

Abstract
Human endogenous retroviruses (HERVs) are remnants of ancient retroviral infections within the human genome. These molecular fossils draw parallels with present-day exogenous retroviruses and have been linked previously with immunopathology within rheumatoid arthritis (RA). Mechanisms of pathogenesis for HERV-K in RA such as molecular mimicry were investigated. To clarify a role for HERVs in RA, potential autoantigens implicated in autoimmunity were scanned for sequence identity with retroviral epitopes. Short retroviral peptides modelling shared epitopes were synthesized, to survey anti-serum of RA patients and disease controls. A novel real-time polymerase chain reaction (PCR) assay was also developed to quantify accurately levels of HERV-K (HML-2) gag expression, relative to normalized housekeeping gene expression. Both serological and molecular assays showed significant increases in HERV-K (HML-2) gag activity in RA patients, compared to disease controls. The real-time PCR assay identified significant up-regulation in HERV-K mRNA levels in RA patients compared to inflammatory and healthy controls. Exogenous viral protein expression and proinflammatory cytokines were also shown to exert modulatory effects over HERV-K (HML-2) transcription. From our data, it can be concluded that RA patients exhibited significantly elevated levels of HERV-K (HML-2) gag activity compared to controls. Additional factors influencing HERV activity within the synovium were also identified. The significant variation in RA patients, both serologically and transcriptionally, may be an indication that RA is an umbrella term for a number of separate disease entities, of which particular HERV polymorphisms may play a role in development.
Keywords: arthritis, autoimmunity, retrovirus, rheumatology, virus
Introduction
Rheumatoid arthritis (RA) is an autoimmune disorder involving chronic inflammation of the synovial membrane, culminating in the pathological destruction of cartilage and bone in affected joints. Overall the resultant pain, loss of function and disability causes significant morbidity and mortality in the western world. The aetiology of RA remains unknown, although possible causes include a genetic predisposition combined with environmental triggers, such as viral and bacterial agents. More recently, human endogenous retroviruses (HERVs) have been implicated as potential aetiological agents within RA [1]. HERVs account for up to 9% of the non-protein coding portion of the genome [2] and represent remnants of previous retroviral infections that have since become integrated into germline cells of their host [3]. Over time, HERVs have been subject to repeated amplification giving rise to single and multi-copy proviruses that have become distributed across the genome, with many becoming defective through the accumulation of mutations, although a limited number retain the ability to produce proteins and functional particles [4]. Retroviral infections in animal models and humans suggest a potential association between exogenous retroviruses and RA [5,6]. Clinical studies have also shown increases in levels of antibodies to HERV proteins [1] and HERV transcriptional activity [7] in RA patients compared to controls. More recent studies have demonstrated significant increases in levels of HERV-K transcriptional activity at the site of disease [8].
In order to clarify a mechanism for the role of HERV-K in RA, bioinformatic methodologies were first used to predict key antigenic regions of HERVs that shared sequence homology with epitopes identified within autoantigens in RA disease pathology, potentially implicating molecular mimicry. Candidate epitopes were then synthesized as short peptides and an enzyme-linked immunosorbent assay (ELISA) was used to test the reactivity of patient serum to shared epitopes. Both non-biotinylated and biotinylated peptides were used to maximize the efficiency of epitope presentation. Finally a quantitative two-step reverse transcription–polymerase chain reaction (RT–PCR) was designed for the accurate and sensitive determination of HERV-K (HML-2) gag1 gene transcription in RA patients and controls and to investigate any potentially influencing factors of HERVs such as inflammatory cytokines and other viral pathogens.
Materials and methods
Patients and samples
Blood samples were taken from 48 RA patients who attended Rheumatology Clinics at New Cross Hospital, Wolverhampton and Heartlands Hospital, Birmingham. All RA patients satisfied the criteria set out by the American Rheumatism Association [9]. Serum samples were also taken from control groups including 27 osteoarthritis (OA); 17 irritable bowel disease (IBD); 23 systemic lupus erythematosus (SLE); and 37 normal healthy donors (NHD). All patient samples were taken with patient consent and full local regional ethics committee (LREC) approval. Fibroblast-like synoviocyte (SF) primary cells were purchased commercially (Cell Applications, San Diego, CA, USA). RA/OA/NHD donors were Caucasian (42, 66 and 58 years of age) and male, female and female, respectively.
Separation of cells and RNA extraction
Peripheral blood mononuclear cells (PBMCs) were separated from heparinized whole blood using Histopaque–1077 (Sigma, Poole, UK). Total RNA was extracted from peripheral blood mononuclear cells extracted from patient samples [10]. RNA quality was confirmed by gel electrophoresis. RNA was extracted from cultured cells using RNeasy RNA extraction kits (Qiagen, Crawley, UK).
Bioinformatic analysis
Protein sequences of autoantigens and HERV families associated with RA and SLE were identified from the National Center for Biotechnology Information (NCBI)/GenBank online database (http://www.ncbi.nlm.nih.gov/gquery/). In-silico analysis was performed using publically available algorithms (http://www.expasy.ch/cgi-bin/protscale.pl). Linear scales were used to assay amino acids for hydrophilicity [11], polarity [12], solvent accessibility [13] and flexibility [14] and location on or adjacent to sites of beta turns using ‘BetaPred2’ (http://www.imtech.res.in/raghava/betatpred2/) [15]. Residues in excess of the threshold value in four (or more) of the five scales/criteria were considered antigenic. Epitopes were scanned for homology with autoantigens reported previously in RA pathogenesis using ‘LALIGN’ (http://www.ch.embnet.org/software/LALIGN_form.html). All analysis was run in comparison with a published algorithm ‘BCEPRED’ (http://www.imtech.res.in/raghava/bcepred/) [16].
Peptide synthesis
Peptide PLSK was synthesized manually using Fmoc solid-phase synthesis as described previously [17] on a 0·2 mM scale. Peptide identity was verified by molecular weight using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF) (Shimadzu UK Ltd, Milton Keynes, UK). Peptides were lyophilized and resuspended in sterile phosphate buffer to a concentration of 10 mM and stored at −20°C. Peptides KPR, KPR-biot and Negcont1 and anti-serum were synthesized commercially (Severn Biotech, Kidderminster, UK).
Indirect ELISA
Non-biotinylated ELISA was carried out using peptides KPR (GPESKPRGTSPLPAG) and PLSK (VGPLESKPRGPSPLSA). Wells on a 96-well plate (HB2 Immunolon, Dynex Technologies, Worthing, UK) were coated with 8 µg/ml peptide and incubated 16 h, at 37°C. Plates were washed three times with phosphate-buffered saline (PBS) using a Well washer 4 MK2 (Thermo Labsystems, Waltham, MA, USA), blocked with 2% bovine serum albumin (BSA) in PBS for 1 h at 37°C. After three washes with PBS/Tween20 (Tw20) (0·1%) patient anti-sera was diluted in PBS/Tw20 (0·05%)/BSA (2%) and 50 µl was added to the wells and incubated at 37°C for 1 h. After washing three times with PBS/Tw20 (0·1%), rabbit anti-human immunoglobulin (Ig)G/IgM conjugate (Dako, Ely, UK) was diluted in PBS/Tw20 (0·1%)/BSA (2%) and incubated for 1 h at 37°C in the dark. Plates were washed with PBS/Tw20 (0·1%) before 3, 3′, 5, 5′-tetramethyl benzidine (TMB) liquid substrate system was added to each well. Reactions were stopped after 5 min using 2 N hydrochloric acid. Absorbance was measured at 450 nm using a Multiscan MS spectrophotometer (Thermo Lab Systems). Biotinylated ELISAs were carried out using streptavadin-coated plates (Pierce Biotechnologies Inc., Rockford, IL, USA) according to the manufacturer's instructions.
Inhibition ELISA
An inhibition assay was performed as described previously [18]. Primary antibody was diluted appropriately with PBS/Tw20 (0·05%)/BSA (2%) and incubated with 0·5 log10 dilution series of the target or control peptides for 1 h at room temperature. Incubated preparations were transferred to a plate precoated with the relevant peptide and ELISA protocols were carried out as outlined above.
Real-time PCR reactions
One microgram of total RNA and was converted into cDNA using oligodeoxythymidylic acid (oligo-dT) primers and the avian myeloblastosis virus (AMV) reverse transcriptase enzyme (Promega, Madison, WI, USA). Reagents [magnesium chloride (50 mM), reverse transcription buffer, deoxyribonucleoside triphosphates (dNTPs) and sterile nuclease-free water] were added to RNA and incubated at 42°C for 80 min and 95°C for 5 min. cDNA template (2·5 µl) was added to PCR mastermix containing 9 µl of nuclease-free dH2O, 0·5 µl of each primer (100 nM) (Gag1 forward: GGGGCCATCAGAGTCTAAACC, Gag1 reverse: TGATAGGCTACTTGCGGTTGG) and 12·5 µl absolute SYBR green master mix (Abgene, Epsom, UK). Results were normalized to the geometric mean CT value of two housekeeping genes (HKG) – hypoxanthine–guanine phosphoribothymidine (HPRT) and beta-actin (ACTB). Reagents were transferred to a 96-well plastic plate (Abgene) and run on an iCycler (Bio-Rad Laboratories, Hemel Hempstead, UK). Cycling conditions were 95°C for 15 min, then 40 cycles of 95°C for 30 s, 56°C for 30°C, 72°C for 30 s and a final cycle of 56°C for 1 min.
Cell culture
Human fibroblast-like synoviocytes (HFLS) were grown in synoviocyte basal medium supplemented with growth supplement (Cell Applications). Cells were grown until 70–90% confluency at 37°C in 5% CO2 before being passaged using the subculture reagent kit according to the manufacturer's instructions. HEK-293T cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum.
Viral protein expression studies
Two × 104 cells (HEK-293 and HFLS) were seeded in 500 µl of supplemented medium until confluent. Expression plasmids psG5 (Stratagene, La Jolla, CA, USA) containing inserts of Epstein–Barr virus (EBV) genes EBNA1, LMP2 and LMP2a (kindly donated by P. G. Murray); 0·2–0·4 µg plasmid DNA was mixed in 25 µl Opti-MEM without serum (Invitrogen, Carlsbad, CA, USA) before 0·5–5 µl of lipofectamine (Invitrogen) was added to both aliquots. After incubating for 30 min at ambient temperature, 0·2 ml was added to cell cultures, before incubating for 12 h at 37°C. After incubation, 0·4 ml growth medium supplemented with 2× normal serum concentration was added to the cells. After 48 h cells were harvested and total RNA was isolated and purified using RNeasy kits (Qiagen). Total RNA was quantified using a spectrophotometer and levels of HERV-K (HML-2) gag expression were determined using quantitative RT–PCR (qRT–PCR). All experiments were performed in triplicate.
Cytokine treatments
Two × 104 cells of HFLS primary cell lines (RA, OA and NHD) and HEK-293 cells were seeded in vented flasks before incubating until confluent in supplemented media. Proinflammatory cytokines [interleukin (IL)-6 and tumour necrosis factor (TNF)-α (Chemicon, Chandlers Ford, UK)] were diluted to required concentrations using unsupplemented medium and incubated with cells at 37°C for 48 h. After 48 h, cell viability was determined using Celltiter 96 AQueous one solution reagent (Promega). Cells were then harvested and genetic material was extracted using RNeasy RNA extraction kits (Qiagen). All experiments were carried out in triplicate.
Statistical analysis
Statistics and graphical analysis were performed using Prism 5.1 (Graphpad Software, San Diego, CA, USA). Statistical significance between groups was determined by t-test (paired and unpaired) and using one-way analysis of variance (anova) (with Bonferroni and Turkeys’ multiple comparison test where appropriate). A P-value of <0·05 was considered to be statistically significant.
Results
Bioinformatics
Bioinformatic analysis predicted a number of antigenic regions, shared between both HERVs and host autoantigens, which may play potential roles in disease development (Table 1). One epitope of interest shared five consecutive residues identity with Collagen II, a target of autoantibodies in RA patients. This epitope (KPR), located on the Gag1 protein of HERV-K (HML-2), was chosen for synthesis as a short peptide, but also correlated with the same region of HERV-K (HML-2) gag1 (M14123) to which the qRT–PCR primers were designed. A second epitope (PLSK) was identified at the same location on HERV-K Gag1 (Y18890), but differed from KPR by five residues. Inhibition analysis indicated no serological cross-reactivity was detectable between peptides (Fig. 2). The study also included a control peptide, identified within a Gypsy/Ty3 element, in Arabidopsis thaliana (Negcont1). No predicted epitopes (retroviral or host autoantigen) exhibited significant similarity with Negcont1.
Table 1.
The highest scoring alignments identified between epitopes identified on human endogenous retroviruses (HERVS) and host autoantigens.
| HERV/protein | Accession | Autoantigen | Accession | Shared identity/non-residues |
|---|---|---|---|---|
| HERV-K10 Gag1* | M14123 | Collagen II | NP_001835 | 5 |
| HERV-K Gag** | M14123 | Calpastatin | NP_001741 | 7+/4+ |
| Collagen I | NP_000080 | |||
| HERV-K10 Gag2 | M14123 | 70 kDa Ku antigen | 5++ | |
| HERV-K10 Pol/Env | M14123 | DNA topoisomerase I | NP_003277 | 5+ |
| HERV-K10 Pol/Env | M14123 | Heat shock protein 90 | NP_005339 | 4+ |
| HERV-K10 Pol/Env | M14123 | U1 | P08621 | 7+ |
| HERV-K10 Pol/Env | M14123 | 70 kDa Ku antigen | AAH72449 | 5+ |
| HERV-K10 Pol/Env | M14123 | AHNAK | NP_001611 | 7 |
| HERV-K Env | Y18890 | DNA topoisomerase I | NP_003277 | 5+ |
| ERV3 Pol | M12140 | 52 kDa SS-A/Ro | NP_003132 | 5 |
| ERV-3 Env | M12140 | G6PI | P06744 | 6++ |
| HERV-E(4-1) Gag | M10976 | Ku 86/90 | P13010 | 6 |
| HERV-E(4-1) Gag | M10976 | MMP-1 | NP_002412 | 5 |
| HERV-E(4-1) Gag | M10976 | Histone 1 | NM_005319 | 5+ |
| HERV-E(4-1) Pol | M10976 | RA-47 | NP_001226 | 5 |
| HRES-1 p25 | X16660 | Collagen II | NP_001835 | 8+++ |
Accession numbers indicate allocations within National Center for Biotechnology Information (NCBI) database. Shared identity numbers indicate the number of amino acid residues shared between both. +: Partial match between residues.
KPR peptide;
PLSK peptide; AHNAK: Desmoykin; G6PI, glucose 6 phosphoisomerase; MMP-1, matrix metalloproteinase 1; RA-A47, rheumatoid arthritis-related antigen 47 kDa.
Fig. 2.
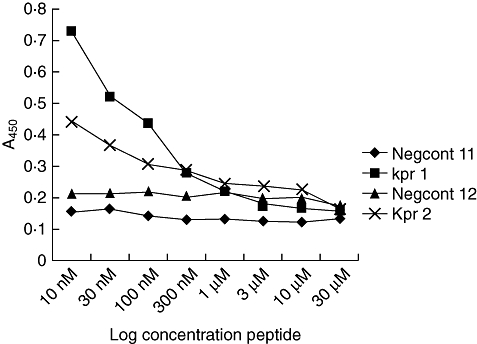
Degree of inhibition shown by two patient bleeds (1 and 2) after incubation with non-biotinylated peptide and negative control peptide.
Serological assays
An ELISA was developed to determine levels of anti-PLSK and anti-KPR in patient anti-serum. Although results to non-biotinylated antigen were indicative of low levels of reactivity [i.e. optical densities (ODs) of <0·1], individual peptides exhibited distinct patterns of activity in different patient cohorts, i.e. OA patients showed increased reactivity to PLSK but not KPR, with marked variation between individuals in each cohort (Fig. 1a,b). Levels of reactivity to a biotinylated KPR peptide were increased significantly compared to non-biotinylated peptide (paired t-test, P < 0·001) and were of increased significance in RA patients (one-way anova with Bonferroni post-test, P < 0·001), in a disease-specific manner (Fig. 1c). Age- and sex-matched RA patients exhibited significantly increased levels of anti-KPR antibodies compared to OA patients (paired t-test, P = 0·0039), although differences between synovial fluid and peripheral blood in paired samples were not significant. The specificity of results was confirmed by competitive inhibition ELISAs, using the two highest responding RA patient samples. Both anti-sera tested exhibited increased OD, with increasing dilutions of incubated peptide (KPR) (Fig. 2). Conversely, the OD values of anti-serum preincubated with a non-related control peptide (Negcont1) remained unchanged.
Fig. 1.
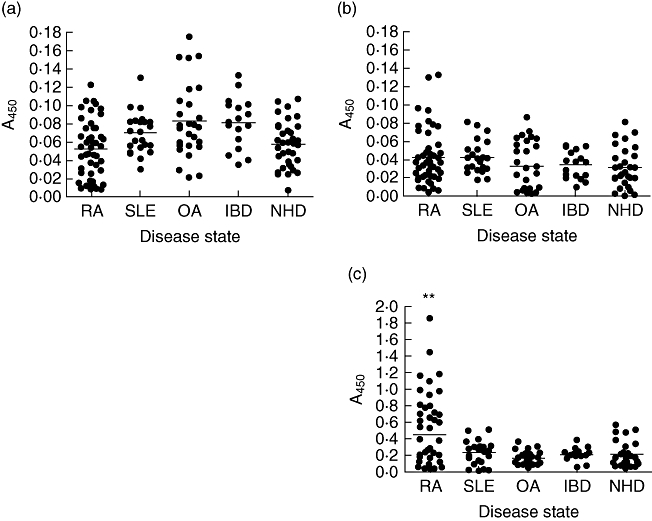
(a) Levels of antibody reactivity to PLSK [human endogenous retrovirus (HERV)-K] peptide in RA patients and disease controls. (b) Levels of antibody reactivity in RA patients and disease controls to KPR [HERV-K (HML-2)] peptide. (c) Levels of antibodies specific for biotinylated KPR peptide in RA patient anti-sera and disease controls. RA, rheumatoid arthritis (n = 48); SLE, systemic lupus erythematosus (n = 23); OA, osteoarthritis (n = 27); IBD, inflammatory bowel disease (n = 17); NHD, normal healthy donors (n = 37).
Molecular analysis
A novel quantitative RT–PCR assay was developed and optimized for the accurate detection and quantification of HERV-K (HML-2) gag1 gene products in peripheral blood mononuclear cell populations extracted from patient whole blood. Results were normalized to the geometric mean of two housekeeping genes (HPRT and ACTB). Levels within RA patients were increased significantly compared to controls (one-way anova, P < 0·001) mirroring that observed in the serological results, although levels were also heterogeneous (Fig. 3). Age- and sex-matched RA patients exhibited significantly increased levels of HERV transcription over OA counterparts (paired t-test, P < 0·0001), with female patients showing the largest increases (paired t-test, P = 0·0009). Female RA patients also exhibited significant increases in transcription over male RA patients (paired t-test, P = 0·0362). Additionally, HERV-K (HML-2) transcripts were quantified in synovial fibroblasts taken from patients with RA, OA and healthy controls (Fig. 4), with RA patients exhibiting significantly increased levels of HERV mRNA compared to those derived from OA and healthy controls (one-way anova with Tukeys’ multiple comparison test, P = 0·0013 and 0·0039, respectively). No significant relationships were identified between HERVs and therapeutic strategies. Further analysis identified a correlation between all RA patients tested by both methodologies, giving an R2 value of 0·953 (Fig. 5). Within this cohort a subset of 10 RA patients were identified whose serological and molecular activity shared an increased R2 value of 0·978.
Fig. 3.
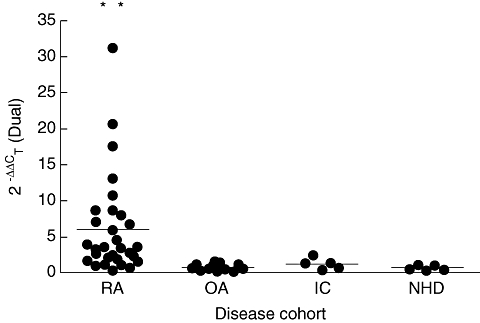
Levels of human endogenous retrovirus (HERV)-K (HML-2) transcription in patient samples as determined by real-time polymerase chain reaction to the HERV-K (HML-2) gag1 gene. Rheumatoid arthritis (RA) patient (n = 34), osteoarthritis patient (OA) (n = 17), non-RA inflammatory controls – irritable bowel disease (IC) (n = 5) and normal healthy donors (NHD) (n = 5).
Fig. 4.
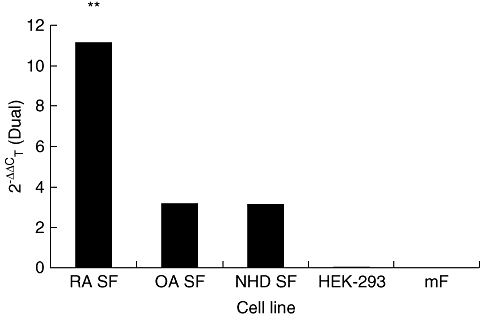
Comparison of human endogenous retrovirus (HERV)-K (HML-2) gag1 expression in different cell lines. Cell lines surveyed include human fibroblast-like synoviocytes (HFLS) derived from a rheumatoid arthritis patient (RA), osteoarthritis patient (OA), normal healthy donor (NHD) and mouse-derived fibroblasts (MF). A human non-synovial fibroblast cell line (HEK-293) was also tested.
Fig. 5.
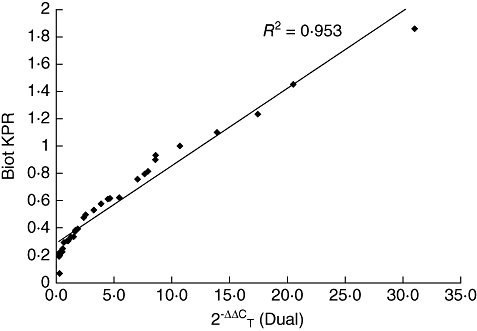
Correlation of all samples tested by both biotinylated enzyme-linked immunosorbent assay with KPR peptide and quantitative reverse transcription–polymerase chain reaction (n = 30). The R2 value for the gradient of the trendline is shown on the graph (0·953).
To explore factors potentially influencing HERV transcriptional activity in RA disease pathology, qRT–PCR was used to determine HERV-K (HML-2) mRNA levels in fibroblast-like synoviocytes treated with inflammatory cytokines. Results in RA-derived synoviocytes showed that treatment with both IL-6 and TNF-α (at 1 ng/ml concentrations) increased HERV transcription significantly over controls (unpaired t-test, P < 0·0001, P = 0·005, respectively) (Fig. 6a). Upon treatment with 10-fold increases of both cytokines, levels of HERV mRNA were decreased significantly (unpaired t-test, P < 0·0001, P < 0·0001, respectively). Treatments with both cytokines gave comparable increases. The modulatory effects of viral proteins on HERVs were also investigated by transfection of fibroblast-like synoviocytes with expression vectors for EBNA1, LMP1 and LMP2A. This resulted in significant increases in the levels of HERV-K10 mRNA transcription compared with untreated and plasmid controls (Fig. 6b).
Fig. 6.
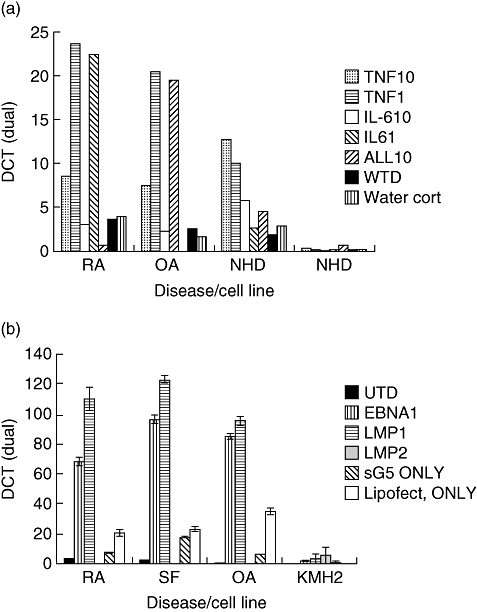
(a) Levels of human endogenous retrovirus (HERV)-K (HML2) gag1 transcriptional activity after incubation with proinflammatory cytokines [interleukin (IL)-1β, IL-6 and tumour necrosis factor-α]. Cell lines incubated included fibroblast-like synoviocytes taken from rheumatoid arthritis (RA) patient, osteoarthritis patient (OA) and normal healthy door (NHD). (b) Levels of HERV-K (HML-2) gag1 after transfection of plasmids encoding Epstein–Barr virus gene products EBNA1 (Epstein–Barr nuclear antigen), LMP1 (late membrane protein 1) and LMP2A (late membrane protein 2a). Cell lines transfected included human fibroblast-like synoviocytes (HFLS) derived from an RA patient, HFLS taken from an OA patient and a healthy donor and a Hodgkin's lymphoma cell line (KMH2).
Discussion
Initial stages of this investigation demonstrated the potential of Gag, Pol and Env proteins of HERVs to generate B cell epitopes. Such predictions were based upon both primary and secondary sequence analysis. A Gag1 sequence (KPR) was identified as a candidate elicitor of anti-HERV antibodies, which also showed potential for cross-reactivity with host proteins through molecular mimicry (Table 1). This epitope was thus selected for further investigation.
From the data presented it was evident that immune responses targeting this epitope were increased significantly in RA patients over controls. Disease pathogenesis through molecular mimicry has been well established, with several examples of retroviruses inducing cross-reactive immunity in other rheumatic diseases [19]. This epitope may also play a potential role in disease development, via processes such as epitope spreading, resulting in the diverse ranges of autoantibody responses observed characteristically in rheumatic patients. When biotinylated peptide was used, levels of anti-KPR antibodies detected increased (significantly in RA patients). Results presented here support those of previous investigations in concluding that the biotinylation of antigen improves assay sensitivity significantly [20,21]. This is due probably to significant modifications to the structural and conformational modelling of antigens leading to the masking of potentially important epitopes during the process of adsorbing the peptide to the solid phase [22]. Biotinylated peptides provided a reliable in vivo model system for testing anti-HERV titres in patients.
Transcriptional activity was also increased significantly in RA patients, compared to controls, in support of the serological data (Fig. 3). The R2 value, indicating correlation between both serological and molecular results (Fig. 5), further supported an association between HERVs and RA disease immunopathology. Of particular interest was a small subset of 10 RA patients who exhibited an increased level of correlation. Female RA patients exhibited the highest levels of activity in comparison to both male RA patients and female age- and sex-matched OA patients, hinting that gender may play an important role in HERV transcriptional activity. This may be reflected in the fact that RA and many of the rheumatic diseases are diagnosed predominantly in women [23]. A number of accounts have documented anti-HERV immune responses previously in patients with a variety of autoimmune diseases and cancers that parallel those reported here. Recent investigations have also identified anti-HERV antibodies in patients with breast cancer, melanoma, germ cell tumours and lymphomas [24–26]. Whether such malignancies predispose these patients to develop autoimmunity is an intriguing question that merits further study. An association between cancer and autoimmunity, however, notably the rheumatic diseases, i.e. RA, SLE and SS, has been reported previously. Increased occurrences of malignancies in patients with established RA have been published in several studies [27,28], with RA patients showing a fivefold increase in development of lymphomas [24]. Additionally a group of cancers known as the paraneoplastic syndromes have been reported in which patients exhibit clinical manifestations of RA and other rheumatic diseases, months and years prior to development of occult cancers [29]. Whether HERVs play an as-yet unknown role in the disease pathology of these patients may well be a topic for future research. The heterogeneous nature of these responses also indicates the potential for polymorphisms to play important roles in predisposed individuals. The presence of HERV polymorphisms and their roles in contribution to disease has been documented previously [30]. Variants of HERV-K (HML-2) found in certain individuals may contribute to the level of variation observed in both serological and molecular results. This may lead to the presence of specific polymorphisms becoming an important factor in small groups of predisposed patients such as those identified within this paper. The heterogenic clinical manifestations of disease in RA suggest that it is still not a single disease entity [31], and this may be reflected in the significant variation of HERV activity exhibited by RA patients in both serological and molecular results. A similar situation has arisen in SLE and its associated disorders [32] and is reflected in the wide range of different pathologies presented by RA patients with the disease [23]. Again, this may suggest a role for HERV-K (HML-2) in a subset of RA patients, which may form an individual disease entity.
HERVs have been reported previously to modulate directly [33], and reside under, the influence of components of the immune response [34]. Both HERV-W and HERV-K have been reported to exhibit increased expression of superantigens in juvenile RA (JRA) [35] and in the presence of exogenous viruses [36]. Several studies investigating the role of the multiple sclerosis retrovirus (MSRV) in MS have reported increased levels of MSRV in the presence of proinflammatory cytokines [34,37]. We demonstrated that levels of HERV-K (HML-2) in synovial fibroblasts were increased in the presence of proinflammatory cytokines (TNF-α and IL-6) compared to treatment controls (Fig. 6a). Data from cultured synoviocytes derived from patient biopsies indicated that when treated with lower concentrations of inflammatory cytokines, HERV-K transcription was increased significantly over controls. This was in comparison to levels in synoviocytes treated with 10-fold increased concentrations of inflammatory cytokines (simulating advanced stages of disease within the synovial microenvironment). These results suggest a role for HERV-K (HML-2) in disease development with levels peaking at low concentrations of cytokines. This may also be indicative as to the effectiveness of immunosuppressive drug regimens used in the treatment of RA. Increases in cytokines responsible for increases in HERV-K transcription could be induced under several conditions ranging from inflammation to infection. This paper investigated the influence of exogenous infections upon levels of HERV mRNA. As with MSRV, it is possible that the presence of viral pathogens such as herpesviruses have influence over HERV activity. We confirmed that proteins of Epstein–Barr virus (EBV), a virus linked previously with RA [38], increased HERV-K (HML-2) gag1 expression significantly, compared to controls in both human synoviocytes and Hodgkins lymphoma cell lines (Fig. 6b). Other herpesviruses have also been shown to modulate HERV transcription [7,39,40]. Recent reports have suggested a synergistic relationship between immune components, i.e. cytokines, and viral pathogens that result in increases in MSRV activity in MS [37]. It may well be that the same rules apply in RA – a complex autoimmune disease with unknown aetiology and numerous possible genetic and environmental factors. From the evidence presented here it seems apparent that RA is not a homogeneous disease and probably has numerous contributing factors including HERVs. It may be that particular polymorphisms are able to cause or contribute to disease mechanisms in subsets of patients with RA. This is complicated further by the heterogeneous nature of individual HERV-K family members, i.e. HERV-K (HML1-6), reflected possibly in those patients tested here. Such individuals with highly transcribed/translated gene products support the hypothesis that induction of HERV-K (HML-2) gag1 influences disease development.
The data described here add to the evidence for a role for HERV-K in RA disease immunopathogenesis; however, the exact capacity of HERV-K in this process remains undefined. It is unclear as to whether HERVs play the triggering agent or are simply up-regulated as a consequence of inflammation and immune responses occurring within the synovial microenvironment. The smoking gun may be in the timing of HERV increases in relation to disease development or whether particular HERV polymorphisms play roles in specific subsets of predisposed patients. Future reports must focus upon shedding more light upon these factors, in order to clarify the true nature of HERVs and their association with disease.
Disclosure
The authors have no conflict of interests and would like to thank the South Staffordshire Medical Foundation, Rotha Abraham Bequest, The Royal Wolverhampton Hospitals NHS Trust Charity and James Beattle Charitable Trust for research funding.
References
- 1.Ejtehadi HD, Freimanis GL, Ali HA, et al. The potential role of human endogenous retrovirus K10 in the pathogenesis of rheumatoid arthritis: a preliminary study. Ann Rheum Dis. 2006;65:612–16. doi: 10.1136/ard.2004.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 3.Nelson PN, Carnegie PR, Martin J, et al. Demystified. Human endogenous retroviruses. Mol Pathol. 2003;56:11–18. doi: 10.1136/mp.56.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boller K, Schonfeld K, Lischer S, et al. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J Gen Virol. 2008;89:567–72. doi: 10.1099/vir.0.83534-0. [DOI] [PubMed] [Google Scholar]
- 5.Michaels FH, Banks KL, Reitz MS., Jr Lessons from caprine and ovine retrovirus infections. Rheum Dis Clin North Am. 1991;17:5–23. [PubMed] [Google Scholar]
- 6.Herrmann M, Neidhart M, Gay S, Hagenhofer M, Kalden JR. Retrovirus-associated rheumatic syndromes. Curr Opin Rheumatol. 1998;10:347–54. doi: 10.1097/00002281-199807000-00012. [DOI] [PubMed] [Google Scholar]
- 7.Nelson PN, Lever AM, Smith S, et al. Molecular investigations implicate human endogenous retroviruses as mediators of anti-retroviral antibodies in autoimmune rheumatic disease. Immunol Invest. 1999;28:277–89. doi: 10.3109/08820139909060862. [DOI] [PubMed] [Google Scholar]
- 8.Ehlhardt S, Seifert M, Schneider J, Ojak A, Zang KD, Mehraein Y. Human endogenous retrovirus HERV-K (HML-2) Rec expression and transcriptional activities in normal and rheumatoid arthritis synovia. J Rheumatol. 2006;33:16–23. [PubMed] [Google Scholar]
- 9.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci USA. 1981;78:3824–8. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–4. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 13.Janin J. Surface and inside volumes in globular proteins. Nature. 1979;277:491–2. doi: 10.1038/277491a0. [DOI] [PubMed] [Google Scholar]
- 14.Bhaskaran R, Ponnuswamy PK. Positional flexibilities of amino-acid residues in globular-proteins. Int J Peptide Protein Res. 1988;32:241–55. doi: 10.1111/j.1399-3011.1984.tb00944.x. [DOI] [PubMed] [Google Scholar]
- 15.Kaur H, Raghava GP. A neural-network based method for prediction of gamma-turns in proteins from multiple sequence alignment. Protein Sci. 2003;12:923–9. doi: 10.1110/ps.0241703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saha S, Raghava GPS. BcePred: prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties. Artificial Immune Systems, Proc. 2004:197–204. [Google Scholar]
- 17.Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int J Pept Protein Res. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 18.Westwood OM, Hay FC. Epitope mapping. 1st. Oxford: Oxford University Press; 2000. [Google Scholar]
- 19.Douvas A, Takehana Y, Ehresmann G, Chernyovskiy T, Daar ES. Neutralization of HIV type 1 infectivity by serum antibodies from a subset of autoimmune patients with mixed connective tissue disease. AIDS Res Hum Retroviruses. 1996;12:1509–17. doi: 10.1089/aid.1996.12.1509. [DOI] [PubMed] [Google Scholar]
- 20.Ivanov VS, Suvorova ZK, Tchikin LD, Kozhich AT, Ivanov VT. Effective method for synthetic peptide immobilization that increases the sensitivity and specificity of ELISA procedures. J Immunol Methods. 1992;153:229–33. doi: 10.1016/0022-1759(92)90326-o. [DOI] [PubMed] [Google Scholar]
- 21.Phumoonna T, Muscatello G, Chicken C, et al. Clinical evaluation of a peptide-ELISA based upon N-terminal B-cell epitope of the VapA protein for diagnosis of Rhodococcus equi pneumonia in foals. J Vet Med B Infect Dis Vet Public Health. 2006;53:126–32. doi: 10.1111/j.1439-0450.2006.00929.x. [DOI] [PubMed] [Google Scholar]
- 22.Shamsuddin AM, Harris CC. Improved enzyme immunoassays using biotin–avidin–enzyme complex. Arch Pathol Lab Med. 1983;107:514–17. [PubMed] [Google Scholar]
- 23.Goronzy JJ, Weyand CM. Rheumatoid arthritis. Immunol Rev. 2005;204:55–73. doi: 10.1111/j.0105-2896.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- 24.Contreras-Galindo R, Kaplan MH, Leissner P, et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol. 2008;82:9329–36. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hahn S, Ugurel S, Hanschmann KM, et al. Serological response to human endogenous retrovirus K in melanoma patients correlates with survival probability. AIDS Res Hum Retroviruses. 2008;24:717–23. doi: 10.1089/aid.2007.0286. [DOI] [PubMed] [Google Scholar]
- 26.Kinlen LJ. Malignancy in autoimmune diseases. J Autoimmun. 1992;5(Suppl. A):363–71. doi: 10.1016/0896-8411(92)90055-u. [DOI] [PubMed] [Google Scholar]
- 27.Andras C, Csiki Z, Ponyi A, Illes A, Danko K. Paraneoplastic rheumatic syndromes. Rheumatol Int. 2006;26:376–82. doi: 10.1007/s00296-005-0005-3. [DOI] [PubMed] [Google Scholar]
- 28.Bernatsky S, Ramsey-Goldman R, Clarke A. Malignancy and autoimmunity. Curr Opin Rheumatol. 2006;18:129–34. doi: 10.1097/01.bor.0000209423.39033.94. [DOI] [PubMed] [Google Scholar]
- 29.Racanelli V, Prete M, Minoia C, Favoino E, Perosa F. Rheumatic disorders as paraneoplastic syndromes. Autoimmun Rev. 2008;7:352–8. doi: 10.1016/j.autrev.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Moyes D, Griffiths DJ, Venables PJ. Insertional polymorphisms: a new lease of life for endogenous retroviruses in human disease. Trends Genet. 2007;23:326–33. doi: 10.1016/j.tig.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Weyand CM, Klimiuk PA, Goronzy JJ. Heterogeneity of rheumatoid arthritis: from phenotypes to genotypes. Springer Semin Immunopathol. 1998;20:5–22. doi: 10.1007/BF00831996. [DOI] [PubMed] [Google Scholar]
- 32.Routsias JG, Tzioufas AG, Moutsopoulos HM. The clinical value of intracellular autoantigens B-cell epitopes in systemic rheumatic diseases. Clin Chim Acta. 2004;340:1–25. doi: 10.1016/j.cccn.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Haraguchi S, Good RA, Day-Good NK. A potent immunosuppressive retroviral peptide: cytokine patterns and signaling pathways. Immunol Res. 2008;41:46–55. doi: 10.1007/s12026-007-0039-6. [DOI] [PubMed] [Google Scholar]
- 34.Mameli G, Astone V, Arru G, et al. Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not human herpesvirus 6. J Gen Virol. 2007;88:264–74. doi: 10.1099/vir.0.81890-0. [DOI] [PubMed] [Google Scholar]
- 35.Sicat J, Sutkowski N, Huber BT. Expression of human endogenous retrovirus HERV-K18 superantigen is elevated in juvenile rheumatoid arthritis. J Rheumatol. 2005;32:1821–31. [PubMed] [Google Scholar]
- 36.Sutkowski N, Conrad B, Thorley-Lawson DA, Huber BT. Epstein–Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity. 2001;15:579–89. doi: 10.1016/s1074-7613(01)00210-2. [DOI] [PubMed] [Google Scholar]
- 37.Brudek T, Christensen T, Hansen HJ, Petersen T, Moller-Larsen A. Synergistic immune responses induced by endogenous retrovirus and herpesvirus antigens result in increased production of inflammatory cytokines in multiple sclerosis patients. Scand J Immunol. 2008;67:295–303. doi: 10.1111/j.1365-3083.2007.02067.x. [DOI] [PubMed] [Google Scholar]
- 38.Sawada S, Takei M. Epstein–Barr virus etiology in rheumatoid synovitis. Autoimmun Rev. 2005;4:106–10. doi: 10.1016/j.autrev.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 39.Nellaker C, Yao Y, Jones-Brando L, Mallet F, Yolken RH, Karlsson H. Transactivation of elements in the human endogenous retrovirus W family by viral infection. Retrovirology. 2006;3:44. doi: 10.1186/1742-4690-3-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brudek T, Luhdorf P, Christensen T, Hansen HJ, Moller-Larsen A. Activation of endogenous retrovirus reverse transcriptase in multiple sclerosis patient lymphocytes by inactivated HSV-1, HHV-6 and VZV. J Neuroimmunol. 2007;187:147–55. doi: 10.1016/j.jneuroim.2007.04.003. [DOI] [PubMed] [Google Scholar]