

Abstract
The dynamics of the tether linking methanethiosulfonate (MTSSL) spin probes to α-helices has been investigated, with the purpose of rationalizing its effects on ESR lineshapes. Torsional profiles for the chain bonds have been calculated ab initio, and steric interactions with the α-helix and the neighboring residues have been introduced at the excluded-volume level. As a consequence of the restrictions deriving from chain geometry and local constraints, a limited number of allowed conformers has been identified, which undergo torsional oscillations and conformational jumps. Torsional fluctuations are described as damped oscillations, while transition rates between conformers are calculated according to the Langer multidimensional extension of the Kramers theory. The time-scale and amplitude of the different motions are compared; the major role played by rotations of the outermost bonds of the side-chain emerges, along with the effects of substituents in the pyrroline ring on the conformer distribution and dynamics. The extent and symmetry of magnetic tensor averaging produced by the side-chain motions are estimated, the implications for the ESR spectra of spin labeled proteins are discussed, and suggestions for the introduction of realistic features of the spin probe dynamics into the lineshape simulation are presented.
I. INTRODUCTION
ESR spectroscopy of nitroxide spin probes introduced by site-directed mutagenesis (site directed spin labeling, SDSL) has become a valuable tool for the investigation of protein structure and dynamics.1–5 Spectra reflect mobility and chemical environment of the spin label; if, as shown in several cases, the mutation does not significantly perturb the system, information on the nitroxide label yields useful insights on the protein. However, given the variety of processes which can produce nitroxide reorientation, this information cannot be easily extracted from lineshapes. Namely, different motions, covering a wide range of amplitudes and different timescales, are simultaneously present in the system: they comprise overall protein tumbling and refolding processes, backbone fluctuations and side-chain isomerizations. Recognition of the role played by the spin label, even though it is not the objective of the investigation, is a prerequisite for extracting from experimental data useful information on structural and dynamical properties of proteins.
In this study we have performed a conformational analysis of the side-chain of a nitroxide spin label, with the purpose of characterizing the geometrical and motional parameters of the tether. In this way we intend to identify any general features, which can be helpful for the interpretation of ESR spectra of spin labeled proteins. We have focussed on the methanethiosulfonate (MTSSL) spin label, whose structure is shown in Figure 1. This is the most widely used spin probe, which has been successfully employed in a variety of studies; a few significant examples are represented by the structural rearrangements associated with the gating mechanisms of the KscA potassium channel6 and the mechanosensitive MscL channel,7 the structure of SNARE proteins involved in membrane fusion,8 the interaction of α-synuclein with membranes,9,10 structure and helix motions in the light-driven proton pump bacteriorhodopsin11 and in the photoreceptor rhodopsin,12,13 and reconstruction of the chemotaxis receptor-kinase assembly.14
Figure 1.

Structure of the spin labels considered in this study. They are obtained by reaction of the sulfhydryl group of a cysteine with l-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-(methyl)methanethiosulfonate (R1) and l-oxyl-2,2,4,5,5-pentamethyl-3-pyrroline-3-(methyl)methanethiosulfonate (R2) The five dihedral angles defining the chain conformation are shown.
We have examined the internal motions of MTSSL attached to a model α-helix. Torsional energy profiles have been obtained by quantum mechanical methods, with a proper account of the constraints imposed by the local environment. Knowledge of the potential energy is a requirement for modeling the internal dynamics; the features of the energy landscape allow the description of the system in terms of a limited number of rotamers, undergoing conformational jumps and librations about the minima of the side-chain torsional potential. A diffusive treatment of the dynamics is used which, although approximate, retains a realistic account of energetic and frictional features of the flexible tail.15–18 As a result of this analysis, the amplitude and time-scale of the chain motions can be estimated, and some general consideration on the effect of the tether dynamics on ESR spectra can be derived. The question might arise whether the results obtained from a model system can be extended to peptides or proteins, where the spin label is inserted into a given amino acid residue pattern. Actually, it has been shown that the main features of the ESR lineshape are preserved under mutation of residues in the proximity of the spin label, with minor differences deriving from the specific environment.19
An account of the dynamics of the spin probe beyond the simplifying assumptions used here could be obtained in the form of trajectories by MD or Brownian Dynamics simulations.20–25 A detailed picture of the local environment probed by the spin label and of the various processes modulating its orientations can be attained by such means, but at a computational cost which can be very great, due to the huge number of degrees of freedom. Conformational transitions in the side-chain occur on the nanosecond time-scale; therefore, even long trajectories can sample just a few conformations. A more effective sampling can be achieved e.g. by suitable Monte Carlo techniques,26,27 or by MD simulations under non-realistic conditions, like very high temperature; with these choices, however, information on the time dependence of the processes is lost. Also the use of MD trajectories with frozen protein degrees of freedom has been proposed for the study of the dynamics of the spin label.28 The present work should not be intended as an alternative to these simulation techniques, but rather as a complementary method. The study of the system under the simpler approach we use directly provides useful physical insights, and these can also yield suggestions for sampling the side-chain conformational space and extracting magnetically relevant parameters from trajectories. The results of this study can also be used to directly introduce the nitroxide dynamics in lineshape analysis: in a companion paper they will be exploited, in the framework of lineshape theory to interpret the ESR spectra of mutants of T4 lysozyme.29
The paper is organized in the following way. In the next Section (II), the model for the conformational dynamics of the nitroxide side-chain is presented. Then the results of the conformational study and the dynamical analysis are reported, and the averaging of magnetic tensors produced by side-chain motions is examined (Section III). In the final Section (IV) the results of our investigation are discussed, stressing their implication for the ESR spectra of spin labeled proteins, and the conclusions of this work are summarized.
II. CONFORMATIONAL DYNAMICS OF A NITROXIDE SIDE-CHAIN LINKED TO AN α-HELIX
A. Torsional potential and distribution
Under the assumption of fixed bond lengths and bond angles, the geometry of the MTSSL spin label, those structure is reported in Figure 1, is specified by the values of the five dihedral angles denoted as χ = (χ1,…, χ5). The potential energy of the nitroxide chain linked to an α-helix is expressed as
| (1) |
where V′ is the torsional potential of the isolated chain, while V″ accounts for interactions of the chain with its neighborhood in the protein, i. e. the α-helix backbone and the side-chains of the nearby residues. The former term is approximated as a sum of single bond contributions:
| (2) |
If the potential energy is characterized by minima separated by sufficiently high barriers, of at least a few kBT units, a simplified description can be adopted, in terms of a finite number of rotational isomers (rotamers),30 each corresponding to a given energy minimum. The geometry of the Jth conformer is defined by the five-dimensional vector , with being the value of the ith dihedral for this conformer. Namely, the torsional angle distribution
| (3) |
can be approximated as the sum:
| (4) |
with δχJ = χ − χJ and pJ(δχJ) being the torsional angle distribution for the Jth conformer:
| (5) |
The function V(δχJ) represents the torsional potential of the chain in the neighborhood of the Jth minimum, which can be approximated by a harmonic expansion:
| (6) |
where VJ is the potential energy of the chain in the conformation specified by the dihedral angles χ = χJ, and is the matrix of the second derivatives of the potential energy at the same point. Thus, the torsional distribution for the Jth conformer, eq 5, can be rewritten as:
| (7) |
where indicates the determinant of the matrix.
By integrating this multivariate Gaussian distribution over the δχJ variables, the probability of the Jth conformer can be defined:
| (8) |
where EJ is the free energy:31
| (9) |
B. Dynamics
In the presence of sufficiently high energy barriers between conformers, the chain dynamics can be described in terms of jumps between stable conformers and torsional oscillations in the minima of the potential.16 In view of the time-scale separation, the two motions can be treated separately. For long chains, simultaneous small rotations around several bonds can produce large amplitude displacements of the tail end, with characteristic times comparable to isomerization times.32 However couplings can be neglected for short chains, like that of MTSSL.18 Thus, conformational jumps can be simply described by the Master Equation:
| (10) |
where PJ(t) is the time dependent probability of the Jth conformer and −WJJ′, with J ≠ J′, represents the J ← J′ transition rate. If multiple transitions are neglected, in view of their high activation energy, the only non-vanishing matrix elements are those between conformers connected by single bond rotations. In the viscous regime characterizing chain motions in water solutions, where inertial effects are damped out, bond rotations can be described as diffusive processes, and transition rates can be expressed in terms of potential energy and friction opposing such rotations, according to the Kramers theory.33 For barrier crossing in a system with several degrees of freedom, the following expression is obtained:15,16,34
| (11) |
Here EJ′ is the free energy of the J′ conformer, defined in eq 9, and Es that at the saddle point, defined in an analogous way, with the matrix of the second derivatives of the potential energy at the saddle point. The parameter λ is the unique negative eigenvalue of the matrix , where Ds is 5×5 diffusion matrix calculated for the chain geometry at the saddle point. It is related to the friction matrix, ξs, by the Stokes-Einstein relation, Ds = kBTξs−1. It is worth emphasizing that transition rates calculated in this way take into account the coupling between reaction coordinate (the rotating dihedral) and non-reactive modes; barrier crossing occurs through cooperative small rotations of bonds adjacent that undergoing the conformational change, in such a way as to optimize frictional and energetic effects at barrier crossing. The diagonal terms of the transition matrix are obtained by the detailed balance condition, which guarantees the existence of a stationary solution, equal to the equilibrium distribution, eq 8:
| (12) |
It is convenient to work with the symmetrized form of the transition matrix, which will be denoted as W̃, and whose elements are defined as:
| (13) |
Within the harmonic approximation, damped oscillations within the potential well corresponding to the Jth conformer are described by the time dependent probability density pJ(δχJ,t), which evolves in time according to the multivariate Fokker-Planck-Smoluchowski equation:
| (14) |
where DJ = kBTξJ −1 is the diffusion matrix calculated for the geometry of the Jth conformer. This equation can be easily solved by a normal mode transformation:35,36
| (15) |
with δχJ = Uy and . Correlation functions for normal modes decay as simple exponentials, with time constants equal to the inverse of the eigenvalues :
| (16) |
C. Orientational distribution of the spin label
Amplitude and symmetry of the orientational distribution of the spin label produced by the side-chain dynamics can be quantified by the order parameters:
| (17) |
where are Wigner matrix elements, p(ΩM) is the orientational distribution function, and ΩM are the Euler angles defining the orientation of the spin label in a frame fixed on the Cα atom bringing the spin lable (the frame of the amino acid residue, according to the notation used in Figure 2). The non-vanishing order parameters depend on the symmetry of the orientational distribution of the spin probe; all order parameters vanish if orientations are isotropically sampled. In principle, Wigner matrices of any rank L could be needed; here we shall limit our consideration to second rank matrices, which are involved in the transformation of the magnetic tensors. Some general considerations on second rank order parameters are presented in the Appendix. In the following, the expressions used to calculate the order parameters of the spin label in the presence of torsional oscillations and conformational jumps are reported. For this purpose, it is convenient to introduce the coordinate transformations shown in Figure 2.
Figure 2.

Reference frames considered in this work.
LF: laboratory frame, with ZL || B0.
DF: Principal Axis System of the rotational diffusion tensor of the protein.
AF: amino acid residue frame, with the origin on the Cα bringing the nitroxide side-chain, the zA-axis perpendicular to the plane of the N-Cα-CO atoms and the xA-axis on the plane containing the zA-axis and Cα-Cβ bond.
Li: local frame, with zLi || i-th chain bond and xLi in the plane of the preceding chain bond when χi−1 = 0°.
MF: magnetic frame, with the origin on the nitroxide N nucleus, the zM-axis along the N-pz orbital and the xM-axis parallel to the N-O bond.
SF: ordering frame, with the axes parallel to the principal ordering axes.
Average values of Wigner rotation matrices for the Jth conformer are obtained by integrating over the torsional distribution eq 7. Under the assumption of independent bond contributions, this can be expressed as the product of the single bond probabilities:
| (18) |
with being the second derivative of the chain potential energy with respect to the χi variable, calculated for . Then, the addition theorem for Wigner matrices38 can be exploited, to decompose the AF → MF rotation into a set of local transformations, and the order parameters for the Jth conformers can be expressed as:
| (19) |
Here the Euler angles and appear, which describe the local transformations Li → Li+1, with the chain in the Jth conformation (see Fig. 2). The single bond averages have the form:
| (20) |
with
| (21) |
where is the mean square amplitude of the single bond torsional distribution:
| (22) |
For a set of conformers which, in addition to torsional oscillations undergo conformational jumps, the order parameters can be obtained as
| (23) |
with the average Wigner matrix elements calculated according to eq 21, and the sum extended to all interconverting conformers.
The magnetic tensors partially averaged by chain motions can be expressed in terms of these order parameters:
| (24) |
where μ = g or μ = A, for the g and the A hyperfine tensor, respectively.
III. RESULTS: NITROXIDE CHAIN MOTIONS
A. Conformational analysis
The single bond contributions to the torsional potential, V(i)(χi), for the MTSSL chain have been obtained considering the subsystems which are shown in Figure 3. Ab initio calculations in a vacuum have been performed, at the HF/6-31G** level (ROHF/6-31G** for the subsystem containing the nitroxide).39 The torsional potential for a given bond was obtained by relaxed scan in the range −180° ÷ +180°. Figure 3 shows the torsional profiles as a function of the dihedral angles. The following convention is used: a right-handed rotation, with the rotation axis directed toward the chain end, is taken as positive. Using the notation adopted for alkyl chains, minima close to 180° will be designed as t states, whereas the labels g± will be used for minima characterized by dihedrals ranging between ±40° and ±120°.30
Figure 3.

Torsional potentials and model subsystems considered for calculating them. Arrows indicate the rotating bonds, with the convention used to define the sense of rotation: a right-handed rotation, with the rotation axis directed toward the chain end, is taken as positive. For χ1 the value χ1 = 0° corresponds to the eclipsed configuration with the Cβ-Sγ bond over the Cα-N bond. For χ5 the value χ5= 0° corresponds to the eclipsed configuration with the C-CH double bond of the pyrroline ring over the CH2-Sδ bond. The dashed circle shows atoms that have been kept frozen in the α-helix configuration in calculations for χ1 Arrows in the torsional profiles show transitions between the allowed conformers, which are listed in Table 2.
For the χ1 dihedral, rotation of the-CH2-S-SH group, linked at the Cα atom of a CH3-NH-CαH-CO-CH3 fragment frozen in the standard α-helix geometry, has been examined. The torsional potential shown in Figure 3-A was obtained taking the Cα-Cβ-Sγ-Sδ dihedral in the t conformation and the Cβ-Sγ-Sδ H dihedral close to +90°; analogous profiles were found with the former in g± states and/or the latter close to −90°. The features of the torsional potential are in accordance with the results of the conformational analysis of the cysteine sulfhydryl in proteins.40,41 There are two roughly equivalent minima corresponding to staggered configurations with the N-Cα-Cβ-Sγ dihedral equal to 180° in one (Cβ-Sγ bond between the Cα-CO and the Cα-H bonds) and to +65° in the other (Cβ-Sγ bond between the Cα-NH and the Cα-CO bonds); a lower minimum is found for a dihedral equal to −60° (Cβ-Sγ bond between the Cα-H and the Cα-NH bonds). The minima are separated by high barriers, corresponding to eclipsed configurations; a significantly lower barrier is obtained for the configuration with the Cβ-Sγ bond over the Cα-H bond.
The rotation of the central bond of the CH3-CH2-S-SH model system has been considered to calculate the torsional profile for the χ2 and χ4 dihedral angles (Figure 3-B). The resulting profile is similar in shape to that for the central C-C bond of n-butane; it is symmetric with respect to χ2(χ4) = 0°, with the absolute minimum at 180° (t), and two equivalent relative minima at ±75° (g±). Barriers of the order of a few kBT units at room temperature separate t and g± minima.
The CH3-S-S-CH3 fragment has been studied to obtain the V(3)(χ3) torsional potential (Figure 3-C). Only two minima are found, for values of the CH3-S-S-CH3 dihedral equal to ±90°, separated by high barriers.
Finally, the torsional potential for the χ5 dihedral has been calculated as a function of the angle about the bond connecting the pyrroline ring to the tether, in the system CH3-S-S-CH2-sl, with sl= l-oxy-2,2,5,5-tetramethylpyrroline (R1 spin probe) and sl= l-oxy-2,2,4,5,5-pentamethylpyrroline (R2 spin probe). Calculations for R1 have been performed with the S-S-CH2-C dihedral angle constrained either in the t or in the g state. In the former case (Figure 3-D), configurations with the S-CH2-C-CH dihedral angle significantly larger than ~ +120° or smaller than ~ −120° have very high energy. For values falling within the range of ±120° the energy profile becomes rather flat, with two minima at χ5 ~ ±77°, separated by a shallow barrier, whose size and shape is very sensitive to the level of calculation. On the contrary, when the S-S-CH2-C dihedral is in the g± state, the torsional profile for S-CH2-C-CH has two minima, at ±10° and ∓100°, with a barrier of a few kBT at room temperature (Figure 3-E). In the case of the R2 spin probe, the presence of the methyl substituent at the 4-position in the pyrroline ring prevents the possibility of configurations with the S-S-CH2-C dihedral in g states. When this angle is in the t state, two minima are found, with χ5 = ±85°, separated by very high barriers (Figure 3-F).
Values of the dihedral angles at the minima of the torsional potentials are collected in Table 1. For the χ1 to χ4 dihedrals, the minima are separated by high barriers, which range from about 4kBT for the g ⇌ t transitions of χ2 or χ4 to more than 10 kBT for the −90° ⇌ +90° transitions of χ3 Analogous considerations hold for the χ5 dihedral of R2, and also for that of R1 if χ4 is in the g state. It follows that in all these cases the nitroxide side-chain can be described in terms of a finite number of stable rotamers.30 From equation 22, we can estimate root mean square fluctuations of about 12.5° for the χ2 and χ4 dihedrals, and 8.5° for χ1 and χ3, at T=298 K. An increase of about 5% can be estimated under a 10° increase in temperature. A root mean square amplitude of 12.5° is appropriate also for the χ5 dihedral of R2, and for that of R1 when χ4 is in the g state (χ5|χ4=g). The situation for the χ5 dihedral of R1 can be quite different when χ4 is in the t state (χ5|χ4=t); from the torsional potential shown in Figure 3-D, a wider distribution of χ5 values about the minima is inferred, and the simple description in terms of a few conformational states may not be fully satisfactory.
TABLE I.
Values of the dihedral angles at the minima of the torsional potentials V(i)(χi).
| χ1 | −60° (g−), +65° (g+), 180° (t) |
| χ2 ≡ χ4 | −75° (g−), +75° (g+), 180° (t) |
| χ3 | −90° (g−), +90° (g+) |
| χ5a | −77° (g−), +77° (g+) |
| χ5b | ∓ 8° (g−), ±100° (g+) |
χ4 in the t state
χ4 in the g± state
Taking the minima of the single bond torsional potentials, a total number of 108 and 54 conformers can be estimated for R1 and R2, respectively. This number is reduced by interactions of the spin label with its environment, i.e. backbone and side-chains of nearby residues. We have introduced such effects, described by the V″ contribution in eq 1, at the level of steric interactions, which have been modeled in terms of excluded volume. For this purpose, each of the possible conformers of the nitroxide side-chain was inserted in a poly-Ala α-helix, and was excluded if the distance between any pair of atoms was shorter than the sum of their van der Waals radii. The following values have been used: rC = 1.5 Å, rO = 1.35 Å, rN = 1.4 Å, rS = 1.8 Å. It is worth mentioning that the choice of these values is not critical for the determination of the excluded conformers, since these do suffer severe hindrance; as an example, Figure 4 shows a forbidden conformer of R1. After the steric control, the number of allowed conformers is reduced from 108 to 18 for R1 and from 54 to 10 for R2; values of the dihedral angles defining their geometry are listed in Table 2. The conformers possible for R2 are denoted by an asterisk in the Table. The geometry of the allowed conformers is displayed in Figure 5; here a compact representation is adopted, with each structure corresponding to a set of conformers connected by χ4 and χ5 jumps. Inspection of table 2 shows that some single bond conformational states, predicted for the isolated chain, are forbidden by the α-helix environment. This is the case, for instance, of the g+ state, which according to Figure 3-A should be as populated as the t state for the χ1 dihedral, but never appears in Table 2. This very low probability of the g+ state of the N-Cα-Cβ-Sγ dihedral in α-helices is confirmed by the analysis of the conformers of cysteine, as shown e.g. in ref.43.
Figure 4.
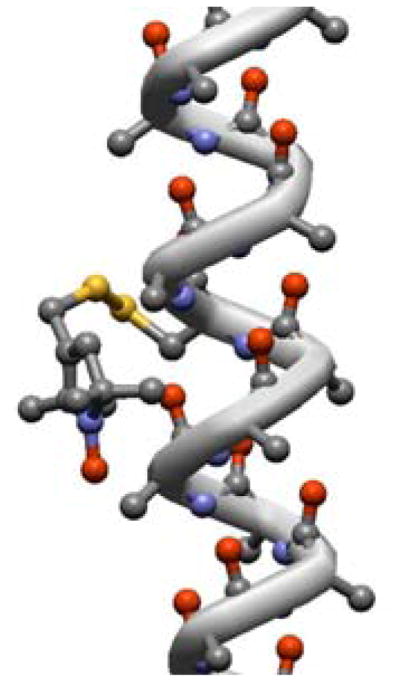
Poly-Ala α-helix with the Rl spin-label in a sterically forbidden conformation (χ1 = −60°, χ2 = −75°, χ3 = −90°, χ4 = +75°).
TABLE II.
Torsional angles and probabilities for the sterically allowed conformers of the nitroxide side-chain of the R1 spin label, at a site located in the middle of a poly-Ala helix.
| conformer | χ1(°) | χ2(°) | χ3(°) | χ4(°) | χ5(°) | PJ |
|---|---|---|---|---|---|---|
| C1* | −60 | −75 | −90 | 180 | +77 (+85) | 0.143 (0.201) |
| C2* | −60 | −75 | −90 | 180 | −77 (−85) | 0.143 (0.201) |
| C3 | −60 | −75 | −90 | −75 | 100 | 0.045 |
| C4 | −60 | −75 | −90 | −75 | −8 | 0.074 |
| C5* | −60 | 180 | 90 | 180 | +77 (+85) | 0.187 (0.263) |
| C6* | −60 | 180 | 90 | 180 | −77 (−85) | 0.187 (0.263) |
| C7 | −60 | 180 | 90 | +75 | −100 | 0.059 |
| C8 | −60 | 180 | 90 | +75 | +8 | 0.098 |
| C9* | 180 | 180 | −90 | 180 | +77 (+85) | 0.009 (0.013) |
| C10* | 180 | 180 | −90 | 180 | −77 (−85) | 0.009 (0.013) |
| C11 | 180 | 180 | −90 | −75 | +100 | 0.003 |
| C12 | 180 | 180 | −90 | −75 | −8 | 0.005 |
| C13* | 180 | 180 | 90 | 180 | +77 (+85) | 0.009 (0.013) |
| C14* | 180 | 180 | 90 | 180 | −77 (−85) | 0.009 (0.013) |
| C15* | 180 | 75 | 90 | 180 | +77 (+85) | 0.007 (0.010) |
| C16* | 180 | 75 | 90 | 180 | −77 (−85) | 0.007 (0.010) |
| C17 | 180 | 75 | 90 | +75 | −100 | 0.002 |
| C18 | 180 | 75 | 90 | +75 | +8 | 0.004 |
Statistical weights were calculated at T=298 K on the basis of the single bond torsional potential, V′ in eq 1. Conformers allowed for the R2 spin probe are denoted by an asterisk; probability and χ5 values for these are reported in parentheses.
Figure 5.

Geometry of the allowed conformers of the Rl spin-label listed in Table 2. Each figure corresponds to a set of interconverting conformers. Only half of the conformers are shown, which correspond to one of the two possible χ5 states compatible with a given χ4 state. Conformers connected by a χ4 rotation are superimposed on the same structure. To avoid crowding, the methyl substituents in the pyrroline ring have been eliminated. The ball and stick representation is used for the side-chain, while simple sticks indicate backbone bonds. The black line shows the orientation of the Y ordering axis; the Z ordering axis is perpendicular to the plane of the figure.
Table 2 also reports the conformer probabilities, PJ, calculated at T=298 K according to eq 8. Root mean square fluctuations were assumed for conformers of R1 with χ4 in the t state. We can see that the overall weight of conformers with χ1 in the g− state greatly exceeds that of conformers having χ1 in the t state. Of course, the probabilities reported in Table 2 should be considered as only a first order estimate; indeed, the primary result of our analysis is the identification of a restricted number of allowed conformers and of their geometry. The actual probabilities for MTSSL at a given helical site could be significantly affected by local effects, depending on the nature of the nearby residues and/or the presence of attractive interactions involving the spin label, which are not taken into account in our present model. In the companion paper,29 where the effects of the chain dynamics on the ESR spectra are investigated, it is shown that the presence of conformers with different mobility do yield the appropriate spectral contributions, but with weights that are different form those predicted by the calculated probabilities.
The geometry we have predicted for the conformers of R1 is in general agreement with the values determined from X-ray structures of four spin labeled T4L mutants.41,42 The g−g−, g−t and tg+ configurations for χ1χ2, which were found in the crystal structures, also appear in Table 2. For the latter we predict a very low probability; actually, the interaction with a nearby residue was suggested as the reason for its stabilization in the crystal. No experimental data confirm the tt configuration, which from our analysis is sterically allowed, although with very low weight.
B. Dynamics
We start by considering the R1 spin label; table 3 reports the elements of the symmetrized transition matrix W̃, calculated according to eq 13, in water solution at T=298 K. Given the 18 conformers listed in Table 2, the possible single bond transitions are those indicated by arrows in Figure 3. Our simple picture of conformational jumps is not strictly valid in the absence of a sufficient high barrier, as in the case of the χ5|χ4=t dihedral, in which case the whole torsional distribution should be taken into account. On the other hand, a detailed description is prevented by the degree of uncertainty affecting the shape of this torsional profile which, as already mentioned, also depends upon the computational method; moreover the possibility of high barriers can be hypothesized, due to interactions involving the pyrroline ring.42 Therefore, we have adopted also for χ5 the description in terms of jumps between the minima of the torsional potential which, in addition to the advantage of simplicity, allows us to obtain quantitative results without the need of a detailed knowledge of the torsional potential, as we shall see below.
TABLE III.
Off-diagonal elements of the symmetrized transition matrix W̃ calculated for water solution at T=298 K. The matrix elements are reported, in units of 109 s−1. Rates higher than 108 s−1 are shown in boldface. Under each transition rate, the dihedral angle involved in the conformational jump is reported in parenthesis.
| C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 | C9 | C10 | C11 | C12 | C13 | C14 | C15 | C16 | C17 | C18 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | > 10 (χ5) | 1.5 (χ4) | ||||||||||||||||
| C2 | > 10 (χ5) | |||||||||||||||||
| C3 | 1.5 (χ4) | 1.5 (χ5) | ||||||||||||||||
| C4 | 1.5 (χ5) | |||||||||||||||||
| C5 | > 10 (χ5) | 0.01 (χ1) | ||||||||||||||||
| C6 | > 10 (χ5) | 0.36 (χ4) | 0.01 (χ1) | |||||||||||||||
| C7 | 0.36 (χ4) | 1.5 (χ5) | ||||||||||||||||
| C8 | 1.5 (χ5) | |||||||||||||||||
| C9 | > 10 (χ5) | 0.36 (χ4) | 0.002 (χ3) | |||||||||||||||
| C10 | > 10 (χ5) | 0.002 (χ3) | ||||||||||||||||
| C11 | 0.36 (χ4) | 1.5 (χ5) | ||||||||||||||||
| C12 | 1.5 (χ5) | |||||||||||||||||
| C13 | 0.01 (χ1) | 0.002 (χ3) | > 10 (χ5) | 0.05 (χ2) | ||||||||||||||
| C14 | 0.01 (χ1) | 0.002 (χ3) | > 10 (χ5) | 0.05 (χ2) | ||||||||||||||
| C15 | 0.05 (χ2) | > 10 (χ5) | ||||||||||||||||
| C16 | 0.05 (χ2) | > 10 (χ5) | 1.5 (χ4) | |||||||||||||||
| C17 | 1.5 (χ4) | 1.5 (χ5) | ||||||||||||||||
| C18 | 1.5 (χ5) |
According to eq 11, calculation of the transition rates requires the evaluation of the friction matrix accounting for the viscous torques opposing bond rotations.15 For a given transition, the 5×5 friction matrix is evaluated for the saddle point geometry of the side-chain.44 A united atom representation has been used: the nitroxide side-chain is described as a collection of spheres, corresponding to the N, O, S, C atoms and the CH, CH3, CH2 groups. For the sake of simplicity, identical spheres of hydrodynamic radius equal to 1 Å have been taken. With these choices, a value of 1010 s−1 is predicted for the g → t transition of n-butane at room temperature.15
It appears clearly from Table 3 that a limited number of single bond jumps is possible. To understand the results reported in the Table, it is worth reminding the reader that the transition rate between a pair of conformers is determined by energetic and frictional effects, i.e. the height of the barrier which has to be crossed and the friction opposing the chain motion in the solution environment. Rotation about a given bond can be slower or faster, depending on the chain geometry, which affects the friction opposing such rotations and the degree of bond cooperativity in the barrier crossing. We can see that interconversions involving the χ3 and χ1 dihedrals are predicted to be very slow, by virtue of the high barriers and the large friction accompanying the displacements of bulky chain portions. Higher rates are obtained for χ2 and χ4 rotations, since lower barriers have to be crossed. The latter are predicted to be about one order of magnitude faster than the former, and this can be explained considering that they produce displacement of a smaller chain portion, therefore they are characterized by lower friction. In view of the arguments presented above, only a lower bound for χ5|χ4=t transitions is reported, which would correspond to a barrier comparable to that for χ5|χ4=g transitions. Anyway, as a consequence of the relatively low friction, high rates are predicted for χ5 jumps.
The χ4 and χ5 dynamics occur on the time-scale of standard X-band ESR; therefore it should have appreciable effects on lineshapes. The χ2 and χ3 transitions are expected to be much less effective, not only because they are slower, but mainly because the large amplitude chain displacements they would produce are likely to be sterically hindered in the α-helix environment. If such transitions are ignored, the 18 conformers of R1 can be grouped into four independent blocks, each containing four rotamers connected by χ4 and χ5 transitions: (C1 to C4), (C5 to C8), (C9 to C12), (C15 to C18), in addition to a block of a pair of conformers, interconverting only through x5 jumps, (C13 and C14).
If the same considerations are applied to the R2 spin label, 10 non-interconverting conformers are found, all having χ4 in the t state and the pyrroline ring perpendicular to the SδCC plane, i.e. χ5 equal to ±85°. The characteristic frequencies of such motions, calculated according to eq. 14, range from 109 s−1 to 1011 s−1 and, given the small amplitude of motions, are expected to have scarce effects on ESR lineshapes.
C. Order parameters and partially averaged magnetic tensors
Order parameters for the spin label orientational distribution and magnetic tensors partially averaged by the chain motions of the spin labels R1 and R2 are reported in Tables 4 and 5, respectively. The cartesian representation is used; the relationships between irreducible spherical and cartesian representation are reviewed in the Appendix, together with some general definitions. Table 4 reports the order parameters calculated for a few representative cases. Capital letters are used for the reference axes of the so called ordering frame (SF), whereas small letters are used for the molecular axes, which correspond to the coordinate axes of the magnetic frame; z = zM is along the N-pz orbital and x=xM is parallel to the N-O bond. Using liquid crystal terminology, the X, Y, Z axes can be denoted as 'directors'. So , with K = X, Y, Z, gives the degree of alignment of the zM-axis, parallel to the N-pz orbital, to the K-director. The order parameter, with i = x, y, z, accounts for the alignment of the ith magnetic axis with respect to the Z-director. According to the usual convention, in the ordering frame we shall denote as Z the axis with the highest order parameter, i.e. the axis to which zM preferentially aligns. If the orientational distribution of the zM-axis. produced by chain dynamics, has axial symmetry, the relation holds, and a single director can be considered, i.e. the Z-director, parallel to the C∞ axis. For a perfectly rigid chain the result and would be obtained, i.e. perfect order, with the Z axis parallel to the N-pz orbital.
TABLE IV.
Order parameters for the orientational distribution of MTSSL produced by the side-chain motions, calculated for selected conformers at T=298 K, under different conditions.
| Conformer | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C13 | −0.44 | −0.37 | 0.81 | −0.44 | −0.38 | 0.02 | −0.01 | 0.01 | ||||||||
| C13–C14 | −0.44 | −0.24 | 0.67 | −0.37 | −0.30 | 0.09 | −0.01 | −0.01 (a) | ||||||||
| C13–C14 | −0.44 | 0.03 | 0.47 | −0.30 | −0.17 | 0.19 | −0.01 | 0.01 (b) | ||||||||
| C1–C4 | −0.28 | −0.22 | 0.51 | −0.25 | −0.26 | 0.14 | −0.05 | 0.02 (a) | ||||||||
| C1–C4 | −0.23 | −0.12 | 0.35 | −0.20 | −0.15 | 0.22 | −0.05 | 0.01 (b) |
The X, Y, Z axes are the so called directors. For C13 only torsional oscillations are assumed, while for C13-C14 and C1 to C4 conformational jumps are also taken into account, as explained in the text. Labels denote different choices for the χ5|χ4=t distribution.
Gaussian distribution of root mean square amplitude ;
distribution in the range 0° to ±120° under the torsional potential shown in Figure 3-D.
TABLE V.
Magnetic tensors partially averaged by side-chain motions, calculated for selected conformers of MTSSL at T=298 K, under different conditions (the same as in Table 4).
| Conformers | gXX | gYY | gZZ | AXX | AYY | AZZ | AXY | AXZ | AYZ |
|---|---|---|---|---|---|---|---|---|---|
| C13 | 2.00767 | 2.00561 | 2.00275 | 7.74 | 8.46 | 32.00 | 0.47 | −0.06 | −0.10 |
| C13–C14 | 2.00708 | 2.00580 | 2.00315 | 7.56 | 11.32 | 29.32 | −0.12 | −1.14 | −0.02 (a) |
| C13–C14 | 2.00710 | 2.00521 | 2.00372 | 7.71 | 15.35 | 25.14 | −0.03 | −1.91 | −0.04 (b) |
| C1–C4 | 2.00628 | 2.00603 | 2.00372 | 11.77 | 10.43 | 26.00 | −0.004 | −1.21 | 0.59 (a) |
| C1–C4 | 2.00632 | 2.00557 | 2.00414 | 11.86 | 13.60 | 22.75 | −0.59 | −1.65 | 0.58 (b) |
Both tensors are expressed in the Principal Axis System of g. The following values before averaging have been assumed: gxx=2.00803, gyy=2.00582, gzz =2.00218, Axx=6.42, G Ayy=5.95 G, Azz=35.83 G.47
The first line of Table 4 reports the order parameters calculated for the C13 conformer experiencing only torsional oscillations; for the χ5 dihedral, fluctuations with root mean square amplitude of 12.5° have been assumed, as in the case of the R2 spin label. The orientation of the Z and Y directors is similar to that shown in Figure 5 for the C13–C14 pair. In view of the strong confinement of motions, the order parameters are very high, with some biaxiality, i.e. the degree of alignment of the N-pz orbital in the plane perpendicular to the Z director is anisotropic, with a preference of this orbital to lie on the Y Z plane.45
The effect of χ5 jumps is illustrated by the results reported in Table 4 for the C13–C14 pair. In this example, all dihedrals experience torsional oscillations; in addition, conformational jumps are possible for χ5. Two different choices for the χ5|χ4=t distribution have been considered. In case (a) a Gaussian distribution centered in the minima of the single bond torsional potential is assumed, with root mean square amplitude ; this value is estimated from the curvature in the minima of the torsional potential, Figure 3-D. In case (b) wider oscillations are allowed: for each minimum the full distribution in the range 0° to ±120°, under the torsional potential shown in Figure 3-D, is taken into account; analogous results would be obtained with a Gaussian distribution of root mean square amplitude to 40°. The orientation of the directors, which is imposed by the chain geometry, is shown in Figure 5; it is similar to that obtained for C13 in the absence of χ5 jumps, with the Z axis not far from parallel to the Sδ-C bond. We can see from Table 4 that the presence of extended χ5 motions has a twofold effect. One is that it reduces the orientational order, and the decrease is greater as the amplitude of fluctuations is greater about the minima of the torsional potential. On the other side, it increases the biaxiality of the distribution: the N-pz orbital is more and more confined on the Y Z plane, i.e. perpendicular to the Sδ-C bond. Considering now the values, we can see that also the difference in alignment of the xM and yM axes to the Z-director increases, with increasing amplitude of χ5 rotations. Moreover, the xM and yM axes no longer correspond to principal alignment directions in the magnetic frame.
The two bottom lines of Table 4 report the order parameters calculated for the CI to C4 set of interconverting conformers. Again, torsional fluctuations occur for all dihedrals, with the additional possibility of χ4 and χ5 conformational jumps. For the χ5|χ4=t distribution, the same two choices denoted above as (a) and (b) have been assumed. Comparison with the results obtained for the C13–C14 pair shows that the occurrence of χ4 jumps has the effect of reducing not only the degree of order, but also the biaxiality in the distribution of both the ordering and magnetic axes. The orientation of the directors can be inferred from Figure 5; the Z and Y axes are roughly parallel to the Sδ-C bond, and the C-Sγ bond, respectively.
The restricted chain motions are not very effective in averaging out the magnetic tensors of the spin probe, as shown in Table 5, which reports the components of the partially averaged g and hyperfine A tensors, both expressed in the PAS of the former. In all cases, the partially averaged A tensor is not far from diagonal in the principal frame of the partially averaged g tensor, and the axes of this frame are close to the X, Y, Z directors. Again a clear dependence on the nature and amplitude of the side chain motions is found. Even conformational jumps, although they are more effective than torsional librations, are not able to fully average out the magnetic anisotropics. Interestingly, as a consequence of the lack of any symmetry in the distribution of the N-pz orbital, the partially averaged hyperfine tensor loses its quasi axial symmetry.
The considerations presented here for C13, for the C13–C14 pair and for the CI to C4 block could be extended to other isolated and interconverting conformers, respectively. Due to the change in the chain geometry, the orientation of the ordering frame (SF) in the molecular frame (AF) depends on the conformer, but both order parameters and partially averaged tensors are similar for all isolated conformers on one side, and for all blocks of interconverting conformers on the other side.
In the context of ESR lineshape analysis, order parameters or coefficients related to them are often introduced to specify the motional restrictions experienced by the spin probe, and in general some simplifying assumptions are made. In all cases axial symmetry of the orientational distribution of zM is assumed. At the lowest level, e.g. in the model of diffusion in a cone, also the equivalence of the xM and yM axes is assumed, and the single order parameter is used. The additional order parameter . is introduced by more general approaches, which however make the simplifying assumptions that the principal alignment axes of the magnetic frame coincide with the xM, yM, zm axes, and that any biaxiality of the zM distribution may be neglected.46–49 The inadequacy of such assumptions to account for the orientational distribution of MTSSL spin probes emerges from our study; analogous conclusions were reached by Budil and coworkers, on the basis of the analysis of MD trajectories.25 We speculate that the features of the nitroxide distribution can influence the ESR lineshapes, especially at high frequency. An accurate lineshape analysis is beyond our present purpose; however, model calculations can be useful to illustrate this point. The ESR spectrum of a spin label in a protein results from the superposition of a variety of motions; for the sake of simplicity, we have assumed the fast motional approximation for the side-chain dynamics, and other motions are simply modeled as isotropic rotational diffusion with an effective correlation time. Thus the ESR spectrum can be calculated by solving the SLE,49 for a rotating body with partially averaged magnetic tensors. Figure 6 shows a series of ESR spectra calculated for the C13 conformer of the R2 spin label at increasing frequencies, with and without account of the biaxiality of order. An isotropic diffusion coefficient has been used, D0 = 108s−1. The partially averaged tensors reported Table 5 have been taken for C13; for the axial case, tensors with identical X and Y components, equal to the average of the values reported in the Table, have been assumed. It clearly appears that, while the 9GHz spectra obtained in the two cases are hardly distinguishable, the differences between spectra become significant at higher frequency.
Figure 6.

ESR spectra at different frequencies calculated for the R2 spin probe with partially averaged magnetic tensors, undergoing overall rotational diffusion with an isotropic diffusion coefficient D0 = 108s−1. Top: the partially averaged tensors reported in Table 5 for C13 have been used. Bottom: axial partially averaged tensors have been used, which have been obtained from the values reported in Table 5 for C13, taking indentical X and Y components, equal to the mean of the values X and Y values appearing in the Table. The examples correspond to 9.4, 95, 170 and 250 GHz from left to right.
IV. DISCUSSION AND CONCLUSIONS
A conformational analysis has been performed for the flexible chain of the MTSSL spin label at a solvent exposed α-helix site. Knowledge of the chain energetics and evaluation of steric constraints, along with a realistic account of the friction opposing bond rotations in a viscous environment, has led to quantitative insight on amplitude and rates of the side-chain motions. A small number of sterically allowed conformers has been identified; and the isomerization rates, provided by the kinetic analysis, support the experimental observation of a rather stiff nitroxide tether.46,48,50 Chain motions are described as jumps between stable conformers and librations about the minima of the chain potential energy. Rotations around the χ1 to χ3 bonds are very unlikely, due to the presence of high torsional barriers and/or steric constraints. So, chain motions relevant for ESR relaxation can be taken as and torsional oscillations of all bonds and χ4 and χ5 conformational transitions. The former are characterized by small amplitudes, of the order of 10° for each bond; for the χ5 dihedral wider fluctuations are possible, with their exact features probably depending on the local environment of the spin probe. Thus, the results of our analysis support the so-called ‘χ4, χ5’ model, which has been suggested by a number of experimental studies46,48,51 and by MD simulations.24,25
In our approach, we have focussed on a simple model, that can be worked out in detail, and it offers the advantage of an easier interpretation of the determining factors of the conformational dynamics. Our description of the conformational dynamics of the side-chain, although approximate, contains substantial realistic features, and it has led to some rather general results on the geometry and kinetics of such motions. More detailed descriptions can be carried out by MD20–25,28 or Monte Carlo simulations.26,27 These techniques can provide a picture of a spin label in its environment in a given protein, so in principle it should be possible to extract from trajectories the information needed to relate ESR lineshape and structural or dynamic characteristics of the different labeling sites. However, our analysis allowed us to single out some key features, which should be considered in applying these techniques for the interpretation of ESR experiments. Since the relevant motions fall in the nanosecond time-scale, there is the obvious requirement that trajectories should not be shorter than tens of nanoseconds. A less obvious caveat derives from the subtle features of the averaging of magnetic tensors produced by tether motions; in particular our investigation highlights the importance of the pattern of substituents in the pyrroline ring and that of the characteristics of the χ5 torsional potential. Therefore, simulations adopting simplified labels cannot provide adequate information which is useful for the analysis of ESR spectra;23 moreover, the parameters appropriate for the spin label, which are not contained in the available force fields, need to be accurately assessed.
The results of our analysis can be exploited to introduce the features of the spin label structure and dynamics into the framework of a lineshape theory. Such a study is presented in a companion paper.29 However, here we simply summarize the implication of the results of our investigation for the interpretation and the simulation of ESR spectra. We have found that, in general, the simultaneous presence of different conformers, undergoing different motions, has to be taken into account. Conformational transitions for the R1 spin label have characteristic times of the order of 1 ns or slower for a protein in water; only χ4 and χ5 jumps are fast on the time-scale of X-band ESR experiments, whereas at higher frequencies the shorter experimental time-scales render the fast motional limit as questionable, even for such motions. Torsional oscillations occur over time-scales shorter than about a nanosecond, and, especially in view of their small amplitude, they are likely to fall in the fast motional regime, even at higher frequency.
We have seen that chain motions produce a limited extent of averaging of magnetic tensors, especially when only torsional oscillations are possible. An anisotropic orientational distribution of the N-pz orbital has been obtained, which is not surprising for the five-bond side-chain of MTSSL, under the further constraints deriving from the presence of the α-helix backbone and the tails of the nearby residues. Namely, in the case of an alkyl chain it is well known that, due to the geometrical bond constraints, conformational motions cannot produce an isotropic distribution of the terminal end, unless the chain is very long.18,52 The presence of three ordering axes, characterized by different degrees of order, emerges from the analysis, in contrast to the common assumption of axial symmetry of the orientational distribution in the protein diffusion frame, e.g. in the popular model of diffusion in a cone.53,54 As we have shown, biaxiality in the ordering could have have non-negligible effects on lineshapes at high frequency;55,56 moreover, it might influence the distance distribution between pairs of nitroxides in doublv labeled systems. 4,57
A significant difference between the R1 and R2 spin labels has been found, which could be exploited for different purposes. For R1, rotamers with different mobility are simultaneously present, and non-trivial effects on ESR spectra are expected. The presence of the methyl substituent at the 4-position of the pyrroline ring reduces the conformational freedom in R2, so only non-interconverting conformers are possible. Since the torsional oscillations experienced by such conformers are shown to have just small averaging effects on the magnetic tensor, and are similar for all conformers, a smaller impact of the chain dynamics on EPR spectra is predicted. In this respect R2 appears to be a more suitable spin label to probe the protein dynamics. On the other hand, the higher mobility of R1 can make it a more sensitive probe of the environment.
Acknowledgments
A.F. and F.T acknowledge financial support by MIUR (PRIN-2005) and Università di Padova (CPDA057391). J.H.F. acknowledges financial support by a grant from NIH/NCRR.
Abbreviations
- PAS
Principal Axis System
- SDSL
Site Directed Spin Labeling
- MTSSL
methanethiosulphonate
- MD
Molecular Dynamics
Appendix. Order parameters
The order parameters defined in eq 17 are complex scalars, quantifying the degree of order which characterizes the distribution of magnetic axes (MF frame) in the AF frame, produced by side-chain motions. The two indices q and k have different meaning; they refer to a frame fixed in the protein and another frame moving with the magnetic tensors, respectively.37 This point will be illustrated by some considerations.
The components specify the distribution of the magnetic zM-axis in the AF frame. Going from the irreducible spherical to the cartesian representation, we can write:
where are the elements of a second rank, traceless tensor ( ), usually denoted as the ordering tensor. The value of gives the degree of order of the zM-axis with respect to the K-axis, with for perfect alignment of zM-to the K-axis, while if zM is perfectly ordered perpendicular to the K-axis. The ordering tensor Szz is generally expressed in its PAS, (ordering frame, SF in Figure 2), whose axes are designed as directors. For a generic orientational distribution of the zM axis, the three principal values of Szz are different; for axial symmetry the relation holds (having chosen the Z principal axis parallel to C∞).
The components specify the distribution of the Z director in the magnetic frame. The following relations hold between cartesian and irreducible spherical components:
Again, are the elements of a second rank, traceless tensor, which accounts for the alignment of the magnetic axes to the Z-director. The value of quantifies the degree of alignment of the k-axis to the Z-director, and for perfect alignment of the k-axis to the Z-director, while if the k-axis is perfectly ordered perpendicular to the Z-director. In general the axes of the magnetic frame will have different propensity to align with respect to the Z-director, i.e. .
Contributor Information
Fabio Tombolato, Dipartimento di Scienze Chimiche, Via Marzolo 1, Università di Padova, 35131 Padova, Italy.
Alberta Ferrarini, Dipartimento di Scienze Chimiche, Via Marzolo 1, Università di Padova, 35131 Padova, Italy.
Jack H. Freed, Baker Laboratory of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853-1301
References and Notes
- 1.Hubbell WL, Altenbach C. Curr Opin Struct Biol. 1994;4:566. doi: 10.1016/j.sbi.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hubbell WL, Gross A, Langen R, Lietzow M. Curr Opin Struct Biol. 1998;8:649. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- 3.Berliner LJ, editor. Biological Magnetic Resonance: the Next Millenium. Vol. 14 Plenum Press; New York: 1998. [Google Scholar]
- 4.Berliner LJ, Eaton SS, Eaton GR, editors. Biological Magnetic Resonance: Distance Measurements in Biological Systems by EPR. Vol. 19 Kluver Academic; New York: 2000. [Google Scholar]
- 5.Borbat PP, Costa-Filho AJ, Earle KA, Moscicki JK, Freed JH. Science. 2001;291:266. doi: 10.1126/science.291.5502.266. [DOI] [PubMed] [Google Scholar]
- 6.Perozo E, Cortes DM, Cuello LG. Science. 1999;285:73. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- 7.Perozo E, Cortes DM, Sompornpisut P, Kloda A, Martinac B. Nature. 2002;418:942. doi: 10.1038/nature00992. [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, Zhang F, Su Z, McNew JA, Shin YK. Nat Struct Mol Biol. 2005;12:417. doi: 10.1038/nsmb921. [DOI] [PubMed] [Google Scholar]
- 9.Jao CC, Der-Sarkissian A, Chen J, Langen R. Proc Nail Acad Sci. 2004;101:8331. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borbat P, Ramlall TF, Freed JH, Eliezer D. J Am Chem Soc. 2006;127:15014. doi: 10.1021/ja063122l. [DOI] [PubMed] [Google Scholar]
- 11.Radzwill N, Gerwert K, Steinhoff HJ. Biophys J. 2001;80:2856. doi: 10.1016/S0006-3495(01)76252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altenbach C, Yang K, Farrens DL, Farahbakhsh ZT, Khorana HG, Hubbell WL. Biochemstry. 1996;35:12470. doi: 10.1021/bi960849l. [DOI] [PubMed] [Google Scholar]
- 13.Cai KW, Langen R, Hubbell WL, Khorana HG. Proc Natl Acad Sci. 1997;94:14267. doi: 10.1073/pnas.94.26.14267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park S, Borbat PP, Gonzalez-Bonet G, Bhatnagar J, Pollard AM, Freed JH, Bilwes AM, Crane BR. Nat Struct Mol Biol. 2006;13:400. doi: 10.1038/nsmb1085. [DOI] [PubMed] [Google Scholar]
- 15.Ferrarini A, Moro G, Nordio PL. Mol Phys. 1988;63:225. [Google Scholar]
- 16.Moro GJ, Ferrarini A, Polimeno A, Nordio PL. In: Reactive and Flexible Molecules in Liquids. Dorfmüller Th., editor. Kluwer Academic; Dordrecht: 1989. p. 107. [Google Scholar]
- 17.Ferrarini A, Moro GJ, Nordio PL, Crepeau RH, Freed JH. J Chem Phys. 1989;91:5707. [Google Scholar]
- 18.Cassol R, Ferrarini A, Nordio PL. J Phys: Cond Matt. 1994;6:A279. [Google Scholar]
- 19.Mchaourab MS, Lietzow MA, Hideg K, Hubbell WL. Bjochemistry. 1996;35:7692. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 20.Robinson BH, Slutsky LJ, Auteri FP. J Chem Phys. 1992;96:2609. [Google Scholar]
- 21.Steinhoff H-J, Hubbell WL. Biophys J. 1996;71:2201. doi: 10.1016/S0006-3495(96)79421-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LaConte LEW, Voelz V, Nelson W, Enz M, Thomas DD. Biophys J. 2002;83:1854. doi: 10.1016/S0006-3495(02)73950-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoica I. J Phys Chem B. 2004;108:1771. [Google Scholar]
- 24.Murzyn K, Rg T, Blicharski W, Dutka M, Pyka J, Szytula S, Froncisz W. Proteins. 2006;62:1088. doi: 10.1002/prot.20838. [DOI] [PubMed] [Google Scholar]
- 25.Budil DE, Sale KL, Khairy KA, Fajer PJ. J Phys Chem B. 2006;110:3703. doi: 10.1021/jp054738k. [DOI] [PubMed] [Google Scholar]
- 26.Sale K, Sr C, Sharp KA, Hideg KA, Fajer PG. J Magn Res. 2002;156:104. doi: 10.1006/jmre.2002.2529. [DOI] [PubMed] [Google Scholar]
- 27.Sale K, Song L, Liu Y-S, Perozo E, Fajer PJ. J Am Chem Soc. 2005;127:9334. doi: 10.1021/ja051652w. [DOI] [PubMed] [Google Scholar]
- 28.Timofeev VP, Nikolsky DO. J Biomol Struct Dyn. 2003;21:367. doi: 10.1080/07391102.2003.10506932. [DOI] [PubMed] [Google Scholar]
- 29.Tombolato F, Ferrarini A, Freed JH. J Chem, Phys B. submitted companion paper. [Google Scholar]
- 30.Flory PJ. Statistical Mechanics of Chain Molecules. Interscience; New York: 1969. [Google Scholar]
- 31.Karplus M, Kushick JN. Macromol. 1981;14:325. [Google Scholar]
- 32.Moro GJ. J Chem Phys. 1991;94:8577. [Google Scholar]
- 33.Kramers HA. Physica. 1940;7:284. [Google Scholar]
- 34.Langer JS. Ann Phys. 1969;54:258. [Google Scholar]
- 35.Shore JE, Zwanzig R. J Chem Phys. 1975;63:5445. [Google Scholar]
- 36.Zientara GP, Freed JH. J Chem Phys. 1983;69:3077. [Google Scholar]
- 37.Zannoni C. In: The Molecular Physics of Liquid Crystals. Luckhurst GR, Gray GW, editors. Academic Press; New York: 1979. p. 51. [Google Scholar]
- 38.Zare NR. Angular Momentum. Wiley; New York: 1987. [Google Scholar]
- 39.Frisch M, et al. Gaussian 03. Gaussian Inc; Pittsburg PA: 2003. [Google Scholar]
- 40.Pal D, Chakrabarti P. J Biomol Struct Dyn. 1998;13:1059. doi: 10.1080/07391102.1998.10509001. [DOI] [PubMed] [Google Scholar]
- 41.Note that the state which we label as g+(g−) corresponds to the state labeled as g−(g+) in refs.40,42.
- 42.Langen R, Joon Oh K, Cascio D, Hubbell WL. Biochemistry. 2000;39:8396. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 43.Lovell SC, Word JM, Richardson JS, Richardson DC. Proteins. 2000;40:389. [PubMed] [Google Scholar]
- 44.Happel J, Brenner H. Low Reynold Number Hydrodynamics. Prentice Hall; Englewood Cliffs, NJ: 1965. [Google Scholar]
- 45.In the context of magnetic resonance, the word ’rhombicity’ is commonly used rather than ’biaxiality’.
- 46.Columbus L, Kámás T, Jekö J, Hideg K, Hubbell WL. Biochemistry. 2001;40:3828. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- 47.Liang Z, Lou Y, Freed JH, Columbus L, Hubbell WL. J Phys Chem B. 2004;108:17649. [Google Scholar]
- 48.Jacobsen K, Oga S, Hubbell WL, Risse T. Biophys J. 2005;88:4351. doi: 10.1529/biophysj.105.059725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Budil DE, Lee S, Saxena S, Freed JH. J Magn Res. 1996;120:155. [Google Scholar]
- 50.Barnes JP, Liang Z, Mchaourab HS, Freed JH, Hubbell WL. Biophys J. 1999;76:3298. doi: 10.1016/S0006-3495(99)77482-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pyka J, Iinicki J, Altenbach C, Hubbell WL, Froncisz W. Biophys J. 2005;89:2059. doi: 10.1529/biophysj.105.059055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferrarini A, Luckhurst GR, Nordio PL. Molec Phys. 1995;85:131. [Google Scholar]; Stocchero M, Ferrarini A, Moro GJ, Dunmur DA, Luckhurst GR. J Chem Phys. 2004;121:8079. doi: 10.1063/1.1794071. [DOI] [PubMed] [Google Scholar]
- 53.Columbus L, Hubbell WL. Trends Biochem Sci. 2002;27:288. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 54.Hustedt EJ, Stein RA, Sethaphong L, Brandon S, Zhou Z, DeSensi SC. Biophys J. 2006;90:340. doi: 10.1529/biophysj.105.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barnes JP, Freed JH. Biophys J. 1998;75:2532. doi: 10.1016/S0006-3495(98)77698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaffney BJ, Marsh D. Proc Natl Acad Sci. 1998;95:12940. doi: 10.1073/pnas.95.22.12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borbat PP, Mchaourab HS, Freed JH. J Am Chem Soc. 2002;124:5304. doi: 10.1021/ja020040y. [DOI] [PubMed] [Google Scholar]