

Abstract
T helper type 1 (Th1)-type polarization plays a critical role in the pathophysiology of acute graft-versus-host disease (aGVHD). The differentiation of T cells into this subtype is dictated by the nature of the donor naive CD4+ T cell–host antigen presenting cell (APC) interaction. Suppressors of cytokine signalling (SOCS) are a family of molecules that act as negative regulators for cytokine signalling, which regulate the negative cytokine signalling pathway through inhibiting the cytokine-induced Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. Studies have shown that SOCS proteins are key physiological regulators of both innate and adaptive immunity. These molecules are essential for T cell development and differentiation. SOCS-3 can inhibit polarization to Th1 and contribute to polarization to Th2. In this study, we found that interleukin (IL)-2 pre-incubation of C57BL/6 naive CD4+ T cells could up-regulate the expression of SOCS-3. Naive CD4+ T cells constitutively expressed low levels of SOCS-3 mRNA. SOCS-3 mRNA began to rise after 4 h, and reached peak level at 6 h. At 8 h it began to decrease. High expression of SOCS-3 mRNA induced by IL-2 could inhibit the proliferation of naive CD4+ T cells following stimulation with allogeneic antigen. IL-2-induced high SOCS-3 expression in naive CD4+ T cells could inhibit polarization to Th1 with stimulation of allogeneic antigens. We have demonstrated that IL-2-induced high SOCS-3 expression in naive CD4+ T cells could reduce the incidence of aGVHD between major histocompatibility complex (MHC) completely mismatched donor and host when high SOCS3 expression of CD4+T cells encounter allogeneic antigen in time. These results show that IL-2-induced high SOCS-3 expression can inhibit aGVHD through inhibiting proliferation and polarization to Th1 with the stimulation of allogeneic antigen.
Keywords: acute graft-versus-host disease, IL-2, suppressors of cytokine signalling (SOCS)-3, T helper 1 cell, T helper 2 cell
Introduction
The curative potential of allogeneic haematopoietic stem cell transplantation (allo-HSCT) relies strongly upon the immune responses against host antigens mediated by donor T lymphocytes as effectors of anti-tumour immune surveillance [1]. The graft-versus-leukaemia (GVL) effect is exploited further by the use of delayed infusions of donor lymphocytes (DLIs) [2]. However, the therapeutic impact of donor lymphocytes is limited by the risk of allogeneic acute graft-versus-host disease (aGVHD). Approximately 55–60% of patients treated with bulk doses of DLIs for recurrent leukaemia develop aGVHD [3]. Recent research has demonstrated that naive but not memory donor T cells are capable of inducing aGVHD [4,5]. aGVHD requires the stimulation of naive donor T cells by antigen-presenting cells (APC). Residual host APCs alone are sufficient for the induction of CD8+ T cell-dependent aGVHD [6]. Following cognate interaction with activated APC, CD4+ T cells are driven towards T helper type 1 (Th1)-biased cytokine production [7], promoting T cell proliferation and further differentiation, so that very large amounts of proinflammatory cytokines are generated which induce tissue damage in a major histocompatibility complex (MHC)-independent fashion [8]. It has been shown that the infusion of purified CD4+ T cells as DLI resulted in the expansion of CD8+ T cells, suggesting the critical importance of CD4+ T cells in regulating the expansion of CD8+ T cells which mediate aGVHD [9], and the differentiation of CD4+ T cells into Th1 is dictated by the nature of the donor T cell–host APC interaction [10–12]. Therefore, Th1 cells not only mediate aGVHD, but also play a critical role in amplifying aGVHD. We hypothesized that inhibiting artificially the Th1-type differentiation of donor naive CD4+ T cells could prevent aGVHD.
Suppressor of cytokine signalling (SOCS) proteins comprise a family of intracellular regulators of cytokine-induced signal transduction. SOCS protein expression is inducible by cytokines, and once expressed, SOCS proteins inhibit cytokine signalling by switching off the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway [13,14]. SOCS expression has been observed at many stages of T cell development and function, and it has been suggested that SOCS-1 and SOCS-3 are important regulators of T cell activation, differentiation and homeostasis [15–19]. It has been shown that naive CD4+ T cells constitutively express low levels of SOCS-1 and SOCS-3 mRNA [15]. Differentiation into Th1 or Th2 phenotypes is accompanied by preferential expression of distinct SOCS mRNA transcripts and proteins. SOCS-1 expression is fivefold higher in Th1 cells than in Th2 cells, whereas Th2 cells contain 23-fold higher levels of SOCS-3. Interleukin (IL)-12-induced STAT4 activation is inhibited in Th2 cells which express high levels of SOCS-3, whereas IL-4/STAT6 signalling is activated constitutively in Th2 cells, but not in Th1 cells with high SOCS-1 expression [15]. Using SOCS-1+/– T cells, Fujimoto et al. showed that SOCS-1 regulated negatively both Th1- and Th2-cell differentiation in response to IL-12 and IL-4, respectively [20]. SOCS-3 can force the Th1/Th2 balance towards a Th2-type but not a Th1-type differentiation [21,22]. In addition, SOCS-3 transgenic mice showed increased Th2 responses. In contrast, dominant-negative mutant SOCS-3 transgenic mice demonstrated decreased Th2 development [21]. This suggests that SOCS-3 has an important role in balancing Th1/Th2 towards Th2-type differentiation. SOCS-3 not only has an influence on the balance of Th1/Th2 differentiation, but can also inhibit lymphocyte proliferation. IL-2-mediated proliferation of BaF3 transfectants expressing SOCS-3 is inhibited [22]. T cells from transgenic mice expressing SOCS3 exhibit a significant reduction in IL-2 production induced by T cell receptor cross-linking when T cells are co-stimulated with CD28 [23]. In addition, SOCS-3-deficient CD8+ T cells show greater proliferation than wild-type cells in response to T cell receptor (TCR) ligation, despite normal activation of signalling pathways downstream from TCR or CD28 receptors [24]. These studies suggest that SOCS-3 could regulate lymphocyte proliferation negatively.
The expression of SOCS-3 proteins has been shown to be highly regulated by IL-2 and other cytokines [22,25–27]. IL-2 can induce the kit-225 cell line to express SOCS-3 proteins highly in a final concentration of 50 U/ml [22], and the proliferation of T cell transfectants expressing SOCS-3 mRNA is inhibited. Therefore, is the proliferation of T lymphocytes inducibly expressing SOCS-3 by IL-2 inhibited? SOCS-3 can force the Th1/Th2 balance towards Th2-type but not Th1-type differentiation [21,22]. Does the SOCS-3 expression induced by IL-2 inhibit Th1-type polarization? Because Th1-type polarization plays a critical role in the pathophysiology of aGVHD, does the SOCS-3 expression induced by IL-2 inhibit aGVHD if it can inhibit the naive CD4+ T cell proliferation and polarization into Th1? In this study, we have demonstrated that IL-2 pre-incubation can induce B6 mouse CD4+ T cells to highly express SOCS-3, and high expression of SOCS-3 can inhibit proliferation and polarization into Th1 and prevent aGVHD between MHC completely mismatched donor and host.
Materials and methods
Animals
Eight to 10-week-old male C57BL/6 (B6, H-2b) and female BALB/c (H-2d) mice were purchased from the Experimental Animal Center of Academia Sinica. All mice were housed in specific pathogen-free (SPF) facilities at Academia Sinica and provided with sterilized food and water.
Purification of naive B6 CD4+ T cells
Spleens were removed from B6 mice to produce a single cell suspension. Red blood cells were lysed with Tris-NH4Cl. Cells were then washed three times with RPMI-1640, and purified with a CD4+CD62+ T cell isolation kit (Miltenyi Biotec, Germany).
IL-2 pre-incubation
Lymphocytes were cultured in RPMI-1640 (Gibco Corporation, Portland, OR, USA) supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin and 10% fetal bovine serum (FBS) (Hyclone Corporation, Logan, UT, USA), referred to here as complete RPMI (cRPMI). T cells isolated from B6 mice were resuspended with cRPMI at a density of 5 × 106/ml and then incubated for 4 h in vitro with IL-2 (Sigma Corporation, Santa Clara, CA, USA) at a final concentration of 50 U/ml at 37°C in 5% CO2.
Reverse transcription polymerase chain reaction (RT-PCR) analysis
RNA isolation and first-strand cDNA synthesis were performed as described previously [28]. Primers used for PCR amplification are as follows: for SOCS3, 5′-TGC GCC ATG GTC ACC CAC AGC AAG TTT-3′ and 5′-GCT CCT TAA AGT GGA GCA TCA TAC TGA-3′. Amplification was carried out for 30 cycles of denaturation for 30 s at 95°C, annealing for 30 s at 60°C, and extension for 30 s at 72°C. After the 30th cycle, the samples were subjected to a final 10-min extension at 72°C. PCR-amplified fragments were fractionated on 1·5% agarose gels and stained with ethidium bromide. Real-time PCR was performed on a LightCycler™ real-time PCR sequence detection system (Roche, Switzerland), as described previously, with the following forward and reverse primers, respectively: for SOCS3, 5′-CAA GTC ATC ACT ATT GGC AAC GA-3′ and 5′-CCC AAG AAG GAA GGC TGG A-3′; for β-actin, 5′-CCA GCC ATG TAC GTT GCT ATC-3′ and 5′-CAG GTC CAG ACG CAG GAT GGC-3′. PCR parameters were recommended for the TaqMan Universal PCR Master Mix kit (Applied Biosystems, Carlsbad, CA, USA). Triplicate samples of twofold serial dilutions of cDNA were assayed and used to construct the standard curves.
Lymphoproliferation assays
Lymphocyte proliferation assays were performed as detailed elsewhere [29]. Briefly, freshly isolated B6 naive CD4+ T cells at a density of 5 × 106/ml were pre-incubated with IL-2 at a final concentration of 50 U/ml for 4 h, and were then stimulated for 72 h with the same quantity of mitomycin-inactivated BALB/c spleen cells at 37°C in 5% CO2. We added the WST-8/Cell Counting Kit-8 (CCK-8 kit, Japan) for 4 h before stopping stimulation with allogeneic antigen, and then detected the optical density (OD) value with a 450 nm microplate reader.
Generation of DO11·10 T cell line transfectants expressing SOCS-3 and assays of IL-2 production
Mouse SOCS3 DNA fragments flanked by BamHI and EcoRI restriction sites were generated from a pMD18-T/SOCS3 plasmid obtained in a preliminary experiment by PCR amplification using the primers (5′-CTG GAA TTC ATG GTC ACC CAC AGC AAG TT-3′ and 5′-CTG GGA TCC TTA AAG TGG AGC ATC ATA CTG ATC-3′) targeting the SOCS3 construct. The fragments were cloned directionally into the BamHI and EcoRI sites of a pLXSN vector (kindly provided by the Laboratory of Immunity, Fudan University), and the identity of the product was confirmed by sequencing. PA317 packaging cells were transfected with pLXSN-SOCS3 (2·0 µg/ml) using Lipofectamine™ 2000, according to the manufacturer's instructions (Invitrogen, Portland, OR, USA), and cultured to generate supernatants containing retrovirus. DO11·10 T cells (kindly provided by the Laboratory of Immunity, Fudan University) were then infected with the supernatant, as described previously [30], and selected using G418 (400 µg/ml). DO11·10 T cells transfected with pLXSN-SOCS3 for DO-SOCS3 T cell were named DS, and DO11·10 T cells transfected with empty pLXSN for DO-pLXSN T cell were named DO. DS and DO cells were added into 96-well plates (at 1·5 × 105/ ml) containing 0·6 µM ovalbumin (OVA) peptide and BALB/c mouse splenic cells (3 × 105/ ml). Cells were cultured for 3 days at 37°C in an atmosphere of 5% CO2. Supernatants were then collected and analysed using a sandwich enzyme-linked immunosorbent assay (ELISA) kit, according to the manufacturer's instructions (Biosource, Portland, OR, USA).
Assay for interferon (IFN)-γ and IL-4 expression in T cells by ELISA
B6 naive CD4+ T cells and spleen cells with IL-2 pre-incubation in 96-well plates at a density of 1 × 106/ml were stimulated for 48 h with 1 × 106 BALB/c spleen cells inactivated by mitomycin at 37°C, 5% CO2. Supernatants were then collected and analysed using the ELISA kit for IL-4 and IFN-γ, according to the manufacturer's instructions (Biosource).
IL-2 pre-incubation programme to inhibit aGVHD
We used female BALB/c recipients and male B6 donors. All recipients received 5 Gy total body irradiation (TBI) (Gammacell 40137 Se γ irradiance system; Canada Nordion International Corporation, Canada) before 3 × 107 B6 spleen cells were injected intraperitoneally. The intraperitoneal injection model for donor lymphocytes has been described previously [31,32]. The rate and duration of irradiation were 0·86 Gy/min and 6 min, respectively. The recipients were divided into four groups: group A (n = 9), 3 × 107 B6 spleen cells transfer; group B (n = 9), 3 × 107 B6 spleen cells transfer with IL-2 pre-incubation; group C (n = 9), 3 × 107 B6 spleen cells transfer stimulated with host allogeneic antigen presented by inactivated BALB/c spleen cells for 72 h before intraperitoneal injection; and group D (n = 9), 3 × 107 B6 spleen cells transfer pre-incubated with IL-2 for 4 h and then stimulated with host allogeneic antigen presented by inactivated BALB/c spleen cells for 72 h before intraperitoneal injection. The four groups were observed for 60 days.
The observation parameters were as follows:
Survival time: register the survival times of each group recipient and draw the survival curve.
Detection of donor chimerism: spleen cells of surviving mice at the end of the observation time were used to detect H-2b – the mark of the B6 mouse chimerism by fluorescence activated cell sorter (FACS) (MHC molecule of BALB/c: H-2d; and MHC molecule of B6: H-2b.). In addition, we detected the B6 mouse chimerism by PCR. The target gene is the SRY sequence of the sex-determining region on the Y chromatosome of B6 mice (Pubmed Entrez Nucleotide: gi: 6755760, NM: 011564). Primer sequence: CTG CTG TGA ACA GAC ACT AC (forward), GAC TCC TCT GAC TTC ACT TG (backward). The predicted length of PCR product is 722 base pairs (bp).
Score of symptoms of aGVHD [33]: the degree of systemic GVHD was assessed by a scoring system described in another mouse model that incorporates five clinical parameters: weight loss, posture (hunching), activity, fur texture and skin integrity. Each mouse from coded cages was evaluated and graded from 0 to 2 for each criterion. A clinical index was generated subsequently by summation of the five item scores (maximum index = 10).
Parameters of histology of aGVHD [34]: formalin-preserved liver and small bowel were embedded in paraffin, and 5-m thick sections were stained with haematoxylin and eosin for histological examination. Six parameters were observed for small bowel (villous blunting crypt regeneration, crypt epithelial cell apoptosis, crypt loss, lumina sloughing of cellular debris, lamina propria inflammatory cell infiltrate and mucosal ulceration). Ten parameters were observed for liver (portal tract expansion by an inflammatory cell infiltrate, lymphocytic infiltrate of bile ducts, bile duct epithelial cell apoptosis, bile duct epithelial cell sloughing, vascular endothelialitis, parenchymal apoptosis, parenchymal microabscesses, parenchymal mitotic figures, hepatocellular cholestasis and hepatocellular steatosis).
Statistics
All data were analysed using SPSS version 13·0 software. Descriptive data for the major variables were presented as mean ± standard deviation. One-way analysis of variance (anova) test and independent t-tests were performed to compare group differences. Survival data were analysed using the Kaplan–Meier method of life-table analysis, and statistical analysis was performed with the log-rank test. P-values < 0·05 were considered statistically significant.
Results
IL-2 pre-incubation allogeneic lymphocytes can up-regulate SOCS-3 expression
Although it has been shown that IL-2 can induce high SOCS-3 kit-225 cell line expression [22], no one has detected inducible SOCS-3 expression by IL-2 in allogeneic lymphocytes, which are the effect cells of aGVHD. To determine if the regularity of expression of SOCS-3 in allogeneic lymphocytes pre-incubated by IL-2 is similar to that of the cell line, we detected the SOCS-3 expression of allogeneic lymphocytes with IL-2 pre-incubation.
Recent data have demonstrated that naive but not memory donor T cells are capable of inducing aGVHD [4,5]. First, we investigated the expression of SOCS-3 in B6 naive CD4+ T cells which were pre-incubated with IL-2 (final concentration of 50 U/ml) every 2 h for periods of up to 10 h by real-time PCR. SOCS-3 expression began to rise at 2 h, and reached its peak level at 4–6 h. It began to decrease 8 h later (Fig. 1). This regularity was similar to the kit-225 cell line, although the peak time was at 2–4 h [22]. The observed peak time difference was due probably to the reason that the cells we used were different from kit-225 and the detection method was also different (Cohney et al.[22] used the Western blot method at the proteic level). Subsequently, we detected SOCS-3 expression in B6 spleen cells which were pre-incubated with IL-2. The regularity of expression was the same as that of B6 naive CD4+ T cells. SOCS-3 expression still began to rise at 2 h, peaked at 4–6 h, and decreased at 8 h (Fig. 1).
Fig. 1.
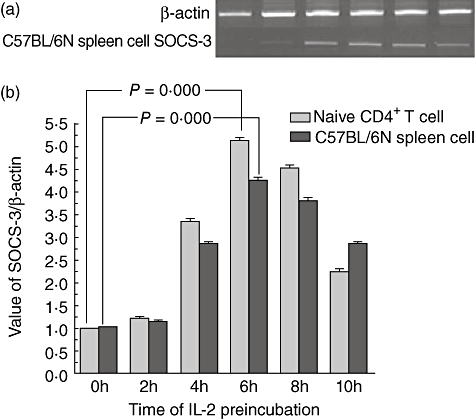
Change of suppressors of cytokine signalling (SOCS-3) mRNA expression of C57BL/6N spleen cells and naive CD4+ T cells during pre-incubation with interleukin (IL)-2. SOCS-3 mRNA expression reached a peak 6 h after pre-incubation and began to decrease after 8 h.
IL-2 pre-incubation can inhibit the proliferation of allogeneic lymphocytes with allogeneic antigen stimulation
It has been shown that IL-2-mediated proliferation of BaF3 transfectants expressing SOCS-3 is inhibited [22]. We investigated whether the proliferation of T lymphocytes inducibly expressing SOCS-3 by IL-2 could be inhibited.
We first established the DO-SOCS3 T cell line by transfecting the SOCS3 gene into a DO11·10 hybridoma cell line and explored whether the proliferation of DO11·10 expressing SOCS-3 was influenced following stimulation with OVA-specific antigen. We used OVA323–329-specific antigen to stimulate DS-SOCS3 and DO11·10 cells which were transfected with empty pMD18T plasmid (DO) and detected the activation and proliferation of DS-SOCS3 following stimulation with OVA323–329. DO11·10 was a hybridoma cell line, so we detected IL-2 secretion as the proliferation activity. The results showed that proliferation of DS-SOCS3 following stimulation with OVA323–329 was inhibited significantly (P = 0·0000, Fig. 2a).
Fig. 2.
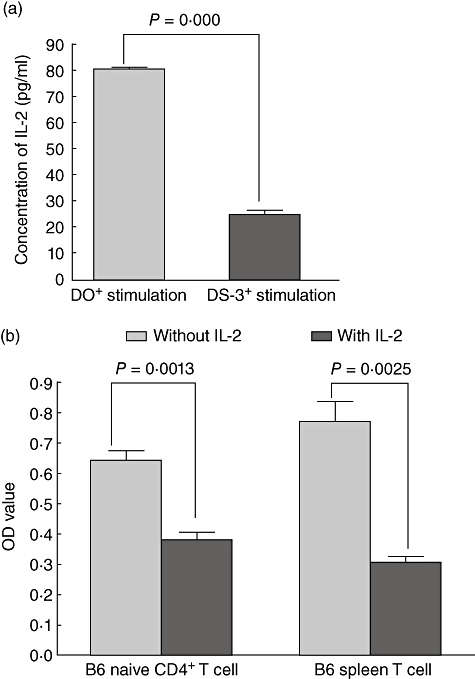
Proliferation of lymphocytes was suppressed with high suppressors of cytokine signalling (SOCS-3) mRNA expression to allogeneic antigen stimulation. (a) Proliferation of the DO11·10 cell line with high SOCS-3 mRNA expression was suppressed by stimulation of ovalbumin (OVA); DO+ stimulation was the group of DO11.10 cells being transferred empty viral vector, and DS-3+ stimulation was the group of DO11.10 cells being transferred viral vector with the SOCS-3 gene. (b) Proliferation of B6 naive CD4+ T cells and B6 spleen T cells after interleukin (IL)-2 pre-incubation for 4 h was suppressed by stimulation of the allogeneic antigen.
Subsequently, we explored the proliferation of B6 naive CD4+ T cells inducibly expressing SOCS-3 mRNA by IL-2 following stimulation with allogeneic antigen. Our results showed that SOCS-3 mRNA peaked 4–6 h after IL-2 pre-incubation, so we pre-incubated B6 naive CD4+ T cells with IL-2 for 4 h, followed by stimulation with allogeneic antigen-BALB/C spleen cells inactivated by mitomycin for 72 h. The results showed that proliferation of B6 naive CD4+ T cells with IL-2 pre-incubation was lower than proliferation in controls that were not pre-incubated with IL-2 (P = 0·0013, Fig. 2b).
Finally, we investigated the proliferation of B6 spleen cells inducibly expressing SOCS-3 mRNA by IL-2 following stimulation with allogeneic antigen. Using the same method, we pre-incubated spleen cells with IL-2 for 4 h before stimulation with allogeneic antigen-BALB/C spleen cells inactivated by mitomycin for 72 h, and detected the proliferation of B6 spleen cells following stimulation with allogeneic antigen with the CCK-8 kit. The results also showed that the proliferation of B6 spleen cells with IL-2 pre-incubation was significantly weaker than that of the controls without IL-2 pre-incubation (P = 0·0025, Fig. 2b).
IL-2 pre-incubation can inhibit Th1-type polarization of B6 naive CD4+ lymphocytes following stimulation with allogeneic antigen
SOCS-3 can inhibit the Th1-type polarization which plays a critical role in the pathophysiology of aGVHD [21,22,35,36]; therefore, we explored whether high SOCS-3 mRNA expression induced by IL-2 pre-incubation can inhibit Th1-type polarization in B6 naive CD4+ lymphocytes. According to the regularity of expression of SOCS-3 mRNA, we pre-incubated B6 naive CD4+ lymphocytes and B6 spleen cells, respectively, with IL-2 for 4 h before stimulation of allogeneic antigen-BALB/c spleen cells inactivated by mitomycin for 48 h. We then collected the supernatants to detect the levels of IFN-γ and IL-4.
The results showed that expression of IFN-γ and IL-4 of B6 naive CD4+ lymphocytes was different between pre-incubation of the two groups with or without IL-2. The IFN-γ level in group pre-incubation with IL-2 was lower than that in group pre-incubation without IL-2 (P = 0·000, Fig. 3a). The IL-4 level in group pre-incubation with IL-2 was higher than that in group pre-incubation without IL-2 (P = 0·000, Fig. 3a).
Fig. 3.
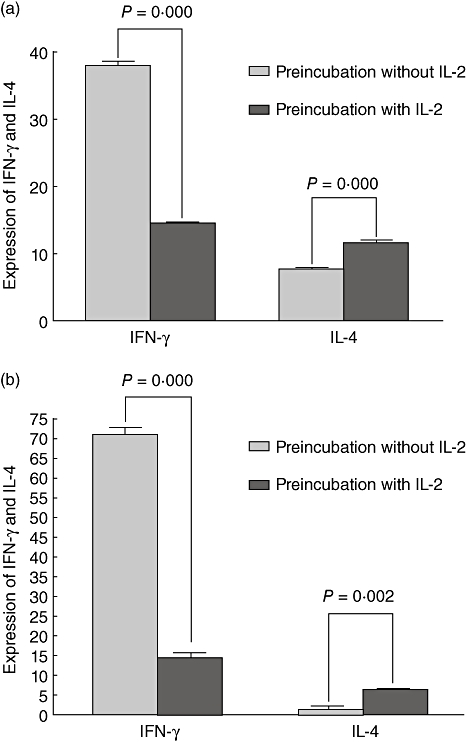
(a) Interferon (IFN)-γ and interleukin (IL)-4 expression of B6 naive CD4+ cells after IL-2 pre-incubation. (b) IFN-γ and IL-4 expression of B6 spleen cells after IL-2 pre-incubation.
The expression of IFN-γ and IL-4 of B6 spleen cells was similar to that of B6 naive CD4+ lymphocytes (P = 0·002, and 0·000, respectively, Fig. 3b)
IL-2 pre-incubation can prevent fully MHC mismatched mice aGVHD
We assessed suppressive function in vivo in an aGVHD mice model. We used female BALB/C recipients and male B6 donors. All recipients received 5 Gy TBI as conditioning regimen. In group A (n = 9), B6 spleen cells (3 × 107 cells) were injected intraperitoneally into recipients as control. We first explored whether aGVHD was inhibited in the recipients (group B, n = 9) which received 3 × 107 B6 spleen cells pre-incubated with IL-2 before intraperitoneal injection. We found that the mean survival time of group B (14·4 ± 1·5 days) was not statistically different from that of group A (12·2 ± 3·1 days) (P = 0·3090, Fig. 4a). The scores of aGVHD symptoms between the two groups were also not different (P = 0·7851). These findings suggest that IL-2 pre-incubation can up-regulate the expression of SOCS-3, but it was a short-lived gene product induced by IL-2 in lymphocytes. If the spleen cells with short-lived SOCS-3 did not receive allogeneic antigen in time, aGVHD could also not be inhibited; therefore, we projected another group (group D, n = 9) in which recipients received 3 × 107 B6 spleen cells which were presented with host-allogeneic antigen-inactivated BALB/C spleen cells for 72 h after IL-2 pre-incubation for 4 h. The results showed that aGVHD was inhibited significantly in group D. The mean survival time of group D was 44·1 ± 23·8 days, which was longer than that of group A (P = 0·0042, Fig. 4b). The score of aGVHD in group D was lower than that in group A (P = 0·0046). In addition, we established another control group (group C, n = 9) in which recipients received 3 × 107 B6 spleen cells which were presented with host-allogeneic antigen-inactivated BALB/C spleen cells for 72 h without IL-2 pre-incubation for 4 h. The mean survival time of group D was longer than that of group C (P = 0·0039, Fig. 4c). The score of aGVHD in group D was lower than that in group C (P = 0·0422).
Fig. 4.
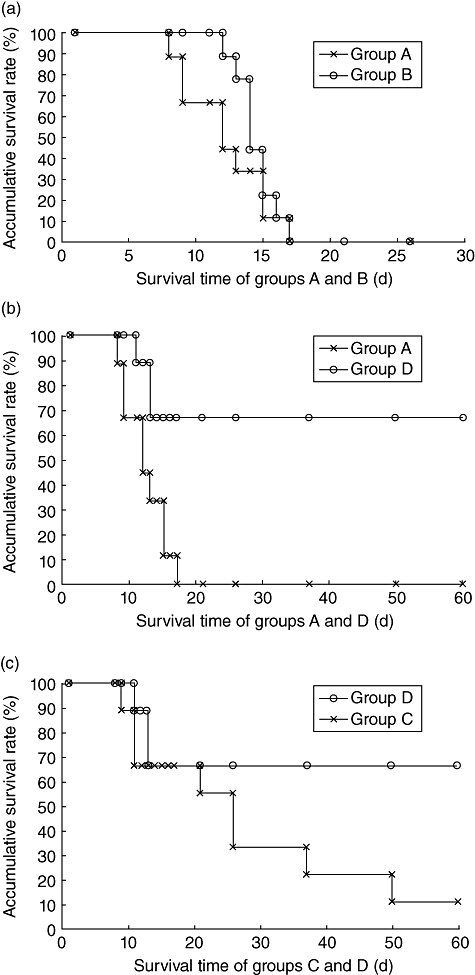
Kaplan–Meier survival curve of four groups. (a) Survival time of groups A and B. The two groups have no statistical difference (P = 0·3090); (b) survival time of groups A and D. The group D survival time was longer than group A (P = 0·0042); (b) survival time of groups C and D. The group D survival time was longer than group C (P = 0·0039).
We detected donor spleen cell chimerism (H-2b) in the long-term surviving mice of group D by FACS. The donor mouse chimerism rate was 3·15 ± 1·59%, which is higher than that of normal BALB/C spleen cells (0·61 ± 0·32%) (P = 0·0062, Fig. 5b). Although the chimerism rate was much lower, we could detected the chimerism by PCR again (Fig. 5a).
Fig. 5.
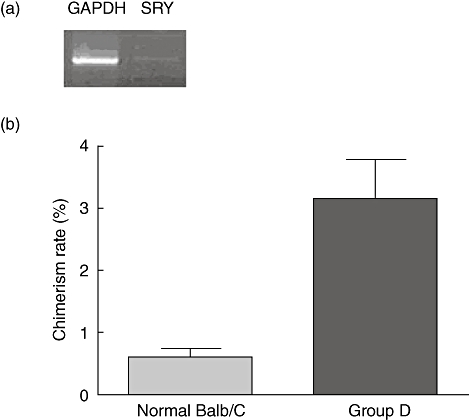
The chimerism rate of group D was higher than the normal BALB/C mouse (P = 0·0062). Electropherogram of the SRY gene of male C57BL/6N mice detected by polymerase chain reaction (PCR), housekeeping gene: glyceraldehyde-3-phosphate-dehydrogenase (GAPDH), 660 base pairs (bp); objective gene: SRY, 722 bp.
The liver and small bowel of dead mice and the long-term surviving mice of group D following the observation period were taken for GVHD histological examination. The aGVHD histological manifestations in the long-term surviving group D mice were slight, such as the damage to sinus hepaticus endothelial cells and anabrosis of the mucous membrane of the small intestine (Fig. 6c and d). However, the histological manifestations in those mice which died of aGVHD were serious, showing diffuse cellular swelling, degeneration of hepatic parenchymal cells and complete damage of the mucous membrane gland of the small intestine (Fig. 6e,f).
Fig. 6.
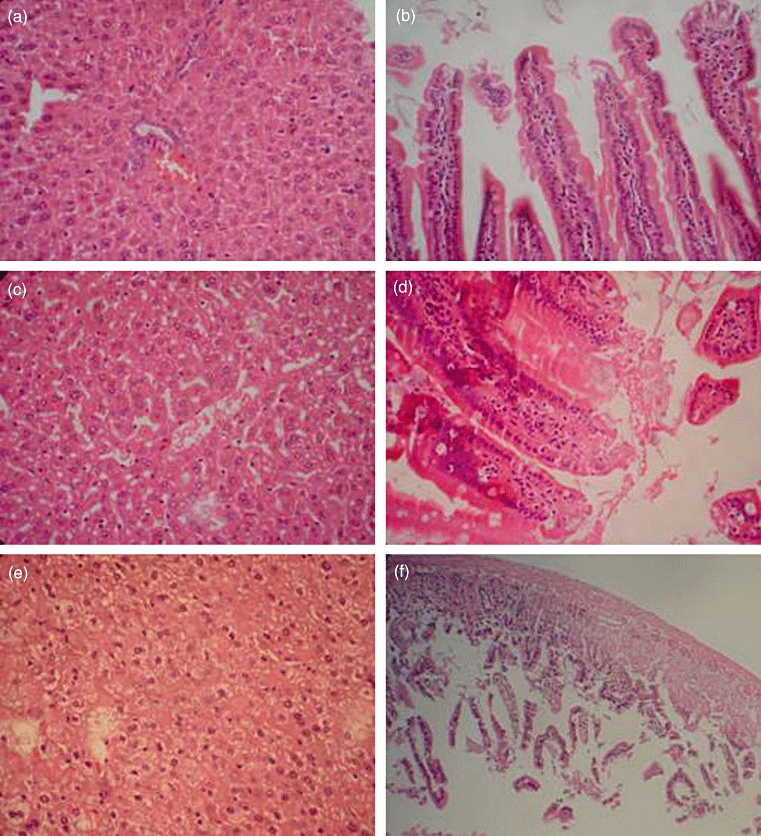
Change of acute graft-versus-host (aGVHD) histology. (a,b) Histology of liver and small intestine after 5 Gy total body irradiation (TBI); (c,d) aGVHD histology of liver and small intestine in group D. Damage to the sinus hepaticus endothelial cells and anabrosis of the mucous membrane of the small intestine are shown; (e,f) liver and small intestine aGVHD histology in a mouse that died of aGVHD. Diffuse cellular swelling and degeneration of hepatic parenchymal cells and complete damage of the mucous membrane of the small intestine are shown.
Discussion
IL-2 is the first T cell growth factor to be cloned molecularly and remains the cytokine of choice for the propagation of T cells in culture [37]. Because IL-2 can induce T cell expansion potently in vitro, it has been assumed for many years that IL-2 played an analogous role in amplifying T cell responses in vivo. This assumption led to the development of therapeutic strategies aimed at modulating IL-2 signal strength for clinical efficacy. On one hand, IL-2 itself is infused in patients with cancer or acquired immune deficiency syndrome (AIDS) to enhance T cell numbers and function [38,39]. On the other hand, antibodies to the IL-2R are used to inhibit IL-2 signalling to suppress rejection of the transplanted organs [40]. These agents show clinical efficacy in some cases, lending support to the notion that IL-2 serves as an important T cell growth factor and can promote immunity in vivo. However, this notion is now being challenged. IL-2 is critical for the development and peripheral expansion of CD4+CD25+ regulatory T cells, which promote self-tolerance by suppressing T cell responses in vivo (for a review, see [41]). A short course of high-dose IL-2 [42], begun on the day of bone marrow transplantation, protects against GVHD. This inhibitory effect is directed against donor CD4+ cells, even though the mechanism has not yet been elucidated.
In this study, our results showed that IL-2 can inhibit T lymphocyte immunity. The up-regulation of SOCS-3 mRNA induced by IL-2 played a critical role during this course. The proliferation of B6 naive CD4+ T lymphocytes inducibly expressing SOCS-3 by IL-2 mRNA is inhibited by the stimulation of allogeneic antigens. The proliferation of the DO11·10 hybridoma cell line transfectants expressing SOCS-3 mRNA is also inhibited by stimulation of specific antigens, which confirms that IL-2 can inhibit T lymphocyte immunity through up-regulating the expression of SOCS-3 mRNA. However, SOCS-3 proteins, not mRNA, have the same effect in lymphocytes, and it would be interesting to perform this at proteic level on primary lymphocyte cells.
SOCS-3 is a critical negative feedback regulatory factor of the JAK/STAT signalling transduction pathway, which plays a critical negative regulatory role in maintaining the balance of immunity. It has been shown that SOCS-3 can inhibit the proliferation of lymphocyte lines to the stimulation of specific antigens [16,19,22,24]. However, inhibition of the proliferation of allogeneic lymphocytes with allogeneic antigen stimulation has not been reported. In this study, our results showed that the proliferation of B6 naive CD4+ T cells inducibly expressing SOCS-3 mRNA by IL-2 to the stimulation of allogeneic antigen was inhibited, suggesting the possibility of the initial inhibition of aGVHD. Further studies also demonstrated that the Th1-type polarization of B6 naive CD4+ T cells inducibly expressing SOCS-3 mRNA by IL-2 to the stimulation of allogeneic antigen was inhibited. These results support further that B6 naive CD4+ T cell inducibly expressing SOCS-3 mRNA by IL-2 could inhibit aGVHD, but we do not know whether B6 naive CD4+ T cell transfectants expressing SOCS-3 can inhibit aGVHD. This will need further study.
These results will help us to understand the mechanisms of the inhibitory effect on aGVHD. We hypothesized that whether IL-2 signalling promotes or inhibits immunity might be related to the state of the CD4+ T cell. If the target cells of IL-2 signalling are activated CD4+ T cells, which express the high-affinity IL-2 receptor (IL-2R) with IL-2Rα (CD25), the IL-2 signal activates the JAK/STAT signalling transduction pathway after IL-2 binds with high-affinity IL-2R. At the same time, down-regulation of SOCS-3 expression induced by antigen-TCR-mediated signals attenuates inhibition to the JAK/STAT signalling transduction pathway [16]. Activation of the JAK/STAT signalling transduction pathway leads to STAT phosphorylation and activation of genetic transcription, which can drive T cell proliferation and promote immunity. If the target cells of IL-2 signalling are naive CD4+ T cells which express low-affinity IL-2R without IL-2Rα (CD25), but with IL-2Rβ and IL-2Rγ, the IL-2 signal up-regulates expression of the negative feedback regulatory factor SOCS-3 when IL-2 binds with low-affinity IL-2R. Up-regulation of SOCS-3 expression can enhance inhibition to the JAK/STAT signalling transduction pathway and inhibit STAT phosphorylation and genetic transcription. This leads to the inhibition of T cell proliferation and immunity. In the earliest stage of bone marrow transplantation (BMT), donor naive lymphocytes do not receive allogeneic antigen stimulation presented by host APC. A short course of high-dose IL-2 starting on the day of BMT can up-regulate the SOCS-3 expression of donor naive CD4+ T cells. The proliferation and Th1-type polarization of donor naive CD4+ T cells inducibly expressing SOCS-3 is inhibited, which inhibits immunity to allogeneic antigen and aGVHD. Our animal experiment provided strong support for this hypothesis.
Clearly, IL-2 pre-incubation can inhibit fully MHC-mismatched mice fatal aGVHD; but donor lymphocytes incubated with IL-2 for 4 h injected immediately into recipients did not inhibit aGVHD. If the lymphocytes inducibly expressing SOCS-3 were stimulated with allogeneic antigen for 72 h, aGVHD could be inhibited significantly. A possible explanation is that it needs time for donor lymphocytes to receive the antigen presented by host APC, but SOCS-3 is a short-lived gene product induced in lymphocytes by IL-2. SOCS-3 could not generate inhibition to aGVHD unless the lymphocytes inducibly expressing SOCS-3 receive allogeneic antigen in time.
Methods of inhibiting aGVHD, such as glucocorticosteroid, anti-thymocyte globulin, cyclosporin A and methylaminopterin, inhibit the whole immune system, and this can lead to the inhibition of graft-versus-tumour effects and serious infections. The aim of this study was to adjust the direction of polarization of Th and to inhibit excessive proliferation during aGVHD; the animal experimental results show the effectiveness of our aim. IL-2 pre-incubation can prevent aGVHD through up-regulating the expression of SOCS-3 and inhibiting the proliferation of Th1-type polarization of naive CD4+ T cells. Hopefully, these will provide new pathways for the inhibition of aGVHD.
Acknowledgments
This paper was supported by the Great Biology and Medicine Foundation of Key Problems in Science and the Technology of Shanghai Science and Technology Committee (no. 06DZ19013). We acknowledge Dr Wan Yin (Department of immunology, Shanghai Medical College, Fudan University), who supported us very much during the initiation of our work.
Disclosure
None.
References
- 1.Appelbaum FR. Hematopoietic cell transplantation as immunotherapy. Nature. 2001;411:385–9. doi: 10.1038/35077251. [DOI] [PubMed] [Google Scholar]
- 2.Kolb HJ, Mittermüller J, Clemm C, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990;76:2462–5. [PubMed] [Google Scholar]
- 3.Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood. 2004;103:767–6. doi: 10.1182/blood-2003-02-0342. [DOI] [PubMed] [Google Scholar]
- 4.Anderson BE, McNiff J, Yan J, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest. 2003;112:101–8. doi: 10.1172/JCI17601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Joe G, Zhu J, et al. Dendritic cell-activated CD44hiCD8+ T cells are defective in mediating acute graft-versus-host disease but retain graft-versus-leukemia activity. Blood. 2004;103:3970–8. doi: 10.1182/blood-2003-09-3135. [DOI] [PubMed] [Google Scholar]
- 6.Shlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285:412–15. doi: 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 7.Antin JH, Ferrara JLM. Cytokine dysregulation and acute graft-versus-host disease. Blood. 1992;80:2964–8. [PubMed] [Google Scholar]
- 8.Teshima T, Ordemann R, Reddy P, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8:575–81. doi: 10.1038/nm0602-575. [DOI] [PubMed] [Google Scholar]
- 9.Zorn E, Wang KS, Hochberg EP, et al. Infusion of CD4+ donor lymphocytes induces the expansion of CD8+ donor T cells with cytolytic activity directed against recipient hematopoietic cells. Clin Cancer Res. 2002;8:2052–60. [PubMed] [Google Scholar]
- 10.Rissoan MC, Soumelis V, Kadowaki N, et al. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999;283:1183–6. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- 11.Rossi M, Arpinati M, Rondelli D, Anasetti C. Plasmacytoid dendritic cells: do they have a role in immune responses after hematopoietic cell transplantation? Hum Immunol. 2002;63:1194–200. doi: 10.1016/s0198-8859(02)00758-9. [DOI] [PubMed] [Google Scholar]
- 12.Arpinati M, Chirumbolo G, Urbini B, Perrone G, Rondelli D, Anasetti C. Role of plasmacytoid dendritic cells in immunity and tolerance after allogeneic hematopoietic stem cell transplantation. Transpl Immunol. 2003;11:345–56. doi: 10.1016/S0966-3274(03)00055-8. [DOI] [PubMed] [Google Scholar]
- 13.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signaling and immune regulation. Nat Rev Immunol. 2007;7:454–65. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 14.Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol. 2002;2:1–7. doi: 10.1038/nri818. [DOI] [PubMed] [Google Scholar]
- 15.Egwuagu CE, Yu CR, Zhang M, Mahdi RM, Kim SJ, Gery I. Suppressors of cytokine signaling proteins are differentially expressed in Th1 and Th2 cells: implications for Th cell lineage commitment and maintenance. J Immunol. 2002;168:3181–7. doi: 10.4049/jimmunol.168.7.3181. [DOI] [PubMed] [Google Scholar]
- 16.Yu CR, Mahdi RM, Ebong S, Vistica BP, Gery I, Egwuagu CE. Suppressor of cytokine signaling 3 regulates proliferation and activation of T-helper cells. J Biol Chem. 2003;278:29752–9. doi: 10.1074/jbc.M300489200. [DOI] [PubMed] [Google Scholar]
- 17.Chong MM, Cornish AL, Darwiche R, et al. Suppressor of cytokine signaling-1 is a critical regulator of interleukin-7-dependent CD8+ T cell differentiation. Immunity. 2003;18:475–87. doi: 10.1016/s1074-7613(03)00078-5. [DOI] [PubMed] [Google Scholar]
- 18.Cornish AL, Davey GM, Metcalf D, et al. Suppressor of cytokine signaling-1 has IFN-gamma-independent actions in T cell homeostasis. J Immunol. 2003;170:878–86. doi: 10.4049/jimmunol.170.2.878. [DOI] [PubMed] [Google Scholar]
- 19.Fletcher J, Starr R. The role of suppressors of cytokine signaling in thymopoiesis and T cell activation. Int J Biochem Cell Biol. 2005;37:1774–86. doi: 10.1016/j.biocel.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Fujimoto M, Tsutsui H, Yumikura-Futatsugi S, et al. A regulatory role for suppressor of cytokine signaling-1 in Th polarization in vivo. Int Immunol. 2002;14:1343–50. doi: 10.1093/intimm/dxf094. [DOI] [PubMed] [Google Scholar]
- 21.Seki Y, Inoue H, Nagata N, et al. SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med. 2003;9:1047–54. doi: 10.1038/nm896. [DOI] [PubMed] [Google Scholar]
- 22.Cohney SJ, Sanden D, Cacalano NA, et al. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol. 1999;19:4980–88. doi: 10.1128/mcb.19.7.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto A, Seki Y, Watanabe R, et al. A role of suppressor of cytokine signaling 3 (SOCS3/CIS3/SSI3) in CD28-mediated interleukin 2 production. J Exp Med. 2003;197:425–36. doi: 10.1084/jem.20020939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brender C, Tannahill GM, Jenkins BJ, et al. Suppressor of cytokine signaling 3 regulates CD8 T-cell proliferation by inhibition of interleukins 6 and 27. Blood. 2007;110:2528–36. doi: 10.1182/blood-2006-08-041541. [DOI] [PubMed] [Google Scholar]
- 25.Endo TA, Masuhara M, Yokouchi M, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–4. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 26.Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–21. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 27.Hilton DJ. Negative regulators of cytokine signal transduction. Cell Mol Life Sci. 1999;55:1568–77. doi: 10.1007/s000180050396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Egwuagu CE, Sztein J, Mahdi RM, et al. IFN-gamma increases the severity and accelerates the onset of experimental autoimmune uveitis in transgenic rats. J Immunol. 1999;162:510–17. [PubMed] [Google Scholar]
- 29.Kim SJ, Zhang M, Vistica BP, et al. Induction of ocular inflammation by T-helper lymphocytes Type 2. Invest Ophthalmol Vis Sci. 2002;43:758–65. [PubMed] [Google Scholar]
- 30.Yoshimoto TM, Furuhata M, Kamiya S, et al. Positive modulation of IL-12 signaling by sphingosine kinase 2 associating with the IL-12 receptor beta 1 cytoplasmic region. J Immunol. 2003;171:1352–9. doi: 10.4049/jimmunol.171.3.1352. [DOI] [PubMed] [Google Scholar]
- 31.Ji YH, Weiss L, Zeira M, et al. Allogeneic cell-mediated immunotherapy of leukemia with immune donor lymphocytes to upregulate antitumor effects and downregulate antihost responses. Bone Marrow Transplant. 2003;32:495–504. doi: 10.1038/sj.bmt.1704150. [DOI] [PubMed] [Google Scholar]
- 32.Morecki S, Lindhofer H, Yacovlev E, Gelfand Y, Slavin S. Use of trifunctional bispecific antibodies to prevent graft versus host disease induced by allogeneic lymphocytes. Blood. 2006;107:1564–9. doi: 10.1182/blood-2005-07-2738. [DOI] [PubMed] [Google Scholar]
- 33.Cooke KR, Kobzik L, Martin TR, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–9. [PubMed] [Google Scholar]
- 34.Hill GR, Cooke KR, Teshima T, et al. Interleukin-11 promotes T cell polarization and prevents acute graft-versus-host disease after allogeneic bone marrow transplantation. J Clin Invest. 1998;102:115–23. doi: 10.1172/JCI3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Couriel D, Caldera H, Champlin R, Komanduri K. Acute graft-versus-host disease: pathophysiology, clinical manifestations, and management. Cancer. 2004;101:1936–46. doi: 10.1002/cncr.20613. [DOI] [PubMed] [Google Scholar]
- 36.Morris ES, Hill GR. Advances in the understanding of acute graft-versus-host disease. Br J Haematol. 2007;137:3–19. doi: 10.1111/j.1365-2141.2007.06510.x. [DOI] [PubMed] [Google Scholar]
- 37.Smith KA. Interleukin-2: inception, impact, and implications. Science. 1988;240:1169. doi: 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- 38.Dutcher J. Current status of interleukin-2 therapy for metastatic renal cell carcinoma and metastatic melanoma. Oncology (Huntingt) 2002;16:4. [PubMed] [Google Scholar]
- 39.Pahwa S, Morales M. Interleukin-2 therapy in HIV infection. AIDS Patient Care STDS. 1998;12:187. doi: 10.1089/apc.1998.12.187. [DOI] [PubMed] [Google Scholar]
- 40.Morris JC, Waldmann TA. Advances in interleukin 2 receptor targeted treatment. Ann Rheum Dis. 2000;59:i109. doi: 10.1136/ard.59.suppl_1.i109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol. 2004;172:3983–8. doi: 10.4049/jimmunol.172.7.3983. [DOI] [PubMed] [Google Scholar]
- 42.Sykes M, Harty MW, Szot GL, Pearson DA. Interleukin-2 inhibits graft-versus-host disease-promoting activity of CD4+ cells while preserving CD4+ and CD8+-mediated graft-versus-leukemia effects. Blood. 1994;83:2560–9. [PubMed] [Google Scholar]