

Abstract
Interstitial lung disease (ILD) is an intractable disease induced by various factors in humans. However, there is no universally effective treatment for ILD. In this study, we investigated the role of transforming growth factor (TGF)-β signalling in the pathogenesis of ILD by using model mice. Injection of interleukin (IL)-18 plus IL-2 in C57BL6 (B6) mice resulted in acute ILD by infiltration of natural killer (NK) cells and a significant increase of TGF-β mRNA in the lung. To examine the pathogenetic role of TGF-β in ILD mice, we used SB-431542 (4-[4-(1,3-benzodioxol-5-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]-benzamide), which is a potent and selective inhibitor of TGF-β receptor I (TβRI), also known as activin receptor-like kinase 5 (ALK5). Treatment of B6-ILD mice with SB-431542 resulted in improvement of ILD, delay in mortality, reduction of the expression of interferon (IFN)-γ and IL-6 in the lungs. The same treatment also decreased significantly the percentage of natural killer (NK) cells in the lungs (P < 0·05) and mRNA expression levels of certain chemokines such as CCL2, CCL3, CCL4, CCL5 and CXCL10 in B6-ILD. These findings were confirmed by IL-18 plus IL-2 treatment of Smad3-deficient (Smad3–/–) mice (P < 0·05). Our results showed that inhibition of TGF-β signalling reduced the percentage of NK cells and the expression of certain chemokines in the lungs, resulting in improvement of ILD. The findings suggest that TGF-β signalling may play an important role in the pathogenesis of IL-18 plus IL-2-induced ILD in mice.
Keywords: activin receptor-like kinase 5, chemokines, interstitial lung disease, pathogenesis, SB-431542, TGF-β signalling
Introduction
Interstitial lung disease (ILD) is an intractable disease induced by various factors such as autoimmune diseases, drugs, occupational and environmental exposure [1]. However, there is no universally effective treatment for ILD. On the other hand, chemotherapy with bleomycin (BLM) and busulphan is reported to cause lung fibrosis in some patients [2]. Histopathologically, diffuse infiltration of mononuclear and polymorphonuclear leucocytes is observed in the lung in the early stages of human ILD. Following the interstitial inflammation, florid fibroblast proliferation within both the interstitium and alveolar space is often detected. The same pathology is observed in BLM-induced ILD in mice [1]. Previous studies suggested that various mediators, such as cytokines and chemokines, including tumour necrosis factor (TNF)-α, transforming growth factor (TGF)-β, interleukin (IL)-1β, macrophage inflammatory factor (MIP)-1α/CCL3, monocyte chemoattractant protein (MCP)-1/CCL2, reactive oxygen species (ROS) and Fas/Fas ligand interactions, are associated with BLM-induced ILD and fibrosis in mice [3–9].
IL-18, a member of the IL-1 family, is a proinflammatory cytokine [10,11] known to induce interferon (IFN)-γ production synergistically by stimulation with IL-12, IL-2, antigens and IFN-α. Previous studies reported that IL-18 can potentially induce Th2 cytokines from T cells, natural killer (NK) cells, NK T cells, basophils and mast cells [11–16]. Thus, IL-18 can act as co-factor for both T helper type 1 (Th1) and Th2 cell development. In BLM-induced ILD mice models, there were two conflicting reports on the effect of IL-18. Nakatani-Okuda et al.[17] reported that IL-18 played a protective role in BLM-induced ILD in mice. In contrast, Hoshino et al.[18] reported that IL-18 played a pathologenetic role in the ILD. Therefore, the intimate role of IL-18 in ILD was controversial. Recently, Okamoto et al.[19] reported a new mouse model of ILD induced by IL-18 plus IL-2 (IL-18/IL-2). Daily administration of IL-18 with IL-2, but not IL-18 or IL-2 alone, produced a synergistic effect and induced ILD in mice. Unlike BLM-induced ILD, lung fibrosis was not caused in IL-18/IL-2-induced ILD. The pathological condition of BLM-induced ILD was mainly fibroblastic proliferation [20]. However, little fibroblastic proliferation was found in IL-18/IL-2-induced ILD. This model of ILD is characterized by severe infiltration of NK cells, mononuclear cells and polymorphonuclear leucocytes in the lung. Furthermore, the mortality in this two ILD mice models was different. Whereas 60% of mice died at 30 days after BLM treatment [21], 100% of mice died at 7 days after IL-18/IL-2 injection. Based on rapid and severe cell infiltration in IL-18/IL-2-induced ILD, the mouse model is considered suitable for early-phase human ILD.
Various chemokines, such as CCL2, CCL3, CCL4, CCL5, CXCL1 and CXCL8, are induced by activated fibroblasts in the lung tissue [22]. Mice with BLM-induced pulmonary fibrosis exhibit up-regulation of CCL2, CCL5, CCL3 and CXCL1 in the lung and such overexpression is associated with enhanced fibroblast proliferation and collagen production [23]. Thus, these chemokines are thought to be involved in the pathogenesis of ILD and subsequent fibrotic process. In contrast, the functional roles of chemokines in IL-18/IL-2-induced ILD in mice remain elusive, although lymphotactin (Ltn), CCL2, CCL3, CCL4, CCL5, CCL11, CXCL1 and CXCL10 are increased in the lung [19].
TGF-β is thought to be one of the pathogenic factors in ILD. Previous reports suggested that TGF-β acts as a regulatory molecule with pleiotropic effects on cell proliferation, differentiation, migration and survival [24]. TGF-β mediates its biological functions via binding to TGF-β receptor II (TβRII) and phosphorylation of TGF-β receptor I (TβRI), also known as activin receptor-like kinase 5 (ALK5). After forming the TGF-β−TβRII-ALK5 complex, ALK5 phosphorylates intracellular signal mediators Smad2/3. The importance of ALK5-mediated Smad2/3 activation in TGF-β signalling has been confirmed both in vitro and in vivo[24–29].
SB-431542 (4-[4-(1,3-benzodioxol-5-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]-benzamide) is a potent and selective inhibitor of ALK-5 [30–32]. Inhibition of ALK-5 suppressed BLM-induced pulmonary fibrosis [33]. However, the therapeutic potential of ALK-5 inhibitor in IL-18/IL-2-induced ILD has not yet been clarified.
In the present study, we examined the role of TGF-β signalling in early-stage ILD. For this purpose, we used SB-431542 and Smad3-deficient (Smad3–/–) mice for IL-18/IL-2-induced ILD. The results demonstrated that inhibition of TGF-β signalling suppressed accumulation of NK cells and reduced mRNA expression of CCL2, CCL3, CCL4, CCL5 and CXCL10 in the lung. The main message of this study is that Smad-mediated TGF-β signalling seems to play an important role in the pathogenesis of IL-18/IL-2-induced ILD.
Materials and methods
Mice
C57BL/6 (B6) mice were purchased from Charles River Japan Inc. (Tokyo, Japan). Smad3-deficient (Smad3–/–) mice were kindly provided by Dr Chuxia Deng (National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, USA) [34]. The genotypes of both B6 and Smad3–/– mice were determined by polymerase chain reaction (PCR) analysis on tail DNA obtained from 4-week-old animals. Female mice were used in this study. The animals were kept under specific pathogen-free conditions and studied at 4–5 weeks of age. The Institutional Animal Care and Use Committee at Tsukuba University approved the experimental protocol.
Cell isolation and purification
Pulmonary lymphocytes were isolated as described previously [35], with the following modifications. The lung was perfused thoroughly with phosphate-buffered saline (PBS) to remove circulating blood cells. The dissected lung was minced in PBS containing 1 mM ethylenediamine tetraacetic acid (EDTA). The minced lung tissue was suspended in RPMI-1640 medium (Sigma-Aldrich, St Louis, MO, USA) containing 10% fetal bovine serum (FBS) (BioWest, FL, USA), 1 mM EDTA and 1 mM dithiothreitol (DTT). The suspension was incubated at 37°C for 45 min with gentle shaking. The resultant suspension was passed through nylon mesh to remove debris. The washed and recovered cells were subjected to Lympholyte (Cedarlane, Ontario, Canada) at 1100 g at room temperature for 20 min. The resultant interface containing pulmonary lymphocytes was recovered and washed with RPMI-1640 medium containing 10% FBS, 100 units/ml penicillin, 100 µg/ml streptomycin and 50 µM 2-mercaptoethanol. Spleens were harvested and haemolyzed with PBS. Single-cell suspensions were prepared in RPMI-1640 medium containing 10% FBS, 100 units/ml of penicillin, 100 µg/ml streptomycin and 50 µM 2-mercaptoethanol.
Antibodies and flow cytometry
All antibodies were used according to the recommendations of the respective manufacturers. For flow cytometric analysis, cells were preincubated with anti-CD16/32 (eBioscience, San Diego, CA, USA) to block Fc receptors. The following antibodies were used in this study: phycoerythrin (PE)-conjugated anti-natural killer (NK)1·1 (PK136) (Biolegend, San Diego, CA, USA) and PE/cyanine 7 (Cy7)-conjugated anti-CD3ε (145-2C11) (Biolegend). The stained cells were analysed on CyAn advanced digital processing (ADP) (Dako, Glostrup, Denmark) and data were processed using Summit4·3 (Dako).
Induction of lung fibrosis with IL-18 and IL-2
Recombinant human IL-2 (rhIL-2) and recombinant mouse IL-18 (rmIL-18) were obtained from MBL (Nagoya, Japan). Mice were treated once a day with an intraperitoneal (i.p.) injection of rhIL-2 (100 000 U) and/or rmIL-18 (1 µg). These cytokines were suspended in sterile 200 µl PBS. Mice treated with 200 µl PBS served as the control group. Following treatment for 3 days, mice were bled and killed. Pulmonary lymphocytes and splenocytes were analysed by flow cytometry.
Treatment of mice with SB-431542
ALK-5 inhibitor, SB-431542 (4-[4-(1,3-benzodioxol-5-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]-benzamide) (SB-431542) was obtained from Tocris Bioscience (Park Ellisville, MO, USA). It was suspended in sterile dimethyl sulphoxide (DMSO) at 20 mg/ml. Mice were treated twice a day (0 h and 12 h after IL-18/IL-2 treated) by i.p. injection of 50 µl (0·2 mg) SB-431542 or vehicle for 3 days.
Reverse transcription polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from the lung, and was reverse transcribed into cDNA using RevertAid™ first-strand cDNA synthesis kit (Fermentas, Burlington, Ontario, Canada), according to the manufacturer's protocol. For amplification of chemokine cDNA, after an initial denaturation step at 94°C for 4 min, 35 cycles were conducted each at 94°C for 30 s followed by 60°C for 30 s and 72°C for 30 s, and further extension at 72°C for 7 min. For amplification of glyceraldehyde-2-phosphate dehydrogenase (GAPDH) cDNA, PCR assays were performed for 30 cycles (94°C for 30 s followed by 60°C for 30 s and 72°C for 30 s). At the end of cycles, samples were stored at 4°C until analysed. After amplification, the PCR products were separated by electrophoresis in 2·0% agarose gels. The primer sequences were as follows and the PCR product sizes [base pairs (bp)] were indicated: CCL2, 5′-AGGTCCCTGTCATGCTTCTG, 3′-TCTGGACCCATTCCTTCTTG (249 bp); CCL3, 5′-AGATTCCACGCCAATTCATC, 3′-CTCAAGCCCCTGCTCTACAC (223 bp); CCL4, 5′-CCCACTTCCTGCTGTTTCTC, 3′-GAGGAGGCCTCTCCCTGAAGT (238 bp); CCL5, 5′-CCCTCACCATCATCCTCACT, 3′-CCTTCGAGTGACAAACACGA (185 bp); CCL11, 5′-TCCACAGCGCTTCTATTCCT, 3′-CTATGGCTTTCAGGGTGCAT (178 bp); CXCL1, 5′-GCTGGGATTCACCTCAAGAA, 3′-TCTCCGTTACTTGGGGACAC (180 bp); CXCL10, 5′-GGATGGCTGTCCTAGCTCTG, 3′-ATAACCCCTTGGGAAGATGG (211 bp); and GAPDH, 5′-CGTCCCGTAGACAAAATGGGT, 3′-GAATTTGCCGTGAGTGGAGT (177 bp).
Quantification of gene expression by RT-PCR
The cDNA samples were amplified with specific primers and fluorescence-labelled probes for the target genes. Specific primers and probes for TGF-β and GAPDH were purchased from Applied Biosystems Japan (Tokyo, Japan). The amplified product genes were monitored on an ABI 7700 sequence detector (Applied Biosystems Japan). The quantitative PCR master mix was purchased from Applied Biosystems Japan. The final concentrations of the primers were 200 nM for each of the 5′ and 3′ primers, and the final probe concentration was 100 nM. The thermal cycler conditions used were 50°C for 2 min, 95°C for 10 min, then 50 cycles of 95°C for 15 s and 60°C for 1 min. Serial dilutions of a standard sample were included in every assay, and standard curves for the genes of interest and GAPDH genes were generated. All measurements were performed in triplicate. The level of gene expression was calculated from the standard curve, and expressed relative to GAPDH gene expression.
Histological examination
For histological analysis, mice were euthanized by isopropanol and lung was fixed with 4% paraformaldehyde. Lung tissues were stained with haematoxylin and eosin (H&E).
Immunohistochemistry
In the present study, anti-phosphorylated Smad3 were used (Rockland, Gilbertsville, PA, USA). For detection of immunocomplexes, Histofine (Nichirei Corporation, Tokyo, Japan) for phosphorylated Smad3 was used using the manufacturer's instructions. Substitution of the primary antibody with irrelevant immunoglobulin G (IgG) served as negative controls. Staining was repeated for each sample at least three times. After counterstaining with haematoxylin, sections were mounted with mounting agent, PARAmount-D (Falma, Tokyo, Japan).
Measurement of cytokines in the lung and serum
For enzyme-linked immunosorbent assay (ELISA), the lung tissue was suspended and homogenized in sterile PBS and centrifuged at 10 000 g for 10 min. The cytokine levels in the lung tissue supernatants and the serum were evaluated by ELISA (R&D Systems, Minneapolis, MN, USA).
Statistical analysis
Data are expressed as median or mean ± standard error of the mean (s.e.m.). Data were analysed using a statistical software package (StatView 5·0; SAS Institute Inc, Cary, NC, USA). The survival rates were analysed by Kaplan–Meier method. Differences between groups were examined for statistical significance using Student's t-test. For multiple group comparisons, one-way analysis of variance (anova) was performed followed by a post-hoc Dunnett's test. A P-value less than 0·05 denoted a statistically significant difference.
Results
Overexpression of TGF-β mRNA in IL-18/IL-2-induced ILD
Figure 1 shows significant up-regulation of TGF-β mRNA in the whole lung tissues from mice at 3, 6 and 12 h after injection of IL-18/IL-2 compared with 0 h (P < 0·05, P < 0·01 and P < 0·05, respectively). However, at 24 h after injection of IL-18/IL-2, the expression of TGF-β mRNA returned to the level at 0 h.
Fig. 1.
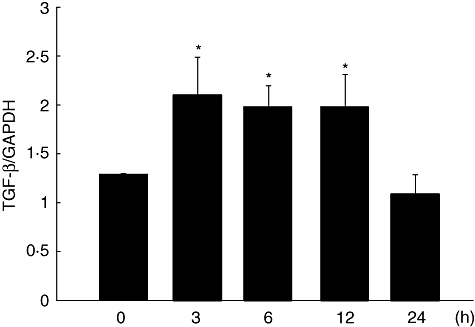
Expression of transforming growth factor (TGF)-β mRNA after injection of interleukin (IL)-18 plus IL-2. Expression of TGF-β mRNA was analysed by real-time–polymerase chain reaction (RT-PCR). C57BL/6 (B6) mice were injected intraperitoneally with a single dose of IL-18/IL-2. At 3, 6, 12 and 24 h after injection, mice were killed and lung mRNA was extracted. Data are mean ± standard error of the mean; n = 3 mice per group. *P < 0·05; one-way analysis of variance.
SB-431542 ameliorated ILD and reduced the expression of IFN-γ and IL-6 in the lung
Treatment with SB-431542 was employed to examine the effect of TGF-β inhibition on IL-18/IL-2-induced ILD. As shown in Fig. 2a, treatment with SB-431542 delayed mortality significantly compared with control on the 11th day (P < 0·05). Histological examination showed inhibition of cell infiltration in the lungs of SB-431542-treated ILD mice compared with the control (Fig. 2b). Furthermore, we analysed the expression of cytokines in sera and the lung tissues from SB-431542 and vehicle-treated ILD-induced mice. In the lung of SB-431542-treated mice, the expression of IFN-γ and IL-6 was significantly lower than control mice (P < 0·05, P < 0·01, Fig. 2c). However, in sera, the expression of IFN-γ and TNF-α was not different in each group (Fig. 2d). The lung wet–dry ratio was not significantly different between SB-431542-treated ILD mice and vehicle-treated ILD mice (data not shown).
Fig. 2.
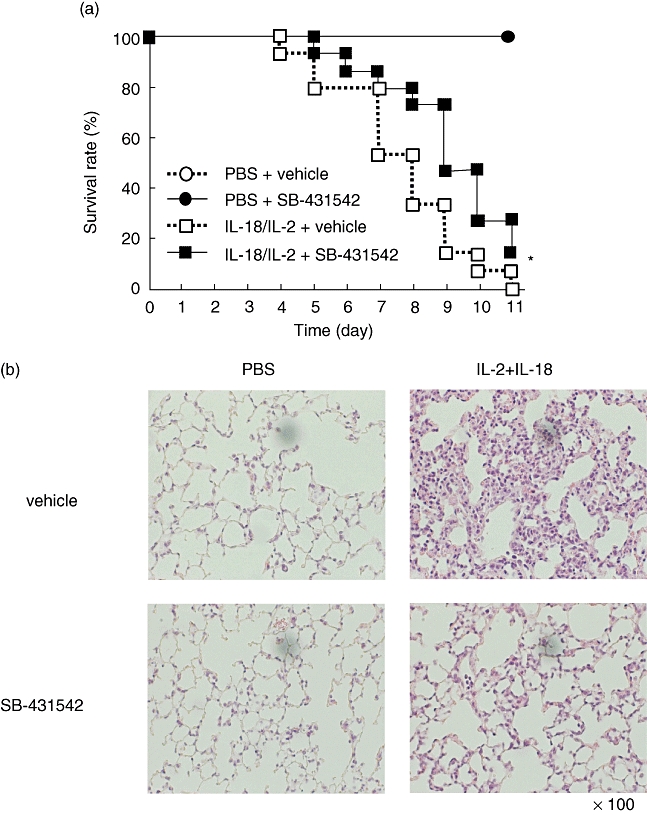
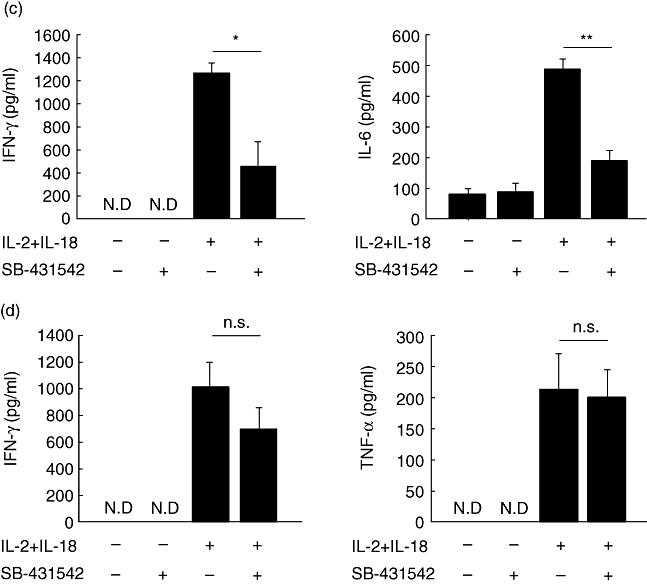
Effects of SB-431542 in interleukin (IL)-18 plus IL-2-induced interstitial lung disease (ILD) mice. (a) B6 mice were injected with IL-18/IL-2 with or without SB-431542 for 10 days, as described in Materials and methods. ○, phosphate = interstitial lung disease (ILD) –buffered saline (PBS) + vehicle; •, PBS + SB-431542; □, IL-18/IL-2 + vehicle;  , IL-18/IL-2 + SB-431542; n = 5 mice per group. Data are representative of three independent experiments and graph shows pooled data of three experiments. *P < 0·05; Kaplan–Meier method. (b) Lungs were harvested from B6 mice at 24 h after injected with IL-18/IL-2 with or without SB-431542 for 3 days. Lung tissues were stained with haematoxylin and eosin. Original magnification: ×100. (c) Lungs were harvested from B6 mice at 6 h after injection with IL-18/IL-2 with or without SB-431542 for 3 days. (d) The sera were harvested from B6 mice at 6 h after injection with IL-18/IL-2 with or without SB-431542 for 3 days. Serum cytokine levels were measured by enzyme-linked immunosorbent assay (ELISA). ELISA assayed the lung tissue supernatant as described in Materials and methods. Date are mean ± standard error of the mean; n = 3 mice per group. *P < 0·05; Student's t-test.
, IL-18/IL-2 + SB-431542; n = 5 mice per group. Data are representative of three independent experiments and graph shows pooled data of three experiments. *P < 0·05; Kaplan–Meier method. (b) Lungs were harvested from B6 mice at 24 h after injected with IL-18/IL-2 with or without SB-431542 for 3 days. Lung tissues were stained with haematoxylin and eosin. Original magnification: ×100. (c) Lungs were harvested from B6 mice at 6 h after injection with IL-18/IL-2 with or without SB-431542 for 3 days. (d) The sera were harvested from B6 mice at 6 h after injection with IL-18/IL-2 with or without SB-431542 for 3 days. Serum cytokine levels were measured by enzyme-linked immunosorbent assay (ELISA). ELISA assayed the lung tissue supernatant as described in Materials and methods. Date are mean ± standard error of the mean; n = 3 mice per group. *P < 0·05; Student's t-test.
SB-431542 ameliorates lowers percentage of phosphorylated Smad3-positive cells
Furthermore, the number of phosphorylated Smad3-positive cells was lower in the lung of B6 mice treated with SB-431542 compared with the control (Fig. 3a). To confirm this result, we counted the percentage of p-Smad3-positive cells relative to the total number of cells. The percentage of p-Smad3-positive cells was significantly lower in SB-431542-treated ILD mice (24·98 ± 6·11%) compared with the control (41·12 ± 4·92%, P < 0·05, Fig. 3b).
Fig. 3.
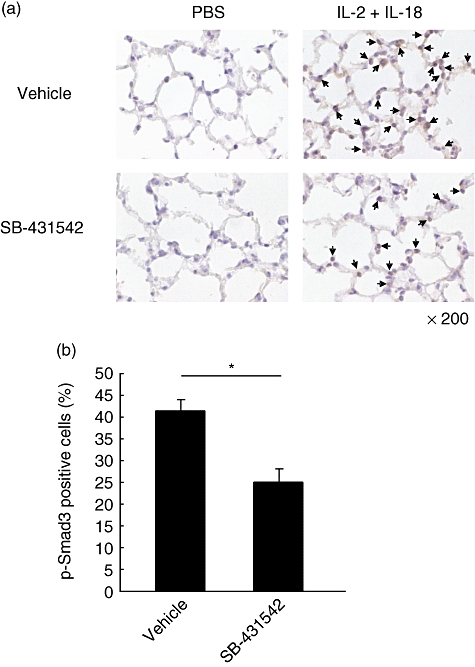
Immunohistochemical findings of phosphorylated Smad3 (p-Smad3) in lungs of B6 mice treated with SB-431542. (a) Lungs were harvested from B6 mice at 6 h after treatment with interleukin (IL)-18/IL-2 with or without SB-431542 for 3 days. The tissues were stained immunohistochemically with anti-pSmad3 antibody. Arrow: pSmad3-positive cells. Original magnification: ×200. (b) In lungs of mice treated with IL-18/IL-2 with or without SB-431542, the percentage of p-Smad3-positive cells per total cells was calculated in five fields under ×200 magnification. Data are mean ± standard error of the mean of three mice per group. *P < 0·05; Student's t-test.
SB-431542 significantly reduces NK cells in the lung
The results showed significantly fewer NK cells (22·53 ± 5·90%) in the lungs of SB-431542-treated B6 mice compared with the control (34·84 ± 4·43%, P < 0·05, Fig. 4a and b). In contrast, the percentage of NK cells in splenocytes of SB-431542-treated B6 mice (11·50 ± 2·13%) tended to be higher than the control (8·99 ± 0·82%, Fig. 4b).
Fig. 4.
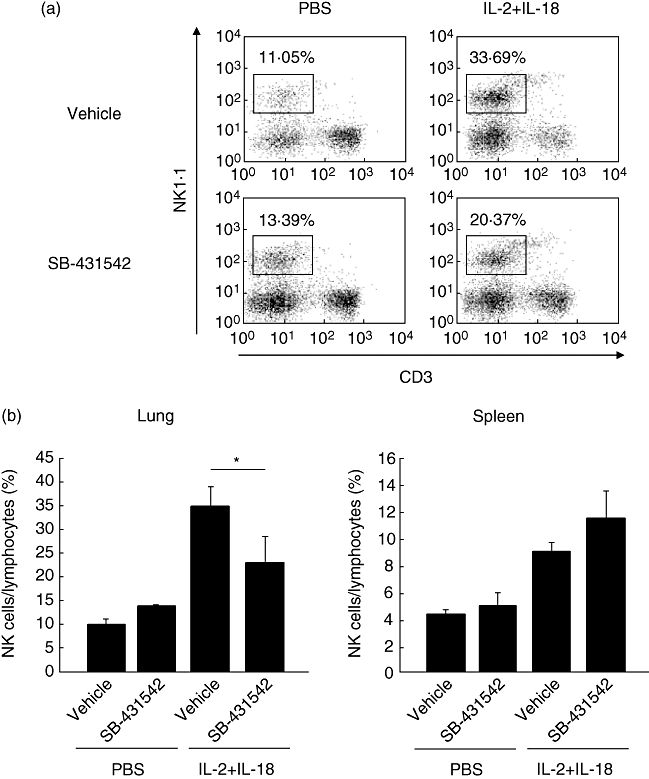
Flow cytometric analysis of pulmonary lymphocytes in B6 mice treated with SB-431542. (a) Pulmonary lymphocytes were harvested from B6 mice at 24 h after treatment with interleukin (IL)-18/IL-2 with or without SB-431542 for 3 days, as described in Materials and methods. Pulmonary lymphocytes were stained with phycoerythrin (PE)-conjugated anti-natural killer (NK)1·1 and PE/cyanine 7 (Cy7)-conjugated anti-CD3ε monoclonal antibodies. (b) Left: proportion of NK cells relative to pulmonary lymphocytes in mice treated with IL-18/IL-2 and with or without SB-431542. Right: proportion of NK cells relative to splenocytes in B6 mice treated with IL-18/IL-2 with or without SB-431542. Data are mean ± standard error of the mean of three mice per group. *P < 0·05; Student's t-test.
Improvement of ILD and reduced cell infiltration in lungs of Smad3–/– mice
We also conducted experiment in Smad3–/– mice for further assessment of the role of TGF-β signalling on IL-18/IL-2-induced ILD [31]. Histological examination showed milder cell infiltration in the lungs of Smad3–/– mice compared with the control (Fig. 5a). Furthermore, flow cytometry showed a significantly small proportion of NK cells in the lung of Smad3–/– mice treated with IL-18/IL-2 (22·77 ± 3·27%) compared with B6 mice (33·89 ± 5·06%, P < 0·05, Fig. 5b and c).
Fig. 5.
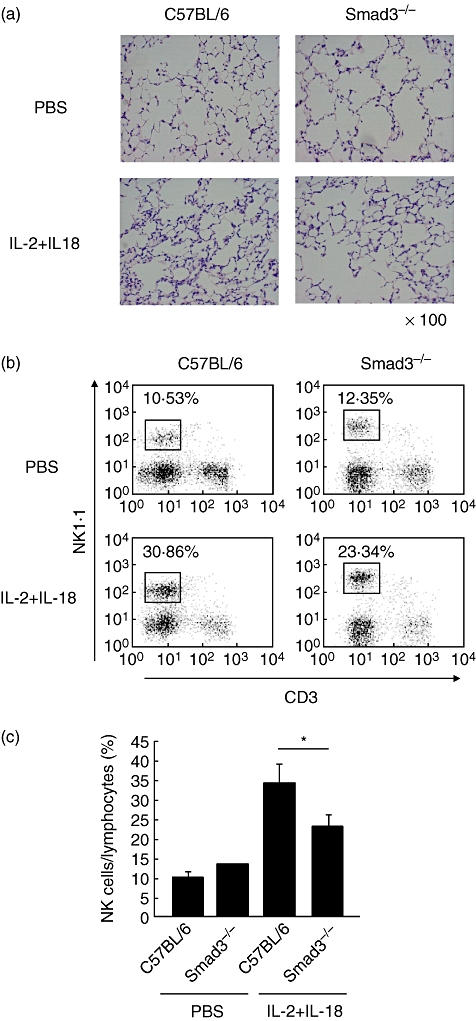
Histological and flow cytometric analysis of pulmonary lymphocytes in Smad3–/– mice. (a) Lungs were harvested from B6 and Smad3–/– mice at 24 h after treated with interleukin (IL)-18/IL-2 for 3 days. Lung tissues were stained with haematoxylin and eosin. Original magnification: ×100. (b) Pulmonary lymphocytes were harvested from B6 and Smad3–/– mice at 24 h after treatment with IL-18/IL-2 for 3 days, as described in Materials and methods. Pulmonary lymphocytes were stained with phycoerythin (PE)-conjugated anti-natural killer (NK)1·1 and PE/cyanine 7 (Cy7)-conjugated anti-CD3ε monoclonal antibodies. (c) Proportion of NK cells relative to pulmonary lymphocytes in B6 and Smad3–/– mice treated with IL-18/IL-2. Data are mean ± standard error of the mean of three mice per group. *P < 0·05; Student's t-test.
Underexpression of chemokine mRNAs in lung of SB-431542-treated and Smad3–/– mice
RT-PCR showed that the expression levels of CCL2, CCL3, CCL4, CCL5 and CXCL10 mRNAs were lower in the lungs of B6 mice treated with SB-431542 than the control (Fig. 6a). Furthermore, the expression levels of CCL2, CCL3, CCL4, CCL5, CCL11, CXCL1 and CXCL10 were lower in the lungs of Smad3–/– mice than the control (Fig. 6b).
Fig. 6.
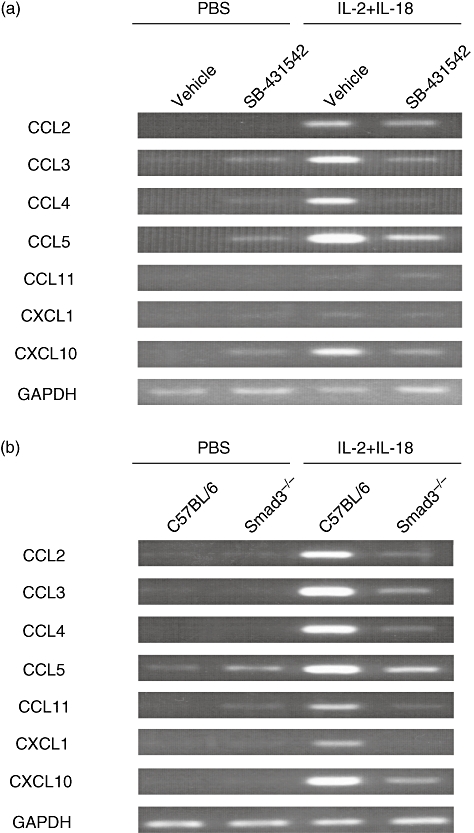
Expression of chemokine mRNAs in lungs of mice deficient in transforming growth factor (TGF)-β. (a) mRNA expression of various chemokines in the lungs of B6 mice treated with SB-431542 and control mice. Lungs were removed at 24 h after the final dose of SB-431542 or vehicle and total RNA was extracted. Chemokine expression (CCL2, CCL3, CCL4, CCL5, CCL11, CXCL1 and CXCL10) was detected by reverse transcription polymerase chain reaction (RT-PCR). (b) mRNA expression of various chemokines in the lungs of Smad3–/– mice and control mice. Lungs were removed at 24 h after the third injection of interleukin (IL)-18/IL-2 and total RNA was extracted. Chemokine expression (CCL2, CCL3, CCL4, CCL5, CCL11, CXCL1 and CXCL10) was detected by RT-PCR. Representative results are shown of three experiments with similar findings.
Discussion
IL-18 and IL-2 are important cytokines that can induce IFN-γ production by NK cells [11]. IL-18 and IL-2 administered daily acted synergistically to induce ILD. ILD mice show severe infiltration of NK cells in the lungs and have high levels of IFN-γ in both serum and lung [19]. Depletion of NK cells by anti-NK1·1 monoclonal antibody or anti-asialo GM1 antibody treatment prevented this effect. Furthermore, the morbid effects of IL-18 and IL-2 were reduced in IFN-γ-deficient mice. These findings suggest that the increase of NK cells and elevation of IFN-γ seem to play a role in the pathogenesis of IL-18/IL-2-induced ILD in mice.
In other mouse models of ILD, BLM induces pulmonary fibrosis and the TGF-β pathway plays an important role in the pathogenesis of ILD [24,27–29,36]. The present study also showed increased expression of TGF-β mRNA in the lung in the early stage of ILD after injection of IL-18/IL-2. Thus, TGF-β seems to be involved in the pathogenesis of IL-18/IL-2-induced ILD and BLM-induced pulmonary fibrosis. Surprisingly, mice treated with SB-431542 delayed mortality in IL-18/IL-2-induced ILD. In addition, IL-18/IL-2-induced NK cell infiltration in the lung was decreased significantly following treatment with SB-431542 and also in Smad3–/– mice. Histological analysis demonstrated that SB-431542 reduced cell infiltration significantly in ILD mice. It was reported that injection of IL-18/IL-2 induced the expression of IFN-γ and TNF-α in sera, and IFN-γ and IL-6 in the lung [19]. In the lung, treatment with SB-431542 reduced the expression of IFN-γ and IL-6 from IL-18/IL-2-induced ILD mice, but in sera the expression of IFN-γ and TNF-α was not changed. Furthermore, IL-18/IL-2-induced ILD was improved in Smad3–/– mice. These findings indicate the involvement of Smad-mediated TGF-β signalling in the pathogenesis of murine ILD.
Bellone et al.[37] reported that TGF-β inhibited activated NK cells in vitro. Our preliminary study confirmed that exogenously added TGF-β suppressed NK cells in vitro (data not shown). However, in the present study, we demonstrated that shutdown of TGF-β signalling by SB-431542 or Smad3 knock-out down-regulated NK cells migration into the lung in vivo, indicating that TGF-β enhanced the infiltration of NK cells into the lung. The discrepancy might be due to the difference between the effects of TGF-β on NK cells in vitro and those in vivo. We proposed that TGF-β could enhance the migration of NK cells into the lung in vivo, whereas TGF-β suppressed the proliferation of NK cells in vitro.
Okamoto et al.[19] showed that certain chemokines, such as CCL2, CCL3, CCL4, CCL5, CCL11, CXCL1 and CXCL10, were up-regulated in the lungs of IL-18/IL-2-induced ILD mice. In contrast, in the lungs of B6 mice treated with SB-431542 and in Smad3–/– mice, chemokine mRNAs were down-regulated. Interestingly, larger proportions of NK cells were noted in the spleens of SB-431542-treated mice with ILD, although their proportion in the lungs was reduced. The latter finding may be due to redistribution and accumulation of NK cells in the spleen. Thus, inhibition of TGF-β signalling could, potentially, be a useful therapeutic strategy in ILD through regulation of NK cells infiltration in the lung.
In conclusion, the present study showed that inhibition of TGF-β signalling regulated IL-18/IL-2-induced ILD through inhibition of NK cells and down-regulation of certain chemokines in the lung. These findings support the notion that TGF-β signalling plays an important role in the pathogenesis of ILD.
Acknowledgments
We thank Dr Chuxla Deng (National Institute of Diabetes and Digestive and Kidney Diseases, MD) for kindly providing Smad3 deficient (Smad3–/–) mice, and Dr F. G. Issa, for the critical reading of the manuscript. This study was supported in part by a Grant-in-Aid for Scientific Research by Japan Society for the Promotion of Science and the Japanese Ministry of Health, Labour and Welfare.
Disclosure
None of the authors has any conflict of interest with the subject matter or materials discussed in the manuscript.
References
- 1.King TE., Jr Clinical advances in the diagnosis and therapy of the interstitial lung diseases. Am J Respir Crit Care Med. 2005;172:268–79. doi: 10.1164/rccm.200503-483OE. [DOI] [PubMed] [Google Scholar]
- 2.Luna MA, Bedrossian CW, Lichtiger B, Salem PA. Interstitial pneumonitis associated with bleomycin therapy. Am J Clin Pathol. 1972;58:501–10. doi: 10.1093/ajcp/58.5.501. [DOI] [PubMed] [Google Scholar]
- 3.Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P. Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J Exp Med. 1989;170:655–63. doi: 10.1084/jem.170.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakao A, Fujii M, Matsumura R, et al. Transient gene transfer and expression of smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest. 1999;104:5–11. doi: 10.1172/JCI6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheule RK, Perkins RC, Hamilton R, Holian A. Bleomycin stimulation of cytokine secretion by the human alveolar macrophage. Am J Physiol. 1992;262:386–91. doi: 10.1152/ajplung.1992.262.4.L386. [DOI] [PubMed] [Google Scholar]
- 6.Smith RE, Strieter RE, Phan SH, et al. Production and function of murine macrophage inflammatory protein-1 alpha in bleomycin-induced lung injury. J Immunol. 1994;153:4704–12. [PubMed] [Google Scholar]
- 7.Zhang K, Gharaee-Kermani M, Jones ML, Warren JS, Phan SH. Lung monocyte chemoattractant protein-1 gene expression in bleomycin-induced pulmonary fibrosis. J Immunol. 1994;153:4733–41. [PubMed] [Google Scholar]
- 8.Hoshino T, Nakamura H, Okamoto M, et al. Redox-active protein thioredoxin prevents proinflammatory cytokine- or bleomycin-induced lung injury. Am J Respir Crit Care Med. 2003;168:1075–83. doi: 10.1164/rccm.200209-982OC. [DOI] [PubMed] [Google Scholar]
- 9.Kuwano K, Hagimoto N, Kawasaki M, et al. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J Clin Invest. 1999;104:13–19. doi: 10.1172/JCI5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamishi K, Yoshimoto T, Tsustumi H, Okamura H. Interleukin-18 regulates both th1 and th2 response. Annu Rev Immunol. 2001;19:423–74. doi: 10.1146/annurev.immunol.19.1.423. [DOI] [PubMed] [Google Scholar]
- 11.Hoshino T, Robert H, Wiltrout RH, Young HA. IL-18 is a potent coinducer of IL-13 in NK and T cells: a new potential role for IL-18 in modulating the immune response. J Immunol. 1999;162:5070–7. [PubMed] [Google Scholar]
- 12.Hoshino T, Yagita H, Ortaldo JR, Wiltrout RH, Young HA. In vivo administration of IL-18 can induce IgE production through TH2 cytokine induction and up-regulation of CD40 ligand (CD154) expression on CD4+ T cells. Eur J Immunol. 2000;30:1998–2006. doi: 10.1002/1521-4141(200007)30:7<1998::AID-IMMU1998>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 13.Hoshino T, Kawase Y, Okamoto M, et al. IL-18- transgenic mice: in vivo evidence of a broad role for IL-18 in modulating immune function. J Immunol. 2001;166:7014–18. doi: 10.4049/jimmunol.166.12.7014. [DOI] [PubMed] [Google Scholar]
- 14.Wild JS, Sigounas A, Sur N, et al. IFN-gamma-inducing factor (IL-18) increases allergic sensitization, serum IgE, Th2 cytokines, and airway eosinophilia in a mouse model of allergic asthma. J Immunol. 2000;164:2701–10. doi: 10.4049/jimmunol.164.5.2701. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimoto T, Min B, Sugimoto T, et al. Nonredundant roles for CD1d-restricted natural killer T cells and conventional CD4+ T cells in the induction of immunoglobulin E antibodies in response to interleukin 18 treatment of mice. J Exp Med. 2003;197:997–1005. doi: 10.1084/jem.20021701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sims JE. IL-1 and IL-18 receptors, and their extended family. Curr Opin Immunol. 2002;14:117–22. doi: 10.1016/s0952-7915(01)00306-5. [DOI] [PubMed] [Google Scholar]
- 17.Nakatani-Okuda A, Ueda H, Kashiwamura S, et al. Protection against bleomycin-induced lung injury by IL-18 in mice. Am J Physiol Lung Cell Mol Physiol. 2005;289:280–7. doi: 10.1152/ajplung.00380.2004. [DOI] [PubMed] [Google Scholar]
- 18.Hoshino T, Okamoto M, Sakazaki Y, Kato S, Young HA, Aizawa H. Role of proinflammatory cytokines IL-18 and IL-1β in bleomycin-induced lung injury in humans and mice. Am J Respir Cell Mol Biol. 2009;41:661–70. doi: 10.1165/rcmb.2008-0182OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okamoto M, Kato S, Oizumi K, et al. Interleukin18 (IL-18) in synergy with IL-2 induced lethal lung injury in mice: a potential role for cytokines, chemokines, and natural killer cells in the pathogenesis of interstitial pneumonia. Blood. 2002;99:1289–98. doi: 10.1182/blood.v99.4.1289. [DOI] [PubMed] [Google Scholar]
- 20.Chen ES, Greenlee BM, Wills-Karp M, Moller DR. Attenuation of lung inflammation and fibrosis in interferon-gamma-deficient mice after intratracheal bleomycin. Am J Respir Cell Mol Biol. 2001;24:545–55. doi: 10.1165/ajrcmb.24.5.4064. [DOI] [PubMed] [Google Scholar]
- 21.Yang HZ, Cui B, Liu HZ, et al. Targeting TLR2 attenuates pulmonary inflammation and fibrosis by reversion of suppressive immune microenvironment. J Immunol. 2009;182:692–702. doi: 10.4049/jimmunol.182.1.692. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki T, Chow CW, Downey GP. Role of innate cells and their products in lung immunopathology. Int J Biochem Cell Biol. 2008;40:1348–61. doi: 10.1016/j.biocel.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Kakizaki T, Kohno M, Watanabe M, et al. Exacerbation of bleomycin-induced injury and fibrosis by pneumonectomy in the residual lung of mice. J Surg Res. 2009;154:336–44. doi: 10.1016/j.jss.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 24.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-β regulation of immune responses. Ann Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 25.Bonniaud P, Kolb M, Galt T, et al. Smad3 null mice develop airspace enlargement and are resistant to TGF-β-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 26.Higashiyama H, Yoshimoto D, Okamoto Y, Kikkawa H, Asano S, Kinoshita S. Receptor-activated Smad localization in bleomycin-induced pulmonary fibrosis. J Clin Pathol. 2007;60:283–9. doi: 10.1136/jcp.2006.037606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venlatesan N, Pini L, Ludwig MS. Changes in Smad expression and subcellular localization in bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1342–7. doi: 10.1152/ajplung.00035.2004. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, Geverd DA. Regulation of Smad3 expression in bleomycin-induced pulmonary fibrosis: a negative feedback loop of TGF-β signaling. Biochem Biophys Res Commun. 2002;294:319–23. doi: 10.1016/S0006-291X(02)00471-0. [DOI] [PubMed] [Google Scholar]
- 29.Zhao J, Shi W, Wang YL, et al. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2002;282:L585–93. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- 30.Laping NJ, Grygielko E, Mathur A, et al. Inhibition of transforming growth factor (TGF)-β1-induced extracellular matrix with a novel inhibitor of the TGF-β type I receptor kinase activity: SB-431542. Mol Pharmacol. 2002;62:58–64. doi: 10.1124/mol.62.1.58. [DOI] [PubMed] [Google Scholar]
- 31.Inman GJ, Nicolás FJ, Callahan JF, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 32.Matsuyama S, Iwadate M, Kondo M, et al. SB-431542 and Gleevec inhibit transforming growth factor-β-induced proliferation of human osteosarcoma cells. Cancer Res. 2003;63:7791–8. [PubMed] [Google Scholar]
- 33.Higashiyama H, Yoshimoto D, Kaise T, et al. Inhibition of activin receptor-like kinase 5 attenuates bleomycin-induced pulmonary fibrosis. Exp Mol Pathol. 2007;83:39–46. doi: 10.1016/j.yexmp.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Letterio JJ, Lechleider RJ, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–91. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li S, Kishihara K, Akashi N, et al. Vδ1+ T cells are crucial for repertoire formation of γδ T cells in the lung. Biochem Biophys Res Commun. 2008;365:246–51. doi: 10.1016/j.bbrc.2007.10.163. [DOI] [PubMed] [Google Scholar]
- 36.Batram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125:754–65. doi: 10.1378/chest.125.2.754. [DOI] [PubMed] [Google Scholar]
- 37.Bellone G, Aste-Amezage M, Trinchieri G, Rodeck U. Regulation of NK cell functions by TGF-β1. J Immunol. 1995;155:1066–73. [PubMed] [Google Scholar]