

Abstract
Malaria remains a globally prevalent infectious disease that leads to significant morbidity and mortality. While there are a number of drugs approved for its treatment, drug resistance has compromised most of them, making the development of new drugs for the treatment and prevention of malaria essential. The completion of the Plasmodium falciparum genome and a growing understanding of parasite biology are fueling the search for novel drug targets. Despite this, few targets have been chemically validated in vivo. The pyrimidine biosynthetic pathway illustrates one of the best examples of successful identification of anti-malarial drug targets. This review focuses on recent studies to exploit the fourth enzyme in the de novo pyrimidine biosynthetic pathway of P. falciparum, dihydroorotate dehydrogenase (PfDHODH), as a new target for drug discovery. Several chemical scaffolds have been identified by high throughput screening as potent inhibitors of PfDHODH and these show strong selectivity for the malarial enzyme over that from the human host. Potent activity against parasites in whole cell models with good correlation between activity on the enzyme and the parasite have also been observed for a number of the identified series. Lead optimization of a triazolopyrimidine-based series has identified an analog with prolonged plasma exposure, that is orally bioavailable, and which shows good efficacy against the in vivo mouse model of the disease. These data provide strong evidence that PfDHODH is a validated target for the identification of new antimalarial chemotherapy. The challenge remains to identify compounds with the necessary combination of potency and metabolic stability to allow identification of a clinical candidate.
Keywords: Malaria, Plasmodium, pyrimidine biosynthesis, dihydroorotate dehydrogenase, drug discovery
Malaria the disease, and the current state of chemotherapy
Malaria is a global disease caused by protozoan Apicomplexan parasites of the Plasmodium species and is transmitted by the Anopheles mosquito [1, 2]. It is endemic in 90 countries, putting over 2 billion people at risk and leading to the deaths of 1–2 million people annually. Pregnant woman and children are the most susceptible to severe disease. Until the early 1950s malaria was endemic in parts of the US and Europe, but it was eliminated through extensive control programs. It is now found primarily in tropical and subtropical countries with the African population most at risk. A number of anti-malarial agents are in clinical use, however the development of resistance to both chloroquine and sulfadoxine-pyrimethamine led to the rapid increase in the number of fatalities due to malaria [1, 3, 4]. Drug resistance has been reported to almost every known anti-malarial agent, underscoring the ease by which parasite populations can adapt and survive. Field isolates from drug-resistant regions of the world appear to acquire resistance to new agents faster than parasites isolated before resistance had developed [5]. The introduction of Artemisinin-based combination therapies (ACT) in conjunction with vector control measures has led to real progress in reducing the burden of the disease [6, 7]. Recent reports of Artemisinin resistance in western Cambodia raise the worrying possibility that this class of drugs may also fall to resistance [8].
The path forward to new drug discovery
A significant effort is underway to identify new anti-malarial agents. The discovery effort is being fostered by partnerships formed between nonprofit organizations such as Medicines for Malaria Venture (http://www.mmv.org/), and academic and industrial collaborators, as well as by public organizations such as the US National Institutes of Health [3, 9, 10]. A number of additional ACTs are in late stage clinical trials, and several novel agents including synthetic trioxalanes (Rbx11160 and OZ439), pyridones developed by GlaxoSmithKline (GSK 932121), and MK4815 licensed to MMV by Merck, are under clinical investigation. The search for new molecular targets is being aided by the completion of the genome sequence for P. falciparum [11]. However despite extensive efforts to identify new essential and druggable targets, few have been chemically validated. The identification of mitochondrial electron transport chain cytochrome bc1 complex as the target for atovoquone represents that most recent discovery of a new target that is validated with clinically proven inhibitors [12–15].
Many of the clinically relevant anti-malarial agents that have a known mechanism of action directly or indirectly affect pyrimidine metabolism. Drugs targeting dihydrofolate reductase (DHFR) or dihydropteroate synthase (e.g. pyrimethamine, cycloguanil, sulfonamides and sulfones) disrupt folate metabolism, which is essential for the formation of thymidine [16]. Atovoquone directly targets the electron transport bc1 complex in the mitochondria, however this too causes toxicity through disrupting pyrimidine metabolism. This link was first suggested by the observation that Atovoquone treatment causes a reduction in cellular UTP and CTP levels [17, 18]. A more recent study showed directly that the activity of the bc1 complex is essential for providing oxidized ubiqinone to DHODH for the formation of pyrimidines [19, 20]. Inhibitors of thymidylate synthase are also potent anti-malarials, though none have yet reached the clinic [21–24]. These studies suggest that the pyrimidine biosynthetic pathway may be a rich source for the discovery of new anti-malarial agents. This review focuses on efforts to exploit the fourth enzyme in the de novo pyrimidine pathway, DHODH for discovery of new chemical species targeting this enzyme for the treatment of malaria.
De novo pyrimidine biosynthesis is essential in malaria
Pyrimidines are essential metabolites that are precursors for DNA and RNA biosynthesis [16]. Cells acquire pyrimidines either through de novo synthesis starting from ammonia (derived from L-glu), bicarbonate, and L-asp, or by salvaging preformed pyrimidine bases (uracil, cytosine and thymine) or nucleosides (uridine, thymidine and cytidine). Plasmodium species are unusual in that they lack pyrimidine salvage enzymes and the de novo pathway provides the only source of pyrimidines for cell growth. In contrast, human cells are able to utilize both pathways. Six enzymes in Plasmodium species are required to synthesize UMP, which is then used to generate UTP, CTP, dTMP, and the subsequent additional metabolites of these nucleotides that are required by the cell. These include bifunctional glutamine amidotransferase/ carbamoylphosphate synthetase (GAT/CPS), aspartate carbamoyltransferase (ACT), dihydroorotase (DHOtase), DHODH, orotate phosphoribosyltransferase (OPRT) and orotidine 5’-monophosphate decarboxylase (OMPDC) (Figure 1). The activities of the enzymes have been detected in parasite extracts [25, 26] and the genes for each have been identified in the parasite genome [11]. PfDHODH has been purified and characterized from the parasite mitochondria [18, 27, 28], and the gene sequence [29] reported prior to the completion of the malaria genome. The organization of the Plasmodium enzymes differs from their mammalian counterparts. In mammals the first four activities (GAT/CPS/ACT/DHOtase) are fused into a single polypeptide, as are the final two (OPRT/OMPDC)[30]. In contrast, for the Plasmodium enzymes, only the first two activities are present in a fused protein (GAT/CPS).
Fig. 1.

De novo pyrimidine biosynthetic pathway in Plasmodium. The pathway involves six enzymes: a bifunctional glutamine amidotransferase (GAT) and carbamoyl-phosphate synthetase (CPS), aspartate carbamoyltransferase (ACT), dihydroorotase (DHOtase), dihydroorotate dehydrogenase (DHODH), orotate phosphoribosyltransferase (OPRT) and orotidine-5’-monophosphate decarboxylase (OMPDC). L-Gln, L-glutamine; CP, carbamoyl phosphate; Asp, L-aspartic acid; CA, carbamoyl aspartate; DHO, dihydroorotate; OA, orotic acid; OMP, orotidylate; UMP, uridylate. Enzyme names are displayed in blue. The rescue pathway used to bypass mitochondrial PfDHODH in transgenic parasites harboring yeast DHODH is displayed in grey.
Plasmodium is the only genus amongst the pathogenic apicoplasts to rely solely on de novo pyrimidine biosynthesis. Toxoplasma gondii has both the de novo and salvage pathways, while Cryptosporidium utilizes only pyrimidine salvage pathways. The absence of pyrimidine salvage pathways in Plasmodium was first biochemically demonstrated by analysis of the incorporation of radiolabeled precursors into nucleic acids [31] and by enzymatic assay [26], and has now been confirmed by the completion of the genome sequence [11]. Orotic acid is the only preformed pyrimidine that can be incorporated by the parasite, and consistent with this observation, 5-fluoroorotate is a potent anti-malarial agent while 5-fluorouracil is not [22, 23]. In the absence of salvage enzymes for the incorporation of preformed bases and nucleosides all of the de novo biosynthetic enzymes are presumably essential to Plasmodium species. However, in order to successfully target an enzyme for a drug discovery program, more than essentiality is required. It is also necessary that the protein target binds small drug-like molecules with high affinity (i.e. the target is “druggable) [32, 33], that inhibitors of the target cause cell growth inhibition, and furthermore that selectivity can be achieved between the host and parasite. As summarized below, Plasmodium DHODH meets these criteria, providing a strong case for the current focus on this target for drug discovery efforts.
DHODH is a “druggable” target
Human mitochondrial-type DHODH is a clinically validated drug target based on the successful development and approval of leflunomide for the treatment of rheumatoid arthritis [34–36]. Leflunomide (1) is an isoxazole-based prodrug, which after administration is rapidly converted to the active metabolite A77 1726 (2) (Figure 2). A77 1726 is a potent inhibitor of mammalian DHODH [37–39] and the immunosuppressive activity of Leflunomide is mediated by pyrimidine depletion [40]. Resting lymphocytes can survive on pyrimidine salvage while activated lymphocytes require de novo pyrimidine biosynthesis to support their enhanced growth rate, providing a level of selective toxicity towards the target cell population. Other inhibitors of human DHODH with immunosuppressive activity have also been reported, including additional analogs of A77 1726 [41], redoxal [42], S-2678[43] and notably the cinchoninic acid derivative brequinar (3) [44–48]. Brequinar was evaluated in clinical trials as a potential anti-cancer agent, however it was never approved for clinical use.
Fig. 2.

Inhibitors of human DHODH
Evolutionary history of DHODH
DHODH belongs to a large super family in the β/α–barrel structural class [30, 49]. In different species DHODH localizes to either the cytoplasm (Class 1) or to the inner mitochondrial membrane (or plasma membrane for bacteria) with the catalytic domain oriented towards the inner membrane space (Class 2). The Class 2 enzymes are bound to the membrane through a short membrane spanning sequence and they have been shown to display detergent-dependent kinetics [50]. Most eukaryotes and gram-negative bacteria encode Class 2 enzymes, however the two classes are distributed unevenly across the evolutionary tree, and no simple generalizations can be made. Both human and Plasmodium contain Class 2 mitochondrial enzymes, thus providing the first indication that malarial DHODH was a “druggable” target. In contrast, the kinetoplastids [51] and some yeast have Class 1 fumartate-dependent enzymes, for which there is no supporting druggability data.
DHODH catalyzes the flavin mononucleotide (FMN)-dependent oxidation of dihydroorotate to produce orotic acid. Two separate half reactions are required to complete the catalytic cycle: 1) oxidation of dihydroorotate driven by the reduction of FMN and, 2) reoxidation of FMNH2 to regenerate the active enzyme. The reoxidation of FMN is accomplished by one of several potential cofactors depending on the species-specific cellular localization of the enzyme. The Class 1 cytoplasmic enzymes use fumarate or NAD+ and the mitochondrial enzymes require oxidized ubiquinone (CoQ), thus coupling pyrimidine biosynthesis to the respiratory chain [20, 52, 53]. The Class 1 enzymes are further classified into Family 1A (fumarate-dependent) and Family 1B (NAD-dependent). For the Family 1A enzymes, fumarate is believed to share the same binding-site as orotate (Type 1A)[54, 55]. In contrast the Family 1B enzymes are composed of proteins from two gene products. Electrons are transferred from FMN bound in the DHODH-homologous subunit to an [1Fe-2S] iron-sulfur cluster followed by FAD and NAD, all three of which are bound on a second subunit with similarity to ferredoxin reductase [56, 57]. In the Class 2 enzymes CoQ is thought to bind on the opposite face of FMN from orotate. This hypothesis is based on the available structural information, and the observation of ping-pong reaction kinetics [49, 58–61]. The kinetic and structural characterization of the family 2 enzymes has been performed with the truncated soluble domain lacking the mitochondrial membrane anchor and the sequence N-terminal to this region. The recombinant protein for the Plasmodium and human enzymes has produced in E. coli (e.g. PfDHODH [62]).
Structural analysis of DHODH
Clardy solved the X-ray structures of human DHODH bound to A77 1726 and brequinar providing the first insight into the nature of the inhibitor binding-site in the Class 2 enzymes [49]. Subsequently a number of additional X-ray structures of the human enzyme bound to various inhibitors have been reported (e.g. [63–65]), and the X-ray structure of PfDHODH has been solved bound to both A77 1726 [59] and to novel malarial inhibitors from the triazolopyrimidine series [66], the latter of which will be discussed in detail below. The central structural element of DHODH from all class types is the core β/α-barrel domain. This domain houses the binding site for the FMN cofactor, which is bound near strand β13 at the top of helix α11 (Figure 3). Orotate forms a stacking interaction with FMN on one side, while the opposite side of the orotate binding-site is formed by β11 and surface loops containing Ser-395 and Thr-459. In addition the class 2 enzymes have a largely hydrophobic N-terminal helical domain (α1 and α2) that presumably sits adjacent to the membrane. The inhibitor binding-site, as illustrated for A77 1726, is formed between the N-terminal helices and the β/α-barrel domain, making interactions with helix α3, α11 and strand β5.
Fig. 3.

X-ray structure of PfDHODH. A. Ribbon diagram of an alignment of the structures bound to A77 1726 (tan; pdb 1TV5) and DSM1 (purple; pdb 3I65). B. A77 1726 and DSM1 inhibitor binding sites. Residues marked with * are conserved in human DHODH, while the remainder of the shown residues are variable between Plasmodium and human DHODH. Structures were displayed in PyMol (Delano Scientific)
Species selective inhibition of DHODH is feasible
Inhibitors of DHODH were recognized early on to show species selective inhibition between even closely related species. Knecht and Loffler found strong species differences in IC50 values for both A77 1726 and brequinar between rat and human DHODH [67], and inhibitors of human DHODH were shown to be poor inhibitors of E. coli DHODH [68]. Capitalizing on these observations, Copeland used HTS approaches to identify two series of DHODH inhibitors that were selective for the bacterial enzymes: the thiadiazolidinediones, which inhibit E. coli DHODH [69], and pyrazoles that have selective activity on the enzyme from Helicobacter pylori [70, 71]. Comparison of the X-ray structures of the unliganded E. coli Class 2 DHODH [61] with the human enzyme bound to A77 1726 or brequinar [49] shows that the amino acid residues in the inhibitor binding-site are not conserved between the two enzymes, explaining the species-selective binding of these inhibitors. Our group showed that species-selective inhibitor binding extended to the P. falciparum enzyme. Potent inhibitors of human DHODH were found to be poor inhibitors of PfDHODH [62]. The subsequent X-ray structure of PfDHODH by Clardy’s group showed that A77 1726 is bound in the same binding pocket in PfDHODH and the human enzyme, but the amino acid residues that contact the ligand are not conserved between the species [59] (Figures 3 and 4).
Fig. 4.

Amino acid sequence alignment of PfDHODH, PbDHODH and human DHODH. Secondary structure elements are indicated by color outline; blue, α-helix and green, β-sheet. Residues within 4Å of DSM1 in the inhibitor binding-site are highlighted by bold lettering.
Genetic evidence that PfDHODH is an essential enzyme
The differences in localization and cofactor specificity between DHODH from divergent species were recently exploited by the Vaidya group to provide important insight into the tight link between the mitochondrial electron transport chain and pyrimidine biosynthesis in the malaria parasite [19, 20]. A transgenic P. falciparum cell line was constructed that expressed yeast fumarate-dependent DHODH in the parasite cytoplasm, thus bypassing the need for the mitochondrial enzyme (Figure 1). This cell line was resistant to atovoquone, suggesting that at least for in vitro growth, the main function of the bc1 complex is to provide oxidized CoQ for the generation of orotic acid by DHODH. These studies provided an elegant genetic confirmation that DHODH is an essential enzyme in P. falciparum.
Summary of the case for PfDHODH as a target for drug discovery efforts. P. faliciparum
DHODH catalyzes an essential reaction and no functional bypass exists in blood stage parasites to overcome the loss of this activity. Based on homology with the human enzyme, malaria DHODH is a “druggable” target, and the lack of conservation in the inhibitor binding-site across species suggests that the identification of species-selective inhibitors was feasible. Furthermore while the lack of pyrimidine salvage pathways in Plasmodium makes it vulnerable to inhibition of this pathway, the presence of salvage pathways in human cells affords additional selectivity. In order to exploit PfDHODH for the development of new anti-malarials, high-throughput screening (HTS), medicinal chemistry on human selective DHODH inhibitors, and structure-based methods have been used to identify species selective inhibitors of PfDHODH. The most successful of these approaches has been HTS.
Novel and selective inhibitors of P. falciparum DHODH discovered by HTS
Our group published the first reported HTS against P. falciparum DHODH [72]. An end point dye-based assay was adapted to 384 well plate format and was used to screen a 220,000 compound library for inhibitors of PfDHODH. This assay couples the reoxidation of CoQ with the reduction of dichloroindophenol (DCIP), which can be monitored at 600 nm. The library was screened using a single concentration point of 3 µM and 1249 hits (defined by >60% inhibition) were identified, representing a hit rate of 0.6%. A preliminary dose response against PfDHODH was determined and the initial hits were counter screened against human DHODH at the same concentrations. The identified hits were almost uniformly selective for the Plasmodium enzyme. We choose compounds with an IC50<0.6 µM for further analysis and our published hits are described below.
Phenylbenzamides, ureas and naphthamides
The most common chemical classes identified by this screen were phenylbenzamides 4, ureas 5 and naphthamides 6 [72] (Figure 5 and Table 1). These compounds inhibit PfDHODH with an IC50 between 0.02 – 0.8 µM and show selectivity versus human DHODH ranging from 70–12,500-fold. The most potent compounds were also the most selective (e.g. compound 4). Despite the potent enzyme activity, these inhibitors have no anti-malarial activity and we chose not to advance the compounds in these series.
Fig. 5.

Species selective inhibitor chemophores of P. falciparum DHODH with submicromolar activity.
Table 1.
Representative early hits derived from HTS and medicinal chemistry programs
| compound |
PfDHODH (IC50) µM |
Human DHODH (IC50) µM |
Fold selectivity |
|---|---|---|---|
| 4 | 0.016 | 200 | 12,500 |
| 5 | 0.23 | 110 | 500 |
| 6 | 0.05 | >50 | >1000 |
| 7 | 0.047 | >200 | >4000 |
| 8 | 0.042 | >30 | >700 |
| 9 | 0.34 | >30 | >90 |
| 10 | 0.083 | >30 | >360 |
| 11 | 0.93 | >30 | >30 |
| 12 | 0.16 | 30 | >190 |
Triazolopyrimidine series
Our screen also identified a potent and selective triazolopyrimidine-based compound DSM1(7), which has similarly potent activity against P. falciparum 3D7 in whole cell assays while displaying stringent species selectivity (IC50 PfDHODH= 0.047 µM; IC50 hDHODH > 200 µM; EC50 Pf 3D7 cells = 0.079 µM [73]) (Table 1 and Figure 5). This compound series was chosen for progression and it has formed the basis of an ongoing lead optimization program by our group. A simple and inexpensive 3-step synthetic route was developed that allowed us to synthesize a number of analogs of DSM1 from commercially available amines to replace the naphthyl portion of the molecule [66, 73, 74] (Table 2). These studies showed that 1) the unsubstituted bridging nitrogen N1 was essential for activity, 2) introduction of heteratoms into the naphthyl ring reduced potency, 3) the naphthyl could be replaced by larger aromatic and hydrophobic ring systems including anthracene and substituted phenyl rings, 4) phenyl rings at the naphthyl position required substitution at the para position (CF3 >Br>Cl>Me>F), with ortho substitution leading to complete loss of activity, and 5) replacement of C6 with ethyl or CF3 reduced potency, as did addition of a Me to C2. A good correlation was observed between enzyme activity and whole cell potency throughout the series providing strong evidence that DHODH is the cellular target of these inhibitors.
Table 2.
Activity of representative triazolopyrimidine-based derivatives against DHODH and P. falciparum
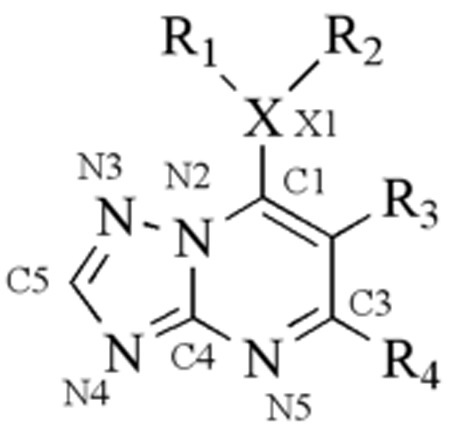 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | R1 | R2 | R3 | R4 | X | IC50(µM) PfDHODH |
IC50(µM) PbDHODH |
EC50(µM) Pf 3D7 cells |
Pred Eh human microsome |
|
| 7 | DSM1 | 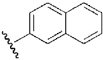 |
H | H | Me | N | 0.047 | 0.23 | 0.079 | 0.84 |
| 13 | DSM2 | 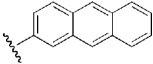 |
H | H | Me | N | 0.056 | 3.7 | 0.19 | 0.7 |
| 14 | DSM12 |  |
H | H | Me | N | >100 | >10 | >100 | n.d. |
| 15 | DSM15/DSM16 | 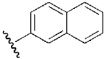 |
H | H | Me | S/ O | >100 | n.d. | >100 | n.d. |
| 16 | DSM6 | 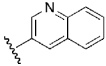 |
H | H | Me | N | 1.7 | n.d. | 1.6 | n.d. |
| 17 | DSM18 |  |
H | Me | Me | N | 0.16 | n.d. | 0.55 | n.d. |
| 18 | DSM9 | 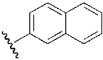 |
Me | H | Me | N | 3.0 | n.d. | 16 | n.d. |
| 19 | DSM20 | 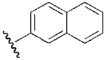 |
H | H | Et | N | 0.19 | n.d. | 0.31 | n.d. |
| 20 | DSM131 | 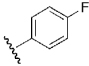 |
H | H | Me | N | 19 | >100 | 50 | n.d. |
| 21 | DSM98 | 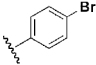 |
H | H | Me | N | 0.78 | 1.3 | 4.2 | n.d. |
| 22 | DSM74 | 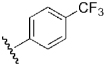 |
H | H | Me | N | 0.28 | 0.38 | 0.34 | 0.29 |
| 23 | DSM125 | 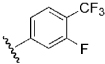 |
H | H | Me | N | 0.077 | 0.26 | 1.3 | 0.21 |
| 24 | DSM72 |  |
H | H | Me | N | 9.2 | >100 | 34 | 0.5 |
| 25 | DSM75 |  |
H | H | Me | N | 1.4 | 31 | 8.5 | 0.68 |
| 26 | DSM106 |  |
H | H | Me | N | >100 | >100 | >50 | n.d. |
| 27 | DSM95 | 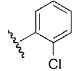 |
H | H | Me | N | >100 | >100 | >25 | n.d. |
The X-ray structures of PfDHODH bound to three inhibitors from the triazolopyrimdine-based series DSM1(7), DSM2 (13) and DSM74 (22) were solved by our group, providing the first structures of PfDHODH bound to a parasite selective, potent inhibitor (Figure 3)[66]. The triazolopyrimidine series inhibitor binding-site is composed of two sites: 1) a completely hydrophobic site, which binds the naphthyl, anthracenyl or phenyl-trifluoromethyl, and 2) the triazolopyrimidine pocket that contains the only residues capable of H-bond interactions. Both sites provide a significant amount of binding energy to the inhibitor series; π-stacking interactions are observed between the naphthyl and two Phe residues (227 and 188), and H-bonds between H185 and R265 and the triazolopyrimidine ring are present in all three structures. The triazolopyrimidine functionality overlaps the binding site of the β-hydroxy enamide portion of A77 1726 observed in the previously reported PfDHODH structure [59]. In contrast, the naphthyl binding-site accesses a previously unidentified pocket created by a significant rotation of Phe188 that moves this residue into a position that overlaps the phenyl portion of A77 1726. These data provide a structural explanation as to how PfDHODH is able to bind multiple chemical classes with high affinity.
The structural basis for the species selective binding of the triazolopyrimidines towards PfDHODH was also evident from these structures (Figures 3 and 4). Some of the key amino acid differences that lead to species selectivity include, the replacement of hAla-59 and hPro-364 with PfPhe-188 and PfMet-536, which closes off the hydrophobic pocket that binds the biphenyl of brequinar in human DHODH, and the substitution of hThr-63 and hMet-111 with PfGly-192 and PfLeu-240, which creates the naphthyl pocket in PfDHODH.
Most importantly, the X-ray structures of the triazolopyrimdine series provided us with significant insight into the emerging structure-activity relationships (SAR), and with a blueprint for further modification of this chemical series. Comparison of the ligand-bound DHODH structures to the small molecule crystal structures provided insight into the importance of the bridging nitrogen (N1). The small molecule structures showed that the intrinsic electronic configuration of the triazolopyrimidine ring favors charge delocalization from the bridging nitrogen (N1) onto the pyridine nitrogen (N5). Intrinsic dipoles in the ring promote formation of energetically favorable H-bond interactions with the protein between His185 and N1, and between Arg265 and N5. Triazolopyrimidine analogs with S or O replacing N1 lack this intrinsic dipole and are inactive as inhibitors, while mutation of His185 or Arg265 to Ala also reduced inhibitor binding, supporting the importance of these interactions. Heteroatom substitution into the naphthyl ring reduced activity, consistent with the completely hydrophobic nature of the naphthyl binding-pocket on the enzyme. Ortho-substituted phenyl rings are inactive and our analysis of the X-ray structure shows that the contacts made between the protein and the ortho positions on the aromatic ring are too close to allow substitution at this position. Finally a narrow channel leading from C5 on the inhibitor towards the FMN cofactor was observed, suggesting that this pocket might be exploited for future optimization of the lead series.
Substituted pyrrole and thiofuran analogs
A second high throughput screen to identify PfDHODH inhibitors was reported by Clardy and Wirth at Harvard using a small molecule library of 208,000 compounds from Genzyme [75]. This screen was also run using the DCIP assay with a fixed inhibitor concentration of 10 µM. The hit rate in this screen was 0.3% and 55 compounds were identified with an IC50 below 1 µM on the enzyme. Five scaffolds inhibited P. falciparum 3D7 growth in whole cell assays at submicromolar concentrations (Figure 5 and Table 1). All 5 inhibitors were selectively active against the parasite enzyme over the human enzyme. The most potent compound 8 has an IC50 = 40 nM on PfDHODH, and they generated a small number of analogs from this lead to establish preliminary SAR (Table 3). While none of the analogs showed greater potency than the original hit, the activity versus the enzyme and the parasite showed similar trends supporting PfDHODH as the cellular target. The cellular target was definitively established by characterizing the DHODH inhibitors against transgenic parasites harboring the cytoplasmic yeast DHODH, as established by the Vaidya Lab [19]. Parasites transfected with yeast DHODH showed significant resistance to the PfDHODH inhibitors, establishing the mechanism of action.
Table 3.
Structure activity of 2,5 substituted thiofurans.
 | |||
|---|---|---|---|
| Structures |
PfDHODH IC50 (µM) |
Pf 3D7 cells EC50 (µM) |
|
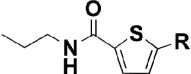
| 8 |  |
0.042 | 0.49 |
| 28 | 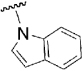 |
0.46 | 0.71 |
| 29 | 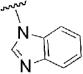 |
0.7 | 0.47 |
| 30 |  |
0.17 | 0.63 |
| 31 | 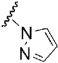 |
>30 | >10 |
| 32 |  |
>30 | >10 |
| 33 | 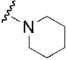 |
>30 | >10 |
Medicinal chemistry to evolve human DHODH inhibitors towards PfDHODH selectivity
Structure-based methods and the synthesis of brequinar and A77 1726 analogs have also been used as strategies to identify inhibitors of PfDHODH by a group at the University of Leeds. The group initially explored a series of brequinar derivatives. Brequinar is an exceptionally poor inhibitor of PfDHODH (IC50 > 500 µM), and the best analog identified in the study only had mid micromolar range activity on PfDHODH (IC50 = 40 µM), and furthermore showed no selectivity versus the human enzyme [76]. Their attempts to generate potent and selective PfDHODH inhibitors from the A77 1726 scaffold were also unsuccessful. They synthesized a series of 26 analogs using the PfDHODH A77 1726 co-crystal structure to aid in analog design. The best inhibitor had micromolar activity against PfDHODH (IC50 = 4 µM), and these compounds showed in almost all cases far better activity against human DHODH [63]. This group also used the molecular design program SPROUT with the PfDHODH structure bound to A77 1726 to identify potential inhibitory scaffolds of PfDHODH [77]. Based on this analysis a series of 6 amide derivatives of anthranilic acid were prepared, with none showing better than mid micromolar activity against PfDHODH. Finally this group utilized a previously described tetracyclic amine that had been shown to have immunosuppressive activity similar to brequinar and A77 1726 as a starting point to generate analogs with potential activity against PfDHODH [78]. Tricyclic amine derivatives of this compound were shown to have submicromolar activity on PfDHODH (IC50 = 0.2 – 0.4 µM) and these inhibitors do show selectivity for the P. falciparum enzyme (200 –1200-fold). The most potent of these 12 (Figure 5 and Table 1) has micromolar activity against P. falciparum 3D7 (EC50 = 2 µM).
The mechanism of PfDHODH inhibition
The location of the CoQ binding site on DHODH is presently a point of speculation. Competitive inhibition kinetics for some human DHODH inhibitors, including brequinar [79], coupled with the structural data showing that the binding-site of A77 1726 and brequinar is adjacent to FMN, led to the suggestion that the CoQ binding-site overlaps the inhibitor binding-site [49]. However, there is no crystallographic data to demonstrate where CoQ binds. Insight into the relationship between the inhibitor binding-site and the CoQ binding-site was obtained by steady-state and pre-steady state kinetic analysis of the PfDHODH inhibitor binding-site mutant enzymes [80]. Our studies suggested that CoQ and inhibitors (e.g. A77 1726, DSM1, 4) do not share an overlapping binding site. A series of mutant enzymes replacing residues in the inhibitor binding-pocket with Ala were evaluated, including mutants of H185, R265, F188 and F227. These mutations decreased inhibitor affinity, consistent with their contribution to the energetics of inhibitor binding. In contrast, steady-state analysis showed that the Kmapp for CoQ was not significantly affected by these mutations. Secondly, the pre-steady-state kinetic analysis showed that the mutations reduced the rate of the CoQ-dependent flavin oxidation step but they did not significantly alter the Kdox for CoQ (the dissociation binding constant for CoQ in the oxidation step of the reaction). The rate of the DHO-dependent reductive half-reaction was also unchanged. DSM1 and 4 also blocked the CoQ-dependent oxidative half-reaction but not the DHO-dependent step. Thus the mutations affect inhibitor binding, and both the inhibitors and the mutations affect the electron transfer between CoQ and FMN, but mutation of this binding site was not coupled to changes in the apparent CoQ affinity. This discrepancy suggests that CoQ and the inhibitors do not bind the same site. Based on these data we proposed two models that explain the inhibition kinetics: 1) the inhibitors block electron transfer between flavin and CoQ or, 2) they act by stabilizing a conformation that excludes CoQ binding. X-ray structure analysis of DHODH bound to CoQ would provide substantial clarification of the mechanism of DHODH inhibition by all inhibitors that bind this site. Clardy and Wirth evaluated this same set of mutant enzymes and found that these mutations also reduced the binding affinity of the inhibitors identified in their screen [75]. This result establishes that all classes of inhibitors identified to date by HTS against PfDHODH share an overlapping binding site.
Identification of PfDHODH inhibitors with in vivo anti-malarial activity
The identification of potent and species selective PfDHODH inhibitors that are able to inhibit parasite growth in whole cell assays has provided several validated hits that can form the basis of lead optimization programs with the goal of identifying a clinical candidate. Of the identified series, our group has progressed the triazolopyrimidines to the early lead stage by the application of an iterative lead optimization strategy [74]. The design of new analogs with better in vivo properties is being informed by in vitro metabolism and pharmacokinetic analysis in mice, in addition to the evaluation of inhibitor potency.
The original HTS hit DSM1, while showing potent activity against P. falciparum in whole cell assays, has no activity against P. berghei in the standard mouse model. Reasonable plasma levels were achieved after a single oral dose, however we found that multiple doses led to decreased plasma levels, suggesting that DSM1 maybe a metabolic inducer. This result explained the lack of in vivo activity. Additionally potency differences between P. falciparum and P. berghei DHODH contributed to the lack of efficacy for some inhibitors in the series. A series of substituted phenyl replacements for the naphthyl moiety were synthesized and analyzed in human liver microsomes to evaluate metabolic stability (Table 2). From these studies we identified DSM74 (22)(Table 2 and Figure 6). This compound shows prolonged plasma exposure after multiple oral doses, and it significantly suppressed parasetimia in a 4-day mouse efficacy test using the P. berghei model (95% and 71% suppression on day five for b.i.d. and q.d. dosing respectively). DSM74 was well tolerated and no signs of overt toxicity were observed. These data provide the first proof of concept that PfDHODH inhibitors can function in vivo to affect parasetimia, and they provide strong validation of PfDHODH as a target for new-antimalarial drug discovery.
Fig. 6.

P. falciparum DHODH inhibitors with demonstrated in vivo activity.
Progress targeting other enzymes in the de novo pyrimidine biosynthetic pathway for drug discovery
In addition to efforts to target PfDHODH some in roads have also been made towards characterizing other enzymes in the pathway for their potential to lead to new chemotherapy. Carbonic anhydrase, which is needed to generate HCO3 − for the first step in pyrimidine biosynthesis has been expressed and purified as the recombinant enzyme. Benzenesulfonamide derivatives with low micromolar activity on the enzyme (Ki = 0.18 µM) and some activity on P. falciparum in whole cell assays (EC50 = 1 µM) and against P. berghei in vivo have been reported [81]. Dihydroorotase, the third enzyme in the de novo pathway, has been expressed as the recombinant protein in E. coli, and characterization of the enzyme shows it is a monofunctional protein differing from the mammalian enzyme, which is part of a multisubunit complex [82]. The most studied enzyme after PfDHODH is orotidine 5’phosphate decarboxylase (OMPDC), which catalyzes the last step in the pathway forming UMP. Both the structure and mechanism of OMPDC have been the subject of intense research because of the difficult chemistry and the high catalytic proficiency of the enzyme [83]. The Plasmodium enzyme has been shown to form a complex with orotate phosphoribosyltransferase, the penultimate step in UMP biosynthesis [84]. Enzymatic properties of both enzymes are affected by formation of the heterotetramer. Several X-ray structures of P. falciparum ODCase have been reported, including the unliganded enzyme [85] and the enzyme bound to several mononucleotide derivatives that are covalent inhibitors of the enzyme [86]. For orotate phosphoribosyltransferase, kinetic isotope effects and computational methods have been used to predict the transition state structures for both the Plasmodium and human enzyme [87]. This is a powerful method that can provide important insight into the design of tight binding inhibitors.
Conclusion
Pyrimidine biosynthesis is a highly privileged pathway for drug discovery against Plasmodium parasites based on the high proportion of known anti-malarials targeting this pathway. The reliance of the parasite on de novo pyrimidine biosynthesis makes it highly vulnerable to inhibitors targeting the pathway. DHODH catalyzes the fourth step in de novo pyrimidine biosynthesis and both genetic and chemical studies have provided conclusive evidence that it is an essential enzyme and a druggable target in malaria. HTS methodology has proved a successful strategy to identify potent and highly species selective inhibitors of PfDHODH. X-ray structures of PfDHODH bound to inhibitors have provided insight into the structural basis for species selectivity and are a valuable asset for continued efforts to optimize the identified leads. The triazolopyrimidine series has been progressed from the hit stage into early lead optimization through an iterative lead optimization strategy that allowed us to identify analogs with good metabolic stability, and which have demonstrated in vivo efficacy. We conclude that PfDHODH is a very promising target for the identification of novel anti-malarial agents, and if the current discovery efforts are successful work on this target will allow chemophores that are entirely different from current drugs to enter the clinics. The tools are now in place, and lead optimization efforts are underway to progress the identified series, several of which are within the MMV portfolio (www.mmv.org). It now remains for compounds with the necessary combination of potency and metabolic stability to be identified so that at least one series can be progressed to a clinical candidate. Final validation of the target awaits the discovery of a clinically useful drug.
Acknowledgements
MAP and PKR are supported by the United States National Institutes of Health grants, U01AI075594 (to MAP and PKR; Ian Bathurst (PI); Medicines for Malaria Venture) and AI053680 (to MAP and PKR). MAP also acknowledges the support of the Welch Foundation (I-1257) and PKR also acknowledges support from a Senior Scholar Award in Global Infectious Diseases from the Ellison Medical Foundation and from the UW Keck Center for Microbial Pathogens. MAP holds the Carolyn R. Bacon Professorship in Medical Science and Education.
References
- 1.Greenwood BM, Fidock DA, Kyle DE, Kappe SH, Alonso PL, Collins FH, Duffy PE. Malaria: progress, perils, and prospects for eradication. J. Clin. Invest. 2008;118(4):1266–1276. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerra CA, Gikandi PW, Tatem AJ, Noor AM, Smith DL, Hay SI, Snow RW. The limits and intensity of Plasmodium falciparum transmission: implications for malaria control and elimination worldwide. PLoS Med. 2008;5(2):e38. doi: 10.1371/journal.pmed.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Craft JC. Challenges facing drug development for malaria. Curr. Opin. Microbiol. 2008;11(5):428–433. doi: 10.1016/j.mib.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 4.White NJ. Antimalarial drug resistance. J. Clin. Invest. 2004;113(8):1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rathod PK, McErlean T, Lee PC. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci., USA. 1997;94(7):9389–9393. doi: 10.1073/pnas.94.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White NJ. The role of anti-malarial drugs in eliminating malaria. Malar. J. 2008;11(7) Suppl 1:S8. doi: 10.1186/1475-2875-7-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenthal PJ. Artesunate for the treatment of severe falciparum malaria. N. Engl. J. Med. 2008;358(17):1829–1836. doi: 10.1056/NEJMct0709050. [DOI] [PubMed] [Google Scholar]
- 8.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009;361(5):455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gelb MH. Drug discovery for malaria: a very challenging and timely endeavor. Curr. Opin. Chem. Biol. 2007;11(4):440–445. doi: 10.1016/j.cbpa.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olliaro P, Wells TN. The global portfolio of new antimalarial medicines under development. Clin. Pharmacol. Ther. 2009;85(6):584–595. doi: 10.1038/clpt.2009.51. [DOI] [PubMed] [Google Scholar]
- 11.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fry M, Pudney M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4'-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80) Biochem. Pharmacol. 1992;43(7):1545–1553. doi: 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- 13.Mather MW, Darrouzet E, Valkova-Valchanova M, Cooley JW, McIntosh MT, Daldal F, Vaidya AB. Uncovering the molecular mode of action of the antimalarial drug atovaquone using a bacterial system. J. Biol. Chem. 2005;280(29):27458–27465. doi: 10.1074/jbc.M502319200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava I, Morrisey J, Darrouzet E, Daldal F, Vaidya A. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Micro. 1999;33(4):704–711. doi: 10.1046/j.1365-2958.1999.01515.x. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava I, Rottenberg H, Vaidya A. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J. Biol. Chem. 1997;272(7):3961–3966. doi: 10.1074/jbc.272.7.3961. [DOI] [PubMed] [Google Scholar]
- 16.Hyde JE. Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr. Drug Targets. 2007;8(1):31–47. doi: 10.2174/138945007779315524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seymour KK, Yeo AE, Rieckmann KH, Christopherson RI. dCTP levels are maintained in Plasmodium falciparum subjected to pyrimidine deficiency or excess. Ann. Trop. Med. Parasitol. 1997;91(6):603–609. doi: 10.1080/00034989760699. [DOI] [PubMed] [Google Scholar]
- 18.Ittarat I, Asawamahasakda W, Meshnick SR. The effects of antimalarials on the Plasmodium falciparum dihydroorotate dehydrogenase. Exp. Parasitol. 1994;79(1):50–56. doi: 10.1006/expr.1994.1058. [DOI] [PubMed] [Google Scholar]
- 19.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446(7131):88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 20.Vaidya AB, Mather MW. Mitochondrial Evolution and Functions in Malaria Parasites. Annu. Rev. Microbiol. 2009;63:249–267. doi: 10.1146/annurev.micro.091208.073424. [DOI] [PubMed] [Google Scholar]
- 21.Jiang L, Lee PC, White J, Rathod PK. Potent and selective activity of a combination of thymidine and 1843U89, a folate-based thymidylate synthase inhibitor, against Plasmodium falciparum. Antimicrob. agents chemother. 2000;44(4):1047–1050. doi: 10.1128/aac.44.4.1047-1050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rathod PK, Khatri A, Hubbert T, Milhous WK. Selective activity of 5-fluoroorotic acid against Plasmodium falciparum in vitro. Antimicrobial. agents chemother. 1989;33(7):1090–1094. doi: 10.1128/aac.33.7.1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rathod PK, Leffers NP, Young RD. Molecular targets of 5-fluoroorotate in the human malaria parasite, Plasmodium falciparum. Antimicrobial. agents chemother. 1992;36(4):704–711. doi: 10.1128/aac.36.4.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rathod PK, Reshmi S. Suceptibility of Plasmodium falciparum to a combination of thymidine and ICI D1694, a quinazoline antifolate directed at thymidylate synthase. Antimicrobial. agents chemother. 1994;38(3):476–480. doi: 10.1128/aac.38.3.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rathod PK, Reyes P. Orotidylate-metabolizing enzymes of the human malarial parasite, Plasmodium falciparum, differ from host cell enzymes. J. Biol. Chem. 1983;258(5):2852–2855. [PubMed] [Google Scholar]
- 26.Reyes P, Rathod PK, Sanchez DJ, Mrema JE, Rieckmann KN, Heidrich HG. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol. Biochem. Parasitol. 1982;5(5):275–290. doi: 10.1016/0166-6851(82)90035-4. [DOI] [PubMed] [Google Scholar]
- 27.Krungkrai J. Purification, characterization and localization of mitochondrial dihydroorotate dehydrogenase in Plasmodium falciparum, human malarial parasite. Biochim. Biophys. Acta. 1995;1243(3):351–360. doi: 10.1016/0304-4165(94)00158-t. [DOI] [PubMed] [Google Scholar]
- 28.Krungkrai J, Krungkrai SR, Phakanont K. Antimalarial activity of orotate analogs that inhibit dihdroorotase and dihydroorotate dehydrogenase. Biochem. Pharmacol. 1992;43(6):1295–1301. doi: 10.1016/0006-2952(92)90506-e. [DOI] [PubMed] [Google Scholar]
- 29.LeBlanc SB, Wilson CM. The dihydroorotate dehydrogenase gene homologue of Plasmodium falciparum. Mol. Biochem. Parasitol. 1993;60(2):349–352. doi: 10.1016/0166-6851(93)90148-q. [DOI] [PubMed] [Google Scholar]
- 30.Nara T, Hshimoto T, Aoki T. Evolutionary implications of the mosaic pyrimidine-biosynthetic pathway in eukaryotes. Gene. 2000;257(2):209–222. doi: 10.1016/s0378-1119(00)00411-x. [DOI] [PubMed] [Google Scholar]
- 31.Gutteridge WE, Trigg PI. Incorporatin of radioactive precursors into DNA and RNA of Plasmodium knowlesi in vitro. J. Protozool. 1970;17(1):89–96. doi: 10.1111/j.1550-7408.1970.tb05163.x. [DOI] [PubMed] [Google Scholar]
- 32.Hopkins AL, Groom CR. The druggable genome. Nat. Rev. Drug Discov. 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 33.Aguero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, Campbell RK, Carmona S, Carruthers IM, Chan AW, Chen F, Crowther GJ, Doyle MA, Hertz-Fowler C, Hopkins AL, McAllister G, Nwaka S, Overington JP, Pain A, Paolini GV, Pieper U, Ralph SA, Riechers A, Roos DS, Sali A, Shanmugam D, Suzuki T, Van Voorhis WC, Verlinde CL. Genomic-scale prioritization of drug targets: the TDR Targets database. Nat. Rev. Drug. Discov. 2008;7(11):900–907. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldenberg MM. Leflunomide, a novel immunomodulator for the treatment of active rheumatoid arthritis. Clin. therapeut. 1999;21(11):1837–1852. doi: 10.1016/S0149-2918(00)86732-6. [DOI] [PubMed] [Google Scholar]
- 35.Herrmann ML, Schleyerbach R, Kirschbaum BJ. Leflunomide: an immunomodulatory drug for the treatment of rheumatoid arthritis and other autoimmune diseases. Immunopharm. 2000;47(2–3):273–289. doi: 10.1016/s0162-3109(00)00191-0. [DOI] [PubMed] [Google Scholar]
- 36.Olsen NJ, Stein CM. New drugs for rheumatoid arthritis. N. Engl. J. Med. 2004;350(21):2167–2179. doi: 10.1056/NEJMra032906. [DOI] [PubMed] [Google Scholar]
- 37.Davis JP, Cain GA, Pitts WJ, Magolda RL, Copeland RA. The immunosuppressive metabolite of leflunomide is a potent inhibitor of human dihydroorotate dehydrogenase. Biochemistry. 1996;35(4):1270–1273. doi: 10.1021/bi952168g. [DOI] [PubMed] [Google Scholar]
- 38.Williamson RA, Yea CM, Robson PA, Curnock AP, Gadher S, Hambleton AB, Woodward K, Bruneau JM, Hambleton P, Moss D, Thomson TA, Spinella-Jaegle S, Morand P, Courtin O, Sautes C, Westwood R, Hercend T, Kuo EA, Ruuth E. Dihydroorotate dehydrogenase is a high affinity binding protein for A77 1726 and mediator of a range of biological effects of the immunomodulatory compound. J. Biol. Chem. 1995;270(38):22467–22472. doi: 10.1074/jbc.270.38.22467. [DOI] [PubMed] [Google Scholar]
- 39.Greene S, Watanabe K, Braatz-Trulson J, Lou L. Inhibition of dihydroorotate dehydrogenase by the immunosppressive agent leflunomide. Biochem. Pharmacol. 1995;50:861–867. doi: 10.1016/0006-2952(95)00255-x. [DOI] [PubMed] [Google Scholar]
- 40.Ruckemann K, Fairbanks LD, Carrey EA, Hawrylowicz CM, Richards DF, Kirschbaum B, Simmonds HA. Leflunomide inhibits pyrimidine de novo synthesis in mitogen-stimulated T-lymphocytes from healthy humans. J. Biol. Chem. 1998;273(34):21682–21691. doi: 10.1074/jbc.273.34.21682. [DOI] [PubMed] [Google Scholar]
- 41.Kuo EA, Hambleton PT, Kay DP, Evans PL, Matharu SS, Little E, McDowall N, Jones CB, Hedgecock CJ, Yea CM, Chan AW, Hairsine PW, Ager IR, Tully WR, Williamson RA, Westwood R. Synthesis, structure-activity relationships, and pharmacokinetic properties of dihydroorotate dehydrogenase inhibitiors: 2-cyano-3-cyclopropyl-3-hydroxy-N-[3'-methyl-4'-(trifluoromethyl)phenyl]propenamide and related compounds. J. Med. Chem. 1996;39(23):4608–4621. doi: 10.1021/jm9604437. [DOI] [PubMed] [Google Scholar]
- 42.Knecht W, Loffler M. Redoxal as a new lead structure for dihydroorotate dehydrogenase inhibitors: a kinetic study of the inhibition mechanism. FEBS Lett. 2000;467(1):27–30. doi: 10.1016/s0014-5793(00)01117-0. [DOI] [PubMed] [Google Scholar]
- 43.Deguchi M, Kishino J, Hattori M, Furue Y, Yamamoto M, Mochizuki I, Iguchi M, Hirano Y, Hojou K, Nagira M, Nishitani Y, Okazaki K, Yasui K, Arimura A. Suppression of immunoglobulin production by a novel dihydroorotate dehydrogenase inhibitor, S-2678. Eur. J. Pharmacol. 2008;601(1–3):163–170. doi: 10.1016/j.ejphar.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 44.Chen SF, Ruben RL, Dexter DL. Mechanism of action of the novel anticancer agent 6-fluoro-2-(2'-fluoro-1,1'-biphenyl-4-yl)-3-methyl-4-quinolinecarbo xylic acid sodium salt (NSC 368390): inhibition of de novo pyrimidine nucleotide biosynthesis. Cancer Res. 1986;46(10):5014–5019. [PubMed] [Google Scholar]
- 45.Peters GJ, Schwartsmann G, Nadal JC, Laurensse EJ, van Groeningen CJ, van der Vijgh WJ, Pinedo HM. In vivo inhibition of the pyrimidine de novo enzyme dihydroorotic acid dehydrogenase by brequinar sodium (DUP-785; NSC 368390) in mice and patients. Cancer Res. 1990;50(15):4644–4649. [PubMed] [Google Scholar]
- 46.Batt DG, Petraitis JJ, Sherk SR, Copeland RA, Dowling RL, Taylor TL, Jones EA, Magolda RL, Jaffee BB. Heteroatom-and carbon-linked biophenyl analogs of brequinar as immunosuppressive agents. Bioorg. and Med. Chem. Lett. 1998;8(13):1745–1750. doi: 10.1016/s0960-894x(98)00308-4. [DOI] [PubMed] [Google Scholar]
- 47.Batt DG, Copeland RA, Dowling RL, Gardner TL, Jones EA, Orwat MJ, Pinto DJ, Pitts WJ, Magolda RL, Jaffee BD. Immunosuppressive structure-activity relationships of Brequinar and related cinchoninic acid derivatives. Bioorg. Med. Chem. Lett. 1995;5(14):1549–1554. doi: 10.1016/s0960-894x(98)00010-9. [DOI] [PubMed] [Google Scholar]
- 48.Pitts WJ, Jetter JW, Pinto DJ, Orwat MJ, Batt DG, Sherk SR, Petraitis JJ, Jacobson IC, Copeland RA, Dowling RL, Jaffee BD, Gardner TL, Jones EA, Magolda RL. Structure-activity relationships of some tetracyclic heterocycles related to the immunosuppressive agent brequinar sodium. Bioorg. and Med. Chem. Lett. 1998;8(3):307–312. doi: 10.1016/s0960-894x(98)00010-9. [DOI] [PubMed] [Google Scholar]
- 49.Liu S, Neidhardt EA, Grossman TH, Ocain T, Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 2000;8(1):25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- 50.Malmquist NA, Baldwin J, Phillips MA. Detergent-dependent kinetics of truncated Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2007;282(17):12678–12686. doi: 10.1074/jbc.M609893200. [DOI] [PubMed] [Google Scholar]
- 51.Arakaki TL, Buckner FS, Gillespie JR, Malmquist NA, Phillips MA, Kalyuzhniy O, Luft JR, Detitta GT, Verlinde CL, Van Voorhis WC, Hol WG, Merritt EA. Characterization of Trypanosoma brucei dihydroorotate dehydrogenase as a possible drug target; structural, kinetic and RNAi studies. Mol. Microbiol. 2008;68(1):37–50. doi: 10.1111/j.1365-2958.2008.06131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagy M, Lacroute F, Thomas D. Divergent evolution of pyrimidine biosynthesis between anaerobic and aerobic yeasts. Proc. Natl. Acad. Sci. USA. 1992;89(19):8966–8970. doi: 10.1073/pnas.89.19.8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bjornberg O, Jordan DB, Palfey BA, Jensen KF. Dihydrooxonate is a substrate of dihydroorotate dehydrogenase (DHOD) providing evidence for involvement of cysteine and serine residues in base catalysis. Arch. Biochem. Biophys. 2001;391(2):286–294. doi: 10.1006/abbi.2001.2409. [DOI] [PubMed] [Google Scholar]
- 54.Bjornberg O, Rowland P, Larsen S, Jensen KF. Active site of dihydroorotate dehydrogenase A from Lactococcus lactis investigated by chemical modification and mutagenesis. Biochemistry. 1997;36(51):16197–16205. doi: 10.1021/bi971628y. [DOI] [PubMed] [Google Scholar]
- 55.Wolfe AE, Thymark M, Gattis SG, Fagan RL, Hu YC, Johansson E, Arent S, Larsen S, Palfey BA. Interaction of benzoate pyrimidine analogues with class 1A dihydroorotate dehydrogenase from Lactococcus lactis. Biochemistry. 2007;46(19):5741–5753. doi: 10.1021/bi7001554. [DOI] [PubMed] [Google Scholar]
- 56.Marcinkeviciene J, Tinney LM, Wang KH, Rogers MJ, Copeland RA. Dihydroorotate dehydrogenase B of Enterococcus faecalis. Characterization and insights into chemical mechanism. Biochemistry. 1999;38(40):13129–13137. doi: 10.1021/bi990674q. [DOI] [PubMed] [Google Scholar]
- 57.Rowland P, Norager S, Jensen KF, Larsen S. Structure of dihydroorotate dehydrogenase B: electron transfer between two flavin groups bridged by a iron-sulphur cluster. Structure. 2000;8(12):1227–1238. doi: 10.1016/s0969-2126(00)00530-x. [DOI] [PubMed] [Google Scholar]
- 58.Copeland RA, Davis JP, Dowling RL, Lombardo D, Murphy KB, Patterson TA. Recombinant human dihydroorotate dehydrogenase: expression, purification, and characterization of a catalytically functional truncated enzyme. Arch. of Biochem. Biophys. 1995;323(1):79–86. doi: 10.1006/abbi.1995.0012. [DOI] [PubMed] [Google Scholar]
- 59.Hurt DE, Widom J, Clardy J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta. Crystallogr. D Biol. Crystallogr. 2006;62(Pt 3):312–323. doi: 10.1107/S0907444905042642. [DOI] [PubMed] [Google Scholar]
- 60.Palfey BA, Bjornberg O, Jensen KF. Insight into the chemistry of flavin reduction and oxidation in Escherichia coli dihydroorotate dehydrogenase obtained by rapid reaction studies. Biochemistry. 2001;40(14):4381–4390. doi: 10.1021/bi0025666. [DOI] [PubMed] [Google Scholar]
- 61.Norager S, Jensen KF, Bjornberg O, Larsen SE. coli dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenases. Structure. 2002;10(9):1211–1223. doi: 10.1016/s0969-2126(02)00831-6. [DOI] [PubMed] [Google Scholar]
- 62.Baldwin J, Farajallah AM, Malmquist NA, Rathod PK, Phillips MA. Malarial dihydroorotate dehydrogenase: substrate and inhibitor specificity. J. Biol. Chem. 2002;277(44):41827–41834. doi: 10.1074/jbc.M206854200. [DOI] [PubMed] [Google Scholar]
- 63.Davies M, Heikkila T, McConkey GA, Fishwick CW, Parsons MR, Johnson AP. Structure-based design, synthesis, and characterization of inhibitors of human and Plasmodium falciparum dihydroorotate dehydrogenases. J. Med. Chem. 2009;52(9):2683–2693. doi: 10.1021/jm800963t. [DOI] [PubMed] [Google Scholar]
- 64.Baumgartner R, Walloschek M, Kralik M, Gotschlich A, Tasler S, Mies J, Leban J. Dual binding mode of a novel series of DHODH inhibitors. J. Med. Chem. 2006;49(4):1239–1247. doi: 10.1021/jm0506975. [DOI] [PubMed] [Google Scholar]
- 65.Walse B, Dufe VT, Svensson B, Fritzson I, Dahlberg L, Khairoullina A, Wellmar U, Al-Karadaghi S. The structures of human dihydroorotate dehydrogenase with and without inhibitor reveal conformational flexibility in the inhibitor and substrate binding sites. Biochemistry. 2008;47(34):8929–8936. doi: 10.1021/bi8003318. [DOI] [PubMed] [Google Scholar]
- 66.Deng X, Gujjar R, El Mazouni F, Kaminsky W, Malmquist NA, Goldsmith EJ, Rathod PK, Phillips MA. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 2009;284(39):26999–27009. doi: 10.1074/jbc.M109.028589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knecht W, Loffler M. Species-related inhibition of human and rat dihydroorotate dehydrogenase by immunosuppressive isoxazol and cinchoninic acid derivatives. Biochem. Pharmacol. 1998;56(9):1259–1264. doi: 10.1016/s0006-2952(98)00145-2. [DOI] [PubMed] [Google Scholar]
- 68.Chen SF, Perrella FW, Behrens DL, Papp LM. Inhibition of dihydroorotate dehydrogenase activity by brequinar sodium. Cancer Res. 1992;52(13):3521–3527. [PubMed] [Google Scholar]
- 69.Marcinkeviciene J, Rogers MJ, Kopcho L, Jiang W, Wang K, Murphy D, Lippy J, Link S, Chung TD, Hobbs F, Haque TS, Trainor GL, Slee A, Stern AM, Copeland RA. Selective inhibition of bacterial dihydroorotate dehydrogenases by thiadiazolidinediones. Biochem. Pharmacol. 2000;60(3):339–342. doi: 10.1016/s0006-2952(00)00348-8. [DOI] [PubMed] [Google Scholar]
- 70.Copeland RA, Marcinkeviciene J, Haque TS, Kopcho LM, Jiang W, Wang K, Ecret LD, Sizemore C, Amsler KA, Foster L, Tadesse S, Combs AP, Stern AM, Trainor GL, Slee A, Rogers MJ, Hobbs F. Helicobacter pylori-selective antibacterials based on inhibition of pyrimidine biosynthesis. J. Biol. Chem. 2000;275(43):33373–33378. doi: 10.1074/jbc.M004451200. [DOI] [PubMed] [Google Scholar]
- 71.Haque T, Tadesse S, Marcinkeviciene J, Rogers J, Sizemore C, Kopcho L, Amsler K, Ecret L, Zhan D, Hobbs F, Slee A, Trainor G, Stern A, Copeland RA, Combs A. Parallel synthesis of potent, pyrazole-based inhibitors of Helicobacter pylori dihydroorotate dehydrogenase. J. Med. Chem. 2002;45(21):4669–4678. doi: 10.1021/jm020112w. [DOI] [PubMed] [Google Scholar]
- 72.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2005;280(23):21847–21853. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 73.Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite, Plasmodium falciparum. J. Med. Chem. 2008;51(12):3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gujjar R, Marwaha A, El Mazouni F, White J, White KL, Creason S, Shackleford DM, Baldwin J, Charman WN, Buckner FS, Charman S, Rathod PK, Phillips MA. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 2009;52(7):1864–1872. doi: 10.1021/jm801343r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patel V, Booker M, Kramer M, Ross L, Celatka CA, Kennedy LM, Dvorin JD, Duraisingh MT, Sliz P, Wirth DF, Clardy J. Identification and characterization of small molecule inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2008;283(50):35078–35085. doi: 10.1074/jbc.M804990200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boa AN, Canavan SP, Hirst PR, Ramsey C, Stead AMW, McConkey GA. Synthesis of brequinar analogue inhibitors of malaria parasite dihydroorotate dehydrogenase. Bioorg. Med. Chem. 2005;13(6):1945–1967. doi: 10.1016/j.bmc.2005.01.017. …..0. [DOI] [PubMed] [Google Scholar]
- 77.Heikkilae T, Thirumalairajan S, Davies M, Parsons MR, McConkey AG, Fishwick CW, Johnson AP. The first de novo designed inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. Bioorg. Med. Chem. Lett. 2006;16(1):88–92. doi: 10.1016/j.bmcl.2005.09.045. [DOI] [PubMed] [Google Scholar]
- 78.Heikkila T, Ramsey C, Davies M, Galtier C, Stead AM, Johnson AP, Fishwick CW, Boa AN, McConkey GA. Design and synthesis of potent inhibitors of the malaria parasite dihydroorotate dehydrogenase. J. Med. Chem. 2007;50(2):186–191. doi: 10.1021/jm060687j. [DOI] [PubMed] [Google Scholar]
- 79.McLean JE, Neidhardt EA, Grossman TH, Hedstrom L. Multiple inhibitor analysis of Brequinar and Leflunomide binding sites on human dihydroorotate dehydrogenase. Biochemistry. 2001;40(7):2194–2200. doi: 10.1021/bi001810q. [DOI] [PubMed] [Google Scholar]
- 80.Malmquist NA, Gujjar R, Rathod PK, Phillips MA. Analysis of flavin oxidation and electron-transfer inhibition in Plasmodium falciparum dihydroorotate dehydrogenase. Biochemistry. 2008;47(8):2466–2475. doi: 10.1021/bi702218c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krungkrai J, Krungkrai SR, Supuran CT. Carbonic anhydrase inhibitors: inhibition of Plasmodium falciparum carbonic anhydrase with aromatic/heterocyclic sulfonamides-in vitro and in vivo studies. Bioorg. Med. Chem. Lett. 2008;18(20):5466–5471. doi: 10.1016/j.bmcl.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 82.Krungkrai SR, Wutipraditkul N, Krungkrai J. Dihydroorotase of human malarial parasite Plasmodium falciparum differs from host enzyme. Biochem. Biophys. Res. Commu. 2008;366(3):821–826. doi: 10.1016/j.bbrc.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 83.Meza-Avina ME, Wei L, Buhendwa MG, Poduch E, Bello AM, Pai EF, Kotra LP. Inhibition of orotidine 5'-monophosphate decarboxylase and its therapeutic potential. Mini Rev. Med. Chem. 2008;8(3):239–247. doi: 10.2174/138955708783744065. [DOI] [PubMed] [Google Scholar]
- 84.Krungkrai SR, DelFraino BJ, Smiley JA, Prapunwattana P, Mitamura T, Horii T, Krungkrai J. A novel enzyme complex of orotate phosphoribosyltransferase and orotidine 5'-monophosphate decarboxylase in human malaria parasite Plasmodium falciparum: physical association, kinetics, and inhibition characterization. Biochemistry. 2005;44(5):1643–1652. doi: 10.1021/bi048439h. [DOI] [PubMed] [Google Scholar]
- 85.Langley DB, Shojaei M, Chan C, Lok HC, Mackay JP, Traut TW, Guss JM, Christopherson RI. Structure and inhibition of orotidine 5'-monophosphate decarboxylase from Plasmodium falciparum. Biochemistry. 2008;47(12):3842–3854. doi: 10.1021/bi702390k. [DOI] [PubMed] [Google Scholar]
- 86.Bello AM, Poduch E, Liu Y, Wei L, Crandall I, Wang X, Dyanand C, Kain KC, Pai EF, Kotra LP. Structure-activity relationships of C6-uridine derivatives targeting plasmodia orotidine monophosphate decarboxylase. J. Med. Chem. 2008;51(3):439–448. doi: 10.1021/jm7010673. [DOI] [PubMed] [Google Scholar]
- 87.Zhang Y, Luo M, Schramm VL. Transition states of Plasmodium falciparum and human orotate phosphoribosyltransferases. J. Am. Chem. Soc. 2009;131(13):4685–4694. doi: 10.1021/ja808346y. [DOI] [PMC free article] [PubMed] [Google Scholar]