessment for extrapolation across dose, route of exposures, and species (Simmons et al., 2002). Early PBPK models consisted of four or five compartments (Krishnan and Andersen, 1994), while current models usually contain more compartments and many now include increasingly sophisticated exposure equations. Compared with classical pharmacokinetic models, there are several advantages are offered by PBPK models including (1) the capacity to provide simultaneous time versus concentration curves for a compound and or metabolite in any organ or tissue; (2) the incorporation of anatomical and physiologic information as well as chemical specific in vitro derived parameters (i.e. Km or Vmax of a metabolizing enzyme); (3) the ability to predict the time versus concentration curve of chemicals across species by allometric scaling; (4) the resultant PBPK model has the potential for extrapolations from observed data to predicted situations, an important application for risk assessment. They also have several disadvantages in that they are calculation intensive, the use of complicated specialized software usually is required, and that physiological input parameters are often ill-defined for various strains and species (Medinsky and Klaassen, 1996).
For the present study, the reliability of the model was tested by simulation with the iv dose experimental data (1.5 and 15 mg/kg molinate). Since in vitro or in vivo derived parameters for absorption of molinate were unavailable, these parameters were obtained by model calibration to two sets of in vivo experimental data after rats were administered single oral dose of 10 and 100 mg/kg molinate. The resultant presentation is a novel, triexponetial exposure simulation for the absorption of molinate following an oral dose. Additionally, adjustments were made to improve the fit; for example, by decreasing or increasing the partition coefficient in each compartment. Generally, results indicate reasonable concordance between model predictions and measured value in all target tissues following both oral and iv administration. Tissue dosimetries of total molinate were reasonably predicted after oral exposures to molinate with the adjustment of the tissues:blood partition coefficients. Additionally, the experimental data of Jewell et al., 1998 which shows good agreement between blood and testes concentrations further supports the flow limited assumption of this compartment.
Switching the fat compartment to a diffusion limited assumption allowed for a better correlation to the measured molinate data, however it still underestimated the elimination phase for molinate sulfoxide. The partition coefficients for the testes and liver had also to be adjusted from the values estimated in order to obtain a good fit with the experimental data (Figures 6A and 6B). The major adjustment was made to the Km for the sulfoxide production. The Km needed to be adjusted much lower in order to produce sufficient levels of sulfoxide in the blood compartment. The data suggests one of three explanations; the original sulfoxidation Km measurement was not perfect, there is some other source of sulfoxide production present besides that in the liver (perhaps in the lung or testes), or that some form of binding, perhaps to a receptor or other less likely but still plausible mechanisms such as diffusion–limitation or enterohepatic recirculation or both occurring in the kidneys and liver (Keys et al., 1999). While the discordance between experimental and optimized Km suggests the need for further research, no attempt was made to further investigate any of the proposed hypotheses.
Sensitivity analyses were implemented to evaluate the relative importance of model parameters on model output at various times. Sensitivity analyses allow for a quantitative assessment of input parameters on the model simulations of tissue concentrations (Simmons et al., 2002). The present results indicate that the influence of PBPK model input parameters on total molinate tissue dosimetry varies, as expected, across parameter. Evaluation of the absolute magnitude of the sensitivity parameters may help to improve future experimental design and or guide decisions regarding which specific parameters require additional experiments to further refine or define.
Future work would consider is the capability of the testis to form and to detoxify the sulfoxide; therefore the importance of testicular metabolism could be ascertained in the rat and incorporated into the human model as needed. The experiments could also explore the primary occupational exposure routes (both dermal and inhalation) for field workers exposed to the herbicide as opposed to oral route demonstrated here which mimics exposures such as drinking water. Finally longer time points would be helpful in addressing the role molinate fat storage plays on both blood concentration levels and elimination of molinate as evidenced recently by Levitt (2007), as well as for discerning distinctions from different routes of administration and for comparison to different data sets used for validation of the model.
Acknowledgments
I thank Dr. Jeffery Fisher from Department of Environmental Health Science, College of Public Health University of Georgia, Athens GA and Drs. Arthur Craigmill and Marion Miller, Department of Environmental Toxicology, UC Davis, California, for their generous contributions of time, resources, and mentorship. I also thank Ms. Betty Brunell and Mr. Robin McDougall of AEGis Technologies Group, Inc., Huntsville, AL for technical support. Lastly I thank Will Phillips for work on the preliminary model and both him and Daniel Medina-Cleghorn for technical assistance. This work was supported by the California Rice Research Board (RP-7) and by the National Institute of Environmental Health Sciences Training Grant 5-T32-EOS- 7059.
Appendix
Abbreviations
Ai: amount of compound in compartment i (nmol)
Ami: Amount metabolized in compartment i (nmol)
Abio: Amount dose available after fecal elimination (nmol)
a0: Starting amount of dose at absorption site at time zero, allocated to rapid phase absorption (nmol)
b0: Starting amount of dose at absorption site at time zero, allocated to slow phase absorption (nmol)
Bmax: Binding Capacity (nmol/L)
c0: Starting amount of dose at absorption site at time zero, allocated to slowest phase absorption (nmol)
Cfree: Free concentration of dose in tissue (nmol/L)
BVFi: Blood volume fraction of compartment i
Ci: Concentration of compartment i (nmol/L)
Dfa: Fraction of starting amount of dose at absorption site at time zero, allocated to rapid phase absorption (nmol)
Dfb: Starting amount of drug in dose at absorption site at time zero, allocated to slow phase absorption (nmol)
Dfc: Starting amount of dose at absorption site at time zero, allocated to slowest phase absorption (nmol)
Dose: Starting amount of dose (nmol/L)
K1: Fast molinate rate constant of absorption (min−1)
K2: Slow molinate rate constant of absorption (min−1)
K3: Slowest molinate rate constant of absorption (min−1)
Kbloodadduct: First order blood adduction rate constant for molinate sulfoxide
Kd: Dissociation Constant (nmol/L)
Kfecal: Fecal elimination constant for molinate
la: Injection Constant
PAi: Tissue permeability constant (L/Hr)
PCi: Compartment i to tissue to plasma partition coefficient
Pfc: Fraction of Tissue Weight that is Cytosolic Protein
Pfl: Fraction of Tissue Weight that is Liver Protein
Ri: Rate of metabolism or excretion from compartment i (nmol/min): Liver CYP elimination rates: (Rgst-L = glutathione transferase –liver Roh = hydroxaylses Rsox =Sulfoxidation) and Rbloodadduction blood adduction elimination rate,
Qi: Plasma flow (Q) to organ i (L/min)
Vi: Volume of compartment i (L)
Vbi: Tissue Blood Volume of compartment i (L)
W: weight liver (gram)
Molinate Equations
Rate differential equation for flow and diffusion limited compartments:
Oral Model
IV Model
min = ACSL coding where the output y is the value of the minimum argument.
Amounts, concentrations, and elimination rates and (metabolism: Roh, sox, ):
Molinate Sulfoxide Equations
Rate differential equation for flow limited compartments:
Concentration and elimination rates (metabolism: Rgst-L, oh, sox, ):
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen ME, Dennison JE. Mode of action and tissue dosimetry in current and future risk assessments. Sci Total Environ. 2001;274:3–14. doi: 10.1016/s0048-9697(01)00744-6. [DOI] [PubMed] [Google Scholar]
- Batten PL, Wollen BH, Loftus NJ, Mash JR, Wilks MF. Report # CTL/R/1099. ICI Central Toxicology Laboratory, Alderly Park; Macclesfield, U.K: 03/06/1992. [Google Scholar]
- Berger T, Miller MG. In vitro fertilization after in vivo treatment of rats with three reproductive toxicants. Horner CM Reprod Toxicol. 2000;14:45–53. doi: 10.1016/s0890-6238(99)00062-3. [DOI] [PubMed] [Google Scholar]
- Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health. 1997;13:407–484. doi: 10.1177/074823379701300401. [DOI] [PubMed] [Google Scholar]
- Campbell AR, Holstege DK, Swezey RL, Medina-Cleghorn D. Detoxification of molinate sulfoxide: Comparison of spontaneous and enzymatic glutathione conjugation using human and rat liver cytosol. J Toxicol Environ Health, Part A. 2008;71:1338–1347. doi: 10.1080/15287390802240975. [DOI] [PubMed] [Google Scholar]
- Casida JE, Tilles H, Gray RA. Thiocarbamate sulfoxides: Potent, selective 22 and biodegradable herbicides. Science. 1974;184:573–574. doi: 10.1126/science.184.4136.573. [DOI] [PubMed] [Google Scholar]
- Chapman DE, Moore TJ, Michener SR, Powis G. Metabolism and covalent binding of 14C-toulene by human and rat liver microsomal fractions and liver slices. Drug Metab Dispos. 1990;18:929–936. [PubMed] [Google Scholar]
- Cochran RC, Formoli TA, Pfeifer KF, Aldous CN. Characterization of risks associated with the use of molinate. Regul Toxicol Pharmacol. 1997;25:146–157. doi: 10.1006/rtph.1997.1082. [DOI] [PubMed] [Google Scholar]
- Coniglio JG, Grogan WM, Rhamy RK. Lipid and fatty acid composition of human testes removed at autopsy. Biol Repro. 1975;12:255–259. doi: 10.1095/biolreprod12.2.255. [DOI] [PubMed] [Google Scholar]
- Craigmill AL. A physiologically based pharmacokinetic model for oxytetracycline residues in sheep. J Vet Pharmacol Therap. 2003;26:55–63. doi: 10.1046/j.1365-2885.2003.00451.x. [DOI] [PubMed] [Google Scholar]
- Dahana A, Mendelmanb A, Amsilib S, Ezovb N, Hoffman A. The effect of general anesthesia on the intestinal lymphatic transport of lipophilic drugs: Comparison between anesthetized and freely moving conscious rat models. Eur J Phar Sci. 2007;32:367–374. doi: 10.1016/j.ejps.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Dean WP. Ordram® 10G: Acute toxicity studies in rats and rabbits: DPR Vol 228-007 # 945329 1977 [Google Scholar]
- DeBaun JR, Bova DL, Tseng CK, Menn JJ. Metabolism of 14C-ordram (molinate) in the rat. 1 Balance and tissue residue study. J Agric Food Chem. 1978;26:1096–1098. doi: 10.1021/jf60219a058. [DOI] [PubMed] [Google Scholar]
- DeBaun JR, Bova DL, Tseng CK, Menn JJ. Metabolism of [ring-14C]-Ordram (Molinate) in the rat. 2 Urinary metabolite identification. J Agric Food Chem. 1978;26:1098–1104. doi: 10.1021/jf60219a007. [DOI] [PubMed] [Google Scholar]
- Duescher RJ, Lawton MP, Philpot RM, Elfarra AA. Flavin-containing monooxygenase (FMO)-dependent metabolism of methionine and evidence for FMO3 being the major FMO involved in methionine sulfoxidation in rabbit liver and kidney microsomes. J Biol Chem. 1994;269:17525–17530. [PubMed] [Google Scholar]
- Ellis MK, Richardson AG, Foster JR, Smith FM, Widdowson PS, Farnworth MJ, Moore RB, Pitts MR, Wickramaratne GA. Molinate: Elucidation of the processes underlying the reproductive effects in the male rat. Toxicol Appl Pharmacol. 1998;151:22–32. doi: 10.1006/taap.1998.8371. [DOI] [PubMed] [Google Scholar]
- Gwynne JT, Mahaffee D, Brewer HB, Jr, Ney RL. Adrenal cholesterol uptake from plasma lipoproteins: Regulation by corticotropin. Proc Natl Acad Sci USA. 1976;73:4329–4333. doi: 10.1073/pnas.73.12.4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C, Leo A. The Fragment Method of calculating partition coefficients. In: Hansch C, Leo A, editors. Substituent constants for correlation analysis in chemistry and biology. New York: John Wiley & Sons; 1979. pp. 18–43. [Google Scholar]
- International Life Sciences Institute (ILSI) Physiological Parameter Values for PBPK Models 1994 [Google Scholar]
- Jewell WT, Miller MG. Comparison of human and rat metabolism of molinate in liver microsomes and slices. Drug Metab Dispos. 1999;27:842 – 847. [PubMed] [Google Scholar]
- Jewell WT, Hess RA, Miller MG. Testicular toxicity of molinate in the rat: Metabolic activation via sulfoxidation. Toxicol Appl Pharmacol. 1998;149:159–166. doi: 10.1006/taap.1998.8380. [DOI] [PubMed] [Google Scholar]
- Jewell WT, Miller MG. Identification of a carboxylesterase as the major protein bound by molinate. Toxicol Appl Pharmacol. 1998;149:226–234. doi: 10.1006/taap.1998.8381. [DOI] [PubMed] [Google Scholar]
- Kaphalia BS, Fritz RR, Ansari GS. Purification and characterization of rat liver microsomal fatty acid ethyl and 2-chloroethyl ester synthase and their relationship with carboxylesterase (pI 6.1) Chem Res Toxicol. 1997;10:211–218. doi: 10.1021/tx960079e. [DOI] [PubMed] [Google Scholar]
- Kavlock R, Cummings A. Mode of action: reduction of testosterone availability--molinate-induced inhibition of spermatogenesis. Crit Rev Toxicol. 2005;35:685–690. doi: 10.1080/10408440591007386. [DOI] [PubMed] [Google Scholar]
- Keys DA, Wallace DG, Kepler TB, Conolly RB. Quantitative evaluation of alternative mechanisms of blood and testes disposition of di(2-ethylhexyl) phthalate and mono(2-ethylhexyl) phthalate in rats. Toxicol Sci. 1999;49:172–185. doi: 10.1093/toxsci/49.2.172. [DOI] [PubMed] [Google Scholar]
- Krieger R, Fong H, Frederickson S, Hernandez B, McChesney M, Ross J, Schneider F, Seiber J. Molinate metabolism differs substantially in humans and rats. The Toxicologist. 1992;12:418. [Google Scholar]
- Krishnan K, Andersen ME. Physiologically based pharmacokinetic modeling in toxicology. In: Hayes AW, editor. Principles and Methods in Toxicology. 3. New York: Raven; 1994. pp. 149–148. [Google Scholar]
- Lake BG. Preparation and characterization of microsomal fractions for studies on xenobiotic metabolism. In: Snell K, Mullock B, editors. Biochemical Toxicology, A Practical Approach. IRL Press; Oxford: 1987. pp. 183–215. [Google Scholar]
- Levitt DG. Heterogeneity of human adipose blood flow. BMC Clinical Pharmacology. 2007;7 doi: 10.1186/1472–6904-7-1. http://www.biomedcentral.com/1472-6904/7/1. [DOI] [PMC free article] [PubMed]
- Lythgoe RE, Jones BK, Macpherson D. Molinate: excretion and blood kinetics in the monkey. 1992;228–132 ICI Study No.CTL/L/4432 DPR. #118003. [Google Scholar]
- Morgan EW, Yan B, Greenway D, Petersen DR, Parkinson A. Purification and characterisation of two rat liver microsomal carboxylesterases (hydrolase A and B) Arch Biochem Biophys. 1994;315:495–512. doi: 10.1006/abbi.1994.1531. [DOI] [PubMed] [Google Scholar]
- Medinsky MA, Klassen CD. Toxicokinetics. In: Klaasen CD, editor. Casarett and Doull’s Toxicology: The Basic Science of Poisons. 5. New York: McGraw-Hill; 1996. pp. 187–198. [Google Scholar]
- Miller MG, Beyer J, Hall G, DeGraffenreid LA, Adams PA. Predictive value of liver slices for metabolism and toxicity in vivo. Toxicol Appl Pharmacol. 1992;122:108–116. doi: 10.1006/taap.1993.1178. [DOI] [PubMed] [Google Scholar]
- Payne AH, Quinn PG, Stalvey JRD. The stimulation of steroid biosynthesis by luteinising hormone. In: Ascoli M, editor. Luteinizing Hormone Action and Receptors. CRC Press: Boca Raton, FL; 1985. pp. 135–173. [Google Scholar]
- Pettersson S, Söderholm B, Persson JE, Eriksson S, Fritjofsson Å. Testicular blood flow in man measured with venous occlusion plethysmography and xenon133. Scand J Urol Nephrol. 1973;7:115–119. doi: 10.3109/00365597309133685. [DOI] [PubMed] [Google Scholar]
- Poulin P, Krishnan K. An algorithm for predicting tissue: blood partition coefficients of organic chemicals from n-octanol: water partition coefficient data. J Toxicol Environ Health. 1995;46:117–129. doi: 10.1080/15287399509532021. [DOI] [PubMed] [Google Scholar]
- Sidelmann UG, Cornett C, Tjornelund J, Hansen SH. A comparative study of precision cut liver slices, hepatocytes, and liver microsomes from the Wistar rat using metronidazole as a model substance. Xenobio. 1996;26:709–722. doi: 10.3109/00498259609046744. [DOI] [PubMed] [Google Scholar]
- Simmons JE, Boyes WK, Bushnell PJ, Raymer JH, Limsakun T, McDonald A, Sey YM, Evans MV. A physiologically based pharmacokinetic model for trichloroethylene in the male long-evans rat. Toxicol Sci. 2002;69:3–15. doi: 10.1093/toxsci/69.1.3. [DOI] [PubMed] [Google Scholar]
- Sundberg K, Dreij K, Seidel A, Jernstrom B. Glutathione conjugation and DNA adduct formation of dibenzo[a,l]pyrene and benzo[a]pyrene diol epoxides in V79 cells stably expressing different human glutathione transferases. Chem Res Toxicol. 2002;15:170–179. doi: 10.1021/tx015546t. [DOI] [PubMed] [Google Scholar]
- Tjeerdema RS, Crosby DG. The biotransformation of molinate in striped bass (Morone saxatilis) Aquat Toxicol. 1987;9:305–317. [Google Scholar]
- Tomenson JA, Taves DR, Cockett AT, McCusker J, Barraj L, Francis M, Pastoor TP, Wickramaratne GA. An assessment of fertility in male workers exposed to molinate. J Occup Environ Med. 1999;41:771–787. doi: 10.1097/00043764-199909000-00009. [DOI] [PubMed] [Google Scholar]
- Zimmerman LJ, Valentine HS, Amarnath K, Valentine WM. Identification of a S-Hexahydro-1H-azepine-1-carbonyl Adduct Produced by Molinate on Rat Hemoglobin β2 and β3 Chains in Vivo . Chem Res Toxicol. 2002;15:209 – 217. doi: 10.1021/tx015564a. [DOI] [PubMed] [Google Scholar]
- Zimmerman LJ, Valentine HL. Characterization of S-(N,N-Dialkylaminocarbonyl)cysteine: Adducts and enzyme inhibition produced by thiocarbamate herbicides in the rat. Chem Res Toxicol. 2004;17:258–267. doi: 10.1021/tx034209c. [DOI] [PubMed] [Google Scholar]