

Abstract
The luminescence properties of lanthanoid ions can be dramatically enhanced by coupling them to antenna ligands that absorb light in the UV/visible and then efficiently transfer the energy to the lanthanoid center. The synthesis and the complexation of Ln(III) cations (Ln=Eu; Gd) for a ligand based on four 1-hydroxy-2-pyridinone (1,2-HOPO) chelators appended to a ligand backbone derived by linking two L-lysine units (3LI-bis-LYS) is described. This octadentate Eu(III) complex ([Eu(3LI-bis-LYS-1,2-HOPO)]−) has been evaluated in terms of its thermodynamic stability, UV/visible absorption and luminescence properties. For this complex the conditional stability constant (pM) is 19.9, which is an order of magnitude higher than diethylenetriaminepentacetic acid (DTPA) at pH= 7.4. This Eu(III) complex also shows an almost two-fold increase in its luminescence quantum yield in aqueous solution (pH= 7.4) when compared to other octadentate ligands. Hence, despite a slight decrease of the molar absorption coefficient, a much higher brightness is obtained for [Eu(3LI-bis-LYS-1,2-HOPO)]−. This overall improvement was achieved by saturating the coordination sphere of the Eu(III) cation, yielding an increased metal centered efficiency by excluding solvent water molecules from the metal’s inner sphere.
Introduction
Chelated lanthanoid ions represent a unique class of luminescent compounds that feature both narrow-band emission in the visible and near infrared (NIR) regions as well as long luminescent excited state lifetimes, due to the Laporte forbidden character of the f-f transitions involved.[2, 3] Their relative resistance toward oxygen quenching allows work in aerated solution, making these chelate complexes ideal candidates for biological applications.[4-7] In particular, terbium and europium are of primary interest since their higher luminescence quantum yields, together with long luminescence lifetimes, enables time gating of their emission,[6-11] dramatically lowering background due to short lived autofluorescence from the sample. To optimize their luminescence properties, the Ln(III) cation also requires saturation of its coordination sphere, using a light absorbing ligand which can also act as antenna.[12] The energy difference between the lanthanoid emitting level and the molecular triplet excited state of the ligand is of paramount importance and sensitization efficiency can strongly depend on the energy gap between these excited states. [13, 14]
For optimal lanthanoid luminescence, octadentate ligands are of particular interest because they generally fill the coordination sphere of the metal, protecting it from its environment (deleterious non-radiative quenching of the Ln(III) ion from O-H and N-H vibrations of the medium) and allowing better luminescence properties (higher luminescence quantum yields and longer luminescence lifetimes).
We report herein the synthesis of a new octadentate ligand comprised of two L-lysine groups covalently linked via their carboxylic acid functional groups with a propyl diamine chain to form the diamide backbone, 3LI-bis-LYS. The resulting four primary amine functional groups of the 3LI-bis-LYS backbone were used to attach four 1-hydroxy-2-pyridinone (1,2-HOPO) groups (see Chart 1) via amidation and this ligand was complexed to Eu(III) to give [Eu(3LI-bis-LYS-1,2-HOPO)]−, which has been evaluated in terms of its thermodynamic stability, and photophysical properties.
Chart 1.

Chemical structure of the discussed ligands
Results and discussion
Synthesis and Design of Ligands
The ligands (Chart 1) feature the 1-hydroxy-2-pyridinone (1,2-HOPO) group, which acts as a bidentate ligand to complex Ln(III) ions efficiently, and the resulting octadentate ligand topology has been shown to form stable ML complexes (where 3LI-bis-LYS-1,2-HOPO ligand is a tetra-bidentate ligand composed of four such 1,2-HOPO moieties). The 1,2-HOPO units can also be linked to form a tetradentate ligand using aliphatic[15, 16] or aromatic spacers[17] via the amide functional groups. For the octadentate topology, we recently reported on the N,N,N’,N’-tetrakis-(2-aminoethyl)-ethane-1,2-diamine (H(2,2)-) backbone bearing four 1,2-HOPO units (Chart 1).[18] In the present case, the backbone of the 3LI-bis-LYS-1,2-HOPO ligand differs in terms of the linking point, by the identity of the linking atom and by the length of the linking central group (i.e. propyl instead of ethyl). Hence, for H(2,2)-1,2-HOPO, the central ethyl chain which connects the two tetradentate moieties is substituted via a nitrogen atom in position 3, while 3LI-bis-LYS-1,2-HOPO is substituted via a carbon atom in position 1. It is not anticipated that this difference will increase the degrees of freedom of the system, but it is expected to change the arrangement of the ligand around the Ln(III) cation. The substitution of the nitrogen by a carbon was done in order to avoid having a protonated nitrogen in the backbone bearing 1,2-HOPO units, which causes lower luminescence quantum yields.[18]
The protected backbone 2 was prepared by coupling the activated L-lysine, protected at its two N-termini, to propylene diamine by activating the carboxylic acid with isobutyl chloroformate. This intermediate was deprotected in acidic medium to give 3.
The benzyl protected 1,2-HOPO chromophore was prepared as reported elsewhere[19] and its acid chloride version (4) was prepared in situ using thionyl chloride.[20] This acid chloride (4, 4.4 equivs.) was combined with one equivalent of 3 to furnish the benzyl protected 1,2-HOPO octadentate ligand (5). Subsequent reflux in 1:1 (v/v) conc. HCl and glacial acetic acid gave the desired ligand (3LI-bis-LYS-1,2-HOPO) in good yields. The Ln(III) complexes (Ln = Eu, Gd) were prepared by refluxing one equivalent of ligand with one equivalent of the appropriate LnCl3·6H2O using pyridine as a base to ensure deprotonation of the N-hydroxyl groups. The complex was then precipitated and washed with diethyl ether to yield an analytically pure hydrated complex. Full characterization of the ligand and complexes and synthetic details are reported in the experimental section.
Complex Stability
The stability of the Eu(III) complex in aqueous medium was determined by competition batch titration (Figure 1) versus DTPA as a known competitor (pEu= 19.04 at pH= 7.4). By analogy to pH, pEu is defined as the negative log of the concentration of free metal in solution (pEu= − log [Eu3+]free) at a specified set of standard conditions (chosen usually as [Eu]T= 1 μM, [L]T= 10 μM, pH= 7.4, 25 °C, and 0.1 M KCl). The pEu values therefore offer a convenient and meaningful way to compare relative chelate stabilities between various ligands, regardless of differing ligand protonation behavior. In the cases of [Eu(5LI-1,2-HOPO)2]− and [Eu(H(2,2)-1,2-HOPO)]−, the pEu’s were determined to be 18.35[16] and 21.2,[18] respectively, an increase of almost three orders of magnitude in stability due to the enhanced chelate effect arising from an octadentate versus bis-tetradentate complex.
Figure 1.

a/ Luminescence spectrum showing the typical decrease of luminescence intensity upon addition of DTPA; b/ DTPA competition batch titration of [Eu(3LI-bis-LYS-1,2-HOPO)]−  and [Eu(H(2,2)-1,2-HOPO)]−
and [Eu(H(2,2)-1,2-HOPO)]−  versus DTPA. The x intercept indicates the difference in pEu between EuDTPA and the two complexes.
versus DTPA. The x intercept indicates the difference in pEu between EuDTPA and the two complexes.
For [Eu(3LI-bis-LYS-1,2-HOPO)]−, the pEu was evaluated to be 19.9, which represents a value almost one order of magnitude higher than the DTPA competitor. A loss in thermodynamic stability when compared to [Eu(H(2,2)-1,2-HOPO)]− (one order of magnitude higher than [Eu(3LI-bis-LYS-1,2-HOPO)]−) can be readily observed. This difference in stability can be attributed to a more ideal geometry adopted by the ligand around the Eu(III) center using the flexible H(2,2) backbone, and suggest that the 3LI-bis-LYS-1,2-HOPO scaffold presents some constraint, yielding a loss of stability.
UV-visible absorption spectroscopy
The UV/visible absorption spectra of the Eu(III) complexes in 0.1 M aqueous TRIS buffer at pH= 7.4 are shown in Figure 2 and the data are summarized in Table 2. The absorption maxima are at ca. 331 nm, 333 nm and 341 nm for the [Eu(5LI-1,2-HOPO)2]−, [Eu(3LI-bis-LYS-1,2-HOPO)]− and [Eu(H(2,2)-1,2-HOPO)]− complexes respectively. The lowest energy transition for these spectra can be assigned to a π–π* transition with some n–π* character as described elsewhere.[16] The maximum absorption of [Eu(3LI-bis-LYS-1,2-HOPO)]− is in accordance with our previously reported results with other Eu(III) bis-tetradentate complexes, as illustrated with [Eu(5LI-1,2-HOPO)2]− (λabs= 331 nm) while the slight bathochromic shift in going from [Eu(3LI-bis-LYS-1,2-HOPO)]− to [Eu(H(2,2)-1,2-HOPO)]− reveals a stabilization of the first excited state using the H(2,2) backbone.
Figure 2.

UV/visible spectra of [Eu(3LI-bis-LYS-1,2-HOPO)]−  and [Eu(H(2,2)-1,2-HOPO)]−
and [Eu(H(2,2)-1,2-HOPO)]−  in 0.1 M aqueous TRIS buffer solution at pH= 7.4.
in 0.1 M aqueous TRIS buffer solution at pH= 7.4.
Table 2.
UV/visible absorption data of the studied Eu(III) complexes in 0.1 M aqueous TRIS buffer (pH= 7.4) and triplet excited state energies determined for the corresponding Gd(III) complexes at 77K.
| 0.1 M aqueous TRIS buffer, pH= 7.4 | 77K a | ||
|---|---|---|---|
| λmaxabs / nm | ε/ M−1.cm−1 | T0-0 / nm (cm−1) | |
| [Eu(5LI-1,2-HOPO)2] − | 331 | 18,800 | 21,260 |
| [Eu(H(2,2)-1,2-HOPO)] − | 341 | 18,200 | 21,980 |
| [Eu(3LI-bis-LYS-1,2-HOPO)] − | 333b | 15,760b | 22,120 |
determined in a solid matrix at 77K (1:4 (v/v) MeOH:EtOH) using the Gd(III) complexes
containing 1% (v/v) DMSO for solubility.
The molar absorption coefficients are within experimental error, with typical values of ca. 18,000 M−1cm−1 for the [Eu(5LI-1,2-HOPO)2]− and [Eu(H(2,2)-1,2-HOPO)]− complexes, whereas a slightly decreased value is apparent for [Eu(3LI-bis-LYS-1,2-HOPO)]− (15,760 M−1cm−1). This 16% decrease of the molar absorption coefficient for [Eu(3LI-bis-LYS-1,2-HOPO)]− compared to [Eu(H(2,2)-1,2-HOPO)]− affects the brightness (here defined as the product of the molar absorption coefficient with the luminescence quantum yield) giving an advantage to [Eu(H(2,2)-1,2-HOPO)]−.
Luminescence of Gd(III) complexes
To estimate the energies of the ligand-based triplet excited state, the Gd(III) complexes were prepared and studied. Gadolinium is a 4f7 lanthanoid ion with the same oxidation state, a similar 4f electronic configuration, and similar size as the europium cation (4f6), but with an inaccessible metal centred electronic excited state (the 6P7/2 is at 32,224 cm−1). For these complexes, at 77K, in a frozen solution, an emission band with a maximum at ca. 500 nm can be seen. This emission is red-shifted compared to the S0 → S1 transitions, observed at ca. 330-340 nm by UV/visible absorption and can be assigned to emission from the triplet excited state, which lies at lower energy than the corresponding singlet excited state. From these spectra, it appears the triplet excited states of the complexes are almost coincident when considering the approximate onset of the emission, varying only slightly from 452 nm for [Gd(3LI-bis-LYS-1,2-HOPO)]− and [Gd(5LI-1,2-HOPO)2]− to 455 nm for [Gd(H(2,2)-1,2-HOPO)]−, respectively. The almost identical triplet excited state energy should therefore give the same energy transfer efficiency in the sensitization process.
The slight decrease of 145 cm−1 in the energy for the triplet excited state follows the trends observed for the first singlet excited state transitions. This should give an intersystem crossing rate slightly in favour of [Eu(3LI-bis-LYS-1,2-HOPO)]−, since the gap between the singlet and the triplet excited states is closer to the 5,000 cm−1 (ΔE(1S*-3T*)= 7,350 cm−1 and 7,900 cm−1 for [Gd(3LI-bis-LYS-1,2-HOPO)]− and [Eu(H(2,2)-1,2-HOPO)]−, respectively) that is the ideal empirical gap predicted by Verhoeven et al..[21]
Luminescence of Eu(III) complexes
The steady state emission spectra, the luminescence quantum yields and luminescence lifetimes of the Eu(III) complexes were measured in 0.1 M aqueous TRIS buffer solution at pH= 7.4. The luminescence lifetimes were also measured in the corresponding deuterated solvents, in order to estimate the number of bound solvent molecules in the inner sphere (i.e. q in water[22]) using the empirical Horrock’s equations. The relevant radiative and non-radiative parameters were also determined following the work of Verhoeven[23] and Beeby[24], and a complete summary of these data are reported in Tables 3 and 4.
Table 3.
Photophysical data of the investigated Eu(III) complexes.
| 0.1 M aqueous TRIS buffer, pH= 7.4 |
77Ka | Brightnessb | ||||
|---|---|---|---|---|---|---|
| φ tot | τH2O / μs | τD2O / μs | q |
τ / μs |
M−1.cm−1 | |
| [Eu(5LI-1,2-HOPO)2] − | 0.207 | 737 | 996 | 0.0 | 778 | 3,900 |
| [Eu(H(2,2)-1,2-HOPO)] − | 0.036 | 480 | 1222 | 1.1 | 914 | 655 |
|
[Eu(3LI-bis-LYS-1,2-
HOPO)] − |
0.070 | 712 | 917 | 0.1 | 754 | 1,100 |
measured in a solid matrix at 77K (1:4 (v/v) MeOH:EtOH).
defined here as the product of the molar extinction coefficient and the overall luminescence quantum yield
Table 4.
Photophysical data of the investigated complexes in 0.1 M aqueous TRIS buffer at pH= 7.4.
| φTot | τ / μs | τrad / μs | kr / s−1 | knr / s−1 | ηEu | ηsens | |
|---|---|---|---|---|---|---|---|
| [Eu(5LI-1,2-HOPO)2]− a | 0.207 | 737 | 1728 | 579 | 778 | 0.426 | 0.485 |
| [Eu(H(2,2)-1,2-HOPO)] − | 0.036 | 480 | 3000 | 333 | 1750 | 0.160 | 0.225 |
| [Eu(3LI-bis-LYS-1,2-HOPO)] − | 0.070 | 712 | 2254 | 444 | 951 | 0.316 | 0.222 |
see reference [18].
The emission spectrum of [Eu(3LI-bis-LYS-1,2-HOPO)]− is typical of our previously reported Eu(III) 1,2-HOPO derivatives, with a very intense 7FJ ← 5D0 (J = 2) transition at ca. 612 nm and a weaker J = 4 transition at ca. 680-700 nm. For [Eu(H(2,2)-1,2-HOPO)]−, the J = 4 peak is slightly more intense (Figure 4). These changes have been shown to originate from differing metal centered symmetries, and in the present case, it is due to the presence of one water molecule in the inner sphere for [Eu(H(2,2)-1,2-HOPO)]−, yielding a nine coordinate Eu(III) complex, versus an eight coordinate structure for complexes such as [Eu(5LI-1,2-HOPO)2]−, which lack a water molecule in the inner sphere. Notably, the crystal field splitting of the 7FJ ← 5D0 (J = 1) transition at ca. 580 nm can be used to infer the site symmetry of the Eu(III) cation. Although the transition in this case is quite broad, which precludes a definitive identification of the exact point group, the observation of only two peaks suggests that from the three most common coordination polyhedra, the best match to the observed luminescence spectra is obtained for the trigonal dodecahedral (D2d) geometry as noted elsewhere for similar derivatives.16
Figure 4.
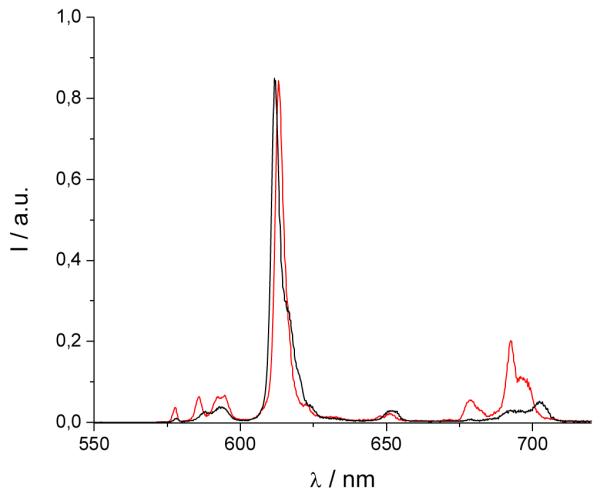
Normalized luminescence spectra of [Eu(3LI-bis-LYS-1,2-HOPO)]− 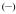 and [Eu(H(2,2)-1,2-HOPO)]−
and [Eu(H(2,2)-1,2-HOPO)]−  in 0.1 M aqueous TRIS buffer solution at pH= 7.4 (λex= 340 nm)
in 0.1 M aqueous TRIS buffer solution at pH= 7.4 (λex= 340 nm)
As can also be seen from Table 3, the luminescence quantum yields are different for each complex. As reported elsewhere, [Eu(5LI-1,2-HOPO)2]− presents an optimum luminescence quantum yield for the bis-tetradentate complexes of around 20.7%[16] while [Eu(H(2,2)-1,2-HOPO)]− has a limited value of 3.6% due to the quenching by a water molecule in the inner sphere of the Eu(III).[18] The measured luminescence quantum yield of [Eu(3LI-bis-LYS-1,2-HOPO)]− reveals a value of 7.0% that is almost a two fold increase when compared to [Eu(H(2,2)-1,2-HOPO)]−.
This improved value of ΦEu, the overall europium quantum yield, will also affect the brightness of the complexes (vide supra) (Table 3) and this time the improvement favors the [Eu(3LI-bis-LYS-1,2-HOPO)]− complex, yielding a maximum brightness of 1,100 M−1cm−1 at 333 nm. Above ca. 358 nm, however, we note that the red-shifted absorption spectrum of the [Eu(H(2,2)-1,2-HOPO)]− complex yields a brightness which reverts to slightly favor the latter.
Indeed, for both eight coordinate complexes, the decrease in luminescence quantum yield (and also of the molar absorption coefficient for [Eu(3LI-bis-LYS-1,2-HOPO)]−) when compared to the bis-tetradentate [Eu(5LI-1,2-HOPO)2]− induces a three-fold decrease in brightness for the former structures, although the gain in stability still remains more important for aqueous measurements, and more specifically for biological measurements.
The luminescence lifetimes were determined in 0.1 M aqueous TRIS buffer and reveal values of 712 μs for [Eu(3LI-bis-LYS-1,2-HOPO)]− that are of the same order as those for [Eu(5LI-1,2-HOPO)2]− (737 μs), and much longer than that of [Eu(H(2,2)-1,2-HOPO)]− which has a decay time of ca. 480 μs. The difference between [Eu(5LI-1,2-HOPO)2]− and [Eu(H(2,2)-1,2-HOPO)]− has been shown to result from the presence of a water molecule in the inner sphere of the latter, and the similarity in lifetimes suggests that [Eu(3LI-bis-LYS-1,2-HOPO)]− does not have an aqua ligand coordinated to the Eu(III) cation (vide infra).
In order to determine whether [Eu(3LI-bis-LYS-1,2-HOPO)]− also possesses an aqua ligand, the luminescence lifetime was also measured in deuterated water yielding a value of 917 μs that is comparable to that of [Eu(5LI-1,2-HOPO)2]− (996 μs). The q number can therefore be obtained (Table 3) using the improved Horrocks equation,[22] revealing a value of 0.1, and hence confirming that the lack of aqueous solvent access toward the metal. This difference as compared with [Eu(H(2,2)-1,2-HOPO)]− mainly explains the large increase of luminescence quantum yield, by minimising solvent induced non-radiative deactivation pathways.
Luminescence lifetimes were also determined at 77K, in a solid matrix (Table 3), to determine whether back energy transfer between the excited state donor triplet and the excited state acceptor manifold of the lanthanoid is present, or alternately whether quenching via a low lying LMCT state can occur. As can be seen from Table 3, no such quenching occurs since there is only a small difference between the luminescence lifetimes at room temperature in solution and in a solid matrix at 77K. Hence, [Eu(3LI-bis-LYS-1,2-HOPO)]− possesses a triplet state ideally located (the 3T* to 5D1 gap is 2,090 cm−1, vide supra) for efficient energy transfer to the europium 5D1 level.[13, 14]
Following the work of Verhoeven et al.[23] and Beeby et al.[24], the efficiency of the sensitization can be estimated using a method that defines the overall europium quantum yield of luminescence (φEu) as the product of the efficiency of the intersystem crossing (ηISC), the efficiency of the energy transfer (ηET) and the efficiency of metal centred luminescence (ηEu).
In this equation, ηEu can be calculated knowing the the ratio of the total integrated emission intensity to the intensity of the 5D0→7F1 transition (yielding kr (and hence τrad)) and knr can then be deduced knowing kr and τobs (the luminescence lifetime of the considered complex). As can be seen from Table 4, the radiative and non-radiative decay rates are rather close for [Eu(3LI-bis-LYS-1,2-HOPO)]− and [Eu(5LI-1,2-HOPO)2]− and are comparable to other 1,2-HOPO based Eu(III) complexes that lack a water molecule in their inner sphere.15,16 The observed differences between these two complexes (higher kr and lower knr for [Eu(5LI-1,2-HOPO)2]−) reveal the better metal centered efficiency for the bis-tetradentate systems (ηEu are 42.6% and 31.6% respectively for [Eu(5LI-1,2-HOPO)2]− and [Eu(3LI-bis-LYS-1,2-HOPO)]−).
For [Eu(H(2,2)-1,2-HOPO)]−, the low value for ηEu obtained (16.0%) can be attributed to the water molecule in the inner sphere, which quenches the emission. More importantly, it should be highlighted that the sensitization efficiencies are effectively the same for both octadentate complexes (ηsens of 22.2% and 22.5% for [Eu(3LI-bis-LYS-1,2-HOPO)]− and [Eu(H(2,2)-1,2-HOPO)]−, respectively) illustrating that while one of the main limitations for [Eu(H(2,2)-1,2-HOPO)]− is attributed to the metal centered efficiency, more generally the octadentate ligand topologies containing 1,2-HOPO chelates result in sensitization efficiencies that are almost two times lower than those of the bis-tetradentate ligands. Unfortunately, the reason for these differences are not yet clear, and remain a topic under current investigation, since further optimization of this crucial parameter will yield highly stable octadentate complexes with superior Eu(III) based luminescence performance.
Conclusion
We have described the synthesis of an octadentate ligand comprising two L-lysine groups attached via amidation of their carboyxlic acid functional groups with a propyl diamine chain. The resulting tetramine was functionalised by 1,2-HOPO chelates, again via amide bond formation, resulting in an a novel octadentate ligand. Upon addition of Eu(III), this ligand was determined to form a complex more thermodynamically stable than DTPA by ca. one order of magnitude, albeit lower than our previously reported [Eu(H(2,2)-1,2-HOPO)]−. The photophysical study of both Eu(III) and Gd(III) complexes has revealed the triplet excited state is ideally located to sensitize the Eu(III) cation, and the luminescence quantum yield presents a two fold increase compared to [Eu(H(2,2)-1,2-HOPO)]−. This improvement has been attributed to a more complete saturation of the metal’s coordination sphere, and also results in a longer luminescence lifetime for [Eu(3LI-bis-LYS-1,2-HOPO)]− similar to other bis-tetradentate ligand topologies comprising the 1,2-HOPO moiety. Despite a slight decrease in the molar absorption coefficient, this increase of luminescence quantum yield also yields a much brighter complex. Finally, since the sensitization efficiency is the same for both octadentate complexes, the main limitation of [Eu(H(2,2)-1,2-HOPO)]− when compared to [Eu(3LI-bis-LYS-1,2-HOPO)]− is attributed to the metal centered efficiency while, when comparing the octadentate chelators to the bis-tetradentate, a current limitation we encounter is a decrease in the sensitization efficiency, which is almost two times less efficient for octadentate versus bis-tetradentate ligands. We attribute this in part to changes in the coordination geometry that affect the efficiency of ligand to metal energy transfer.
Experimental
General
Thin-layer chromatography (TLC) was performed using precoated Kieselgel 60 F254 plates. Flash chromatography was performed using EM Science Silica Gel 60 (230- 400 mesh). NMR spectra were obtained using either Bruker AM-300 or DRX-500 spectrometers operating at 300 (75) MHz and 500 (125) MHz for 1H (or 13C) respectively. 1H (or 13C) chemical shifts are reported in ppm relative to the solvent resonances, taken as δ 7.26 (δ 77.0) and δ 2.49 (δ 39.5) respectively for CDCl3 and (CD3)2SO while coupling constants (J) are reported in Hz. The following standard abbreviations are used for characterization of 1H NMR signals: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m = multiplet, dd = doublet of doublets. Fast-atom bombardment mass spectra (FABMS) were performed using 3-nitrobenzyl alcohol (NBA) or thioglycerol/glycerol (TG/G) as the matrix. Elemental analyses were performed by the Microanalytical Laboratory, University of California, Berkeley, CA.
Synthesis
3LI-bis(Boc-Nε-Z-Lysine) (2)
Under N2 a solution of Boc-Nε-Z- Lysine (1.14 g, 3 mmol) in dry THF (25 mL) was cooled to −15 °C and neutralized with N-methylmorpholine (0.33 mL, 3 mmol). Isobutyl chloroformate (0.40 mL, 3 mmol) was added under stirring. After two minutes, 1,3-diaminopropane (0.13 mL, 0.15 mmol) was added. The reaction mixture warmed to room temperature and evaporated to dryness; the residue was dissolved in DCM and extracted with 5% citric acid solution and brine successively. The organic phase was separated and loaded on a flash silica column. Elution with 2-7% methanol in DCM allows the separation of 3LI-bis(Boc-Nε-Z- Lysine) (2) (1.0 g, 84% based on the free amine) as a thick pale yellow oil which was solidified upon standing overnight.
1H-NMR (300MHz, CDCl3): δ 1.36 (s,br, 4H), 1.41 (s, 18H), 1.48 (m, 4H), 1.62 (s,br, 4H), 3.16 (s,br, 6H), 3.40 (s,br, 2H), 4.04 (m, 2H), 5.07 (s,br, 6H), 5.30 (s,br, 2H), 7.09 (s,br, 2H), 7.29-7.40 (m, 10H); 13C-NMR (75MHz, DMSO-d6): δ 22.3, 27.9, 29.0, 31.7, 36.4, 40.1, 54.2, 65.9, 79.2, 127.2, 127.5,128.0, 136.3, 155.6, 156.3, 172.7; HR-MS(FAB+): 799.4575(MH+).
3LI-bis-LYS-1,2-HOPOBn (5)
3LI-bis(Boc-Nε-Z- Lysine) (0.80 g, 1 mmol) was dissolved in TFA/DCM (1:1) solution (10 mL) and stirred at room temperature for 3 hrs. The volatiles were removed under vacuum, the residual was mixed with Pd/C (10%, 100 mg) in methanol (20 mL) and hydrogenated at 400 Psi H2 overnight in a Parr bomb. The catalyst was filtered off with a fine glass frit and the filtrate was evaporated to dryness yielding 3LI-bis-LYS amine (3) (2,6-Diamino-hexanoic acid [3-(2,6-diamino-hexanoylamino)-propyl]-amide). 1H-NMR (300MHz, D2O): δ 1.31 (qint, 3J = 7.5 Hz, 4H), 1.57 (m, 6H), 1.75 (m, 4H), 2.86 (t, 3J = 7.5 Hz, 4H), 3.14 (t, 3J = 7.5 Hz, 4H), 3.81 (t, 3J = 6.6 Hz, 2H); 13C-NMR (75MHz, DMSO-d6): δ 21.5, 2 6.5, 27.9, 30.6, 37.0, 39.2, 53.3, 169.7.
Compound 3 was dissolved in DCM (20 mL) and a biphasic solution of potassium carbonate (4 g of K2CO3 in 10 mL water) was added. This biphasic solution was cooled in an ice-water bath. A solution of 1,2-HOPOBn chloride (4) (1.25 g, 5 mmol) in DCM (40 mL) was added drop wise to the above stirred mixture over 1 h and the mixture was allowed to warm up overnight. The organic phase was separated and the evaporated residue was loaded onto a flash silica column. Elution with 2-7% methanol in DCM allows the separation of 3LI-bis-LYS-1,2-HOPOBn (5) (0.89 g, 72%) (based on 3LI-bis(Boc-Nε-Z- Lysine)) giving a thick pale yellow oil that solidified overnight. 1H-NMR (300MHz, DMSO-d6): δ 6.56 (d, 2H, 3J = 7.2 Hz, HOPO-H)), 6.67 (dd, 2H, 3J = 9.0; 1.5 Hz, HOPO-H), 7.44 (d+d, 2H, 3J = 9.0 Hz, HOPO-H), 7.90 (s,br, 3H, Py-H), 11.39 (s, 2H, amideH); 13C-NMR (75 MHz, CDCl3): δ 105.2, 110.7, 119.6, 136.9, 140.9, 141.2, 149.8, 157.3, 159.3.
3LI-bis-LYS-1,2-HOPO
3LI-bis-LYS-1,2-HOPOBn (5) (0.5 g, 0.4 mmol) was dissolved in concentrated HCl (12 M)/glacial acetic acid (1:1, 20 mL), and stirred at room temperature for 2 days. Removal of the solvent gives a beige foam as the deprotected product, Yield 0.27 g, 75%. 1H-NMR (300 MHz, DMSO-d6): δ 1.20-1.80 (m, 14H, CH2), 3.08 (m. 4H, CH2), 3.16 (m, 4H, CH2), 4.31 (q, 2H, 3J = 4.5 Hz, CH2), 6.25(dd, 2H, 3J = 6.9 Hz, 4J = 1.5 Hz, HOPO-H)), 6.41 (dd, 2H, 3J = 6.9 Hz, 4J = 1.5, HOPO-H)), 6.55(dd, 2H, 3J = 6.0 Hz, 4J = 1.5 Hz, HOPO-H)), 6.60 (dd, 2H, 3J = 6.3 Hz, 4J = 1.5 Hz,, HOPO-H)), 7.32-7.42 (m, 4H, HOPO-H)), 7.95 (s,br, 2H, amideH)), 8.74 (s,br, 2H, amideH)), 9.04 (d, 2H, 3J = 5.1 Hz, amide-H); 13C-NMR (75 MHz, CDCl3): δ 22.8, 28.4, 29.2, 31.6, 36.4, 48.7, 53.4, 103.9, 105.0, 119.4, 137.2, 137.5, 141.7, 142.5, 157.6, 160.2, 160.3, 171.0. Anal. Calc’d. (Found) for C39H46N10O14·HCl·2.5H2O. C; 48.77 (48.86), H; 5.87 (5.73), N; 14.59 (14.36).
[Eu(3LI-bis-LYS-1,2-HOPO)]−
In a 25mL round bottom flask, 3LI-bis-LYS-1,2-HOPO (1 Eq) was suspended in 10mL of methanol. Europium(III) chloride hexahydrate (1.02 Eq) in 10mL of methanol and two drops of pyridine were added. The solutions were heated to reflux for 4 hours, then cooled to room temperature. Slow evaporation of the methanol at room temperature overnight afforded the desired complexes, as their pyridinium salts, which were collected by filtration and washed thoroughly with diethyl ether, resulting in the pure hydrated complex as a white solid in ca. 60% yield; Anal. Calc’d. (Found) for C44H48N11O14Eu·5H2O. C; 44.15 (43.87), H; 4.88 (4.58), N; 12.87 (13.28).
Optical spectroscopy
UV-Visible absorption spectra were recorded on a Varian Cary 300 double beam absorption spectrometer. Emission spectra were acquired on a HORIBA Jobin Yvon IBH FluoroLog-3 spectrofluorimeter, equipped with 3 slit double grating excitation and emission monochromators (2.1 nm/mm dispersion, 1200 grooves/mm). Spectra were reference corrected for both the excitation light source variation (lamp and grating) and the emission spectral response (detector and grating). Luminescence lifetimes were determined on the same HORIBA Jobin Yvon IBH FluoroLog-3 spectrofluorimeter, adapted for time-correlated single photon counting (TCSPC) and multichannel scaling (MCS) measurements. A sub-microsecond Xenon flashlamp (Jobin Yvon, 5000XeF) was used as the lightsource, with an input pulse energy (100 nF discharge capacitance) of ca. 50 mJ, yielding an optical pulse duration of less than 300 ns at FWHM. Spectral selection was achieved by passage through the same double grating excitation monochromator. Emission was monitored perpendicular to the excitation pulse, again with spectral selection achieved by passage through the double grating emission monochromator (2.1 nm/mm dispersion, 1200 grooves/mm). A thermoelectrically cooled single photon detection module (HORIBA Jobin Yvon IBH, TBX-04-D) incorporating fast rise time PMT, wide bandwidth preamplifier and picosecond constant fraction discriminator was used as the detector. Signals were acquired using an IBH DataStation Hub photon counting module and data analysis was performed using the commercially available DAS 6 decay analysis software package from HORIBA Jobin Yvon IBH. Goodness of fit was assessed by minimizing the reduced chi squared function, χ2, and a visual inspection of the weighted residuals. Each trace contained at least 10,000 points and the reported lifetime values resulted from at least three independent measurements. Typical sample concentrations for both absorption and fluorescence measurements were ca. 10−5-10−6 M and 1.0 cm cells in quartz suprasil or equivalent were used for all measurements. Quantum yields were determined by the optically dilute method (with optical density <0.1) using the following equation;
where A is the absorbance at the excitation wavelength (λ), I is the intensity of the excitation light at the same wavelength, n is the refractive index and D is the integrated luminescence intensity. The subscripts ‘x’ and ‘r’ refer to the sample and reference respectively. For quantum yield calculations, an excitation wavelength of 340 nm was utilized for both the reference and sample, hence the I(λr)/I(λx) term is removed. Similarly, the refractive indices term, nx2/nr2, was taken to be identical for the aqueous reference and sample solutions. Hence, a plot of integrated emission intensity (i.e. Dr) vs. absorbance at 340 nm (i.e. Ar(λr)) yields a linear plot with a slope which can be equated to the reference quantum yield Φr. Quinine sulfate in 0.5 M (1.0 N) sulfuric acid was used as the reference (Φr = 0.546).[25] By analogy, for the sample, a plot of integrated emission intensity (i.e. Dx) versus absorbance at 340 nm (i.e. Ax(λx)) yields a linear plot and Φx can then be evaluated. The values reported in the manuscript are the average of four independent measurements.
Competition Batch Titrations for pEu Determination
The general procedure used to determine the pEu values of the three ligands was adapted from a previously described study of gadolinium[26, 27] and are similar to those reported for other complexes.[18] Different volumes of a standardized DTPA stock solution were added to solutions of constant ligand, metal, and electrolyte concentrations. In the current work the pH of all solutions was kept constant at 7.4 with 0.1M aqueous TRIS buffer instead of adjusting the pH to 6.0 as was done in past studies,[26] and the solutions were diluted to identical volumes. After stirring the solutions for 24 h to ensure thermodynamic equilibrium was reached, the pH was again checked just before analyzing the samples. The concentrations of each ligand relative to DTPA used in the final data analysis ranged from 1:1 to 1:1,000 (L:DTPA). Concentrations of complexed ligand in each solution were determined from the luminescence spectra, using the emission spectra of the fully complexed ligand as a standard. These concentrations were used for the log/log plots (Figures 1b) to yield the difference in pEu between the competing DTPA and ligand of interest.
Scheme 1.

Synthetic pathway for 3LI-bis-LYS-1,2-HOPO.
Figure 3.
Triplet excited state emission spectra for [Gd(3LI-bis-LYS-1,2-HOPO)]−  and [Gd(H(2,2)-1,2-HOPO)]−
and [Gd(H(2,2)-1,2-HOPO)]−  in solid matrix (77K, 1:4 (v/v) MeOH:EtOH) (λex= 340nm).
in solid matrix (77K, 1:4 (v/v) MeOH:EtOH) (λex= 340nm).
Figure 5.

Brightness of [Eu(3LI-bis-LYS-1,2-HOPO)]−  and [Eu(H(2,2)-1,2-HOPO)]−
and [Eu(H(2,2)-1,2-HOPO)]−  in 0.1 M aqueous TRIS buffer at pH= 7.4.
in 0.1 M aqueous TRIS buffer at pH= 7.4.
Table 1.
pEu’s of [Eu(5LI-1,2-HOPO)2]—, [Eu(H(2,2)-1,2-HOPO)]— and [Eu(3LI-bis-LYS-1,2-HOPO)]—, determined by competitions batch titration using DTPA (pEu= 19.04) in 0.1 M aqueous TRIS buffer, pH= 7.4 and 0.1 M KCl.
Acknowledgement
This work was partially supported by the NIH (Grant HL69832) and supported by the Director, Office of Science, Office of Basic Energy Sciences, and the Division of Chemical Sciences, Geosciences, and Biosciences of the U.S. Department of Energy at LBNL under Contract No. DE-AC02-05CH11231. This technology is licensed to Lumiphore, Inc. in which some of the authors have a financial interest. Prof. Gilles Muller is thanked for access to the low temperature time-resolved luminescence facilities.
References and notes
- [1].This paper is dedicated to a giant of Australian chemistry, Alan Sargeson. The senior author had the great good fortune to spend his first sabbatical leave in Australia, much of it at the ANU. It left a lasting influence regarding coordination chemistry and its interface to biology and medicine.
- [2].Bünzli J-CG, Piguet C. Chem. Soc. Rev. 2005;34:1048. doi: 10.1039/b406082m. [DOI] [PubMed] [Google Scholar]
- [3].Parker D. Chem. Soc. Rev. 2004;33:156. doi: 10.1039/b311001j. [DOI] [PubMed] [Google Scholar]
- [4].Terai T, Kikuchi K, Iwasawa S, Kawabe T, Hirata Y, Urano Y, Nagano T. J. Am. Chem. Soc. 2006;128:6938. doi: 10.1021/ja060729t. [DOI] [PubMed] [Google Scholar]
- [5].O’Hara PB. Photochem. Photobiol. 2008;46:1067. doi: 10.1111/j.1751-1097.1987.tb04894.x. [DOI] [PubMed] [Google Scholar]
- [6].Picot A, D’Aléo A, Baldeck PL, Grishine A, Duperray A, Andraud C, Maury O. J. Am. Chem. Soc. 2008;130:1532. doi: 10.1021/ja076837c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yu J, Parker D, Pal R, Poole RA, Cann MJ. J. Am. Chem. Soc. 2006;128:2294. doi: 10.1021/ja056303g. [DOI] [PubMed] [Google Scholar]
- [8].Mariott G, Clegg RM, Arndt-Jovin DJ, Jovin TM. Biophys. J. 1991;60:1374. doi: 10.1016/S0006-3495(91)82175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beeby A, Botchway SW, Clarkson IM, Faulkner S, Parker AW, Parker D, Williams JAG. J. Photochem. Photobiol. B. 2000;57:83. doi: 10.1016/s1011-1344(00)00070-1. [DOI] [PubMed] [Google Scholar]
- [10].Faulkner S, Pope SJA, Burton-Pye BP. Appl. Spec. Rev. 2004;39:1. [Google Scholar]
- [11].Charbonnière LJ, Hildebrandt N, Ziessel R, Löhmannsröben HG. J. Am. Chem. Soc. 2006;128:12800. doi: 10.1021/ja062693a. [DOI] [PubMed] [Google Scholar]
- [12].Moore EG, Samuel APS, Raymond KN. Acc. Chem. Res. 2009;42:542. doi: 10.1021/ar800211j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Latva M, Takalob H, Mukkala V-M, Matachescuc C, Rodriguez-Ubisd JC, Kankarea J. J. Lumin. 1997;75:149. [Google Scholar]
- [14].D’Aléo A, Picot A, Beeby A, Williams JAG, Le Guennic B, Andraud C, Maury O. Inorg. Chem. 2008;47:10258. doi: 10.1021/ic8012969. [DOI] [PubMed] [Google Scholar]
- [15].Moore EG, Xu J, Jocher CJ, Werner EJ, Raymond KN. J. Am. Chem. Soc. 2006;128:10648. doi: 10.1021/ja062597+. [DOI] [PubMed] [Google Scholar]
- [16].Moore EG, Xu J, Jocher CJ, Castro-Rodriguez I, Raymond KN. Inorg. Chem. 2008;47:3105. doi: 10.1021/ic702144n. [DOI] [PubMed] [Google Scholar]
- [17].D’Aléo A, Xu J, Moore EG, Jocher CJ, Raymond KN. Inorg. Chem. 2008;47:6109. doi: 10.1021/ic8003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moore EG, Jocher CJ, Xu J, Werner EJ, Raymond KN. Inorg. Chem. 2007;46:5468. doi: 10.1021/ic700364t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Scarrow RC, Riley PE, Abu-Dari K, White DL, Raymond KN. Inorg. Chem. 1985;24:954. [Google Scholar]
- [20].Xu J, Durbin PW, Kullgren B, Ebbe SN, Uhlir LC, Raymond KN. J. Med. Chem. 2002;45:3963. doi: 10.1021/jm010564t. [DOI] [PubMed] [Google Scholar]
- [21].Steemers JF, Verboom W, Reinhoudt DN, van der Tol EB, Verhoeven JW. J. Am. Chem. Soc. 1995;117:9408. [Google Scholar]
- [22].Supkowski RM, Horrocks WD. Inorg. Chimica. Acta. 2002;340:44. [Google Scholar]
- [23].Werts MHV, Jukes RTF, Verhoeven JW. Phys. Chem. Chem. Phys. 2002;4:1542. [Google Scholar]
- [24].Beeby A, Bushby LM, Maffeo D, Williams JAG. J. Chem. Soc. Dalton Trans. 2002:48. [Google Scholar]
- [25].Demas JN, Crosby GA. J. Phys. Chem. 1971;75:991–1024. [Google Scholar]
- [26].Doble DMJ, Melchior M, O’Sullivan B, Siering C, Xu J, Pierre VC, Raymond KN. Inorg. Chem. 2003;42:4930. doi: 10.1021/ic026240s. [DOI] [PubMed] [Google Scholar]
- [27].Pierre VC, Botta M, Aime S, Raymond KN. Inorg. Chem. 2006;45:8355. doi: 10.1021/ic061262q. [DOI] [PMC free article] [PubMed] [Google Scholar]