

Abstract
Understanding clinical phenotypes through their corresponding genotypes is one of the principal goals of genetic research. Though achieving this goal is relatively simple with single gene syndromes, more complex diseases often consist of varied clinical phenotypes that may be the result of interactions among multiple genetic loci. Microarray technology has brought the phenotype –genotype relationship to the molecular level, using differently behaving cancers, for example, as the basis for comparing patterns of gene expression. With this feasibility study, we attempted to use similar methods of analysis at the clinical level, in order to evaluate our hypothesis that the clustering of clinical phenotypes would provide information that would be useful in elucidating their underlying genotypes. Because of its breadth of content and detailed descriptions, we used OMIMa as our source material for phenotypic and genetic information. After processing the source material, we then performed self-organizing map and hierarchical clustering analysis on representative diseases by phenotypic category. Through pre-determined queries over this analysis, we made two findings of potential clinical significance, one concerning diabetes and another concerning progressive neurologic diseases. Our methods provide a formal approach to analyzing phenotypes among diverse diseases, and may help indicate fruitful areas for further research into their underlying genetic causes.
INTRODUCTION
The production of increasing amounts of data as a result of the “molecular revolution” in biology has changed the approach to scientific discovery. With so much information available, the challenge has moved from obtaining experimental results to providing new methods of analysis of results. Additionally, as tools for genetic analysis become more sophisticated, important discoveries are moving from the gene level to the process level. Understanding interactions among genes, from specific molecular pathways to the whole organ systems, should ultimately provide clinically useful insights into disease processes, including diseases that are influenced by multiple genetic loci1. Analyzing the molecular processes underlying phenotypically similar diseases may provide a window into these complex interactions.
One of the major difficulties in analyzing biological systems is the fact that, unlike other basic sciences, investigators have yet to discover general principles that can serve as the basis for predicting a system’s behavior2. The ever-increasing amounts of available biological data makes the discovery of these rules more likely; however, data alone does not equal knowledge. Indeed, the goal of bioinformatics is to provide the tools to eventually uncover these rules and, ultimately, to “predict computationally systems of higher complexity… such as the phenotypes of whole organisms2”.
Methods for studying complex phenotypes can be divided into two basic approaches: gene-driven, which focus on certain genes in order to discover the phenotypes they influence; and trait-driven, which focus on phenotypes and look to find the causative genes1. Knockout models are the traditional method for proving and analyzing traits influenced by single genes; however, more complex phenotypes, affected by multiple, potentially unknown, genetic loci, as well the epistatic relations among them, require more complicated, multivariate methods of analysis1. We hypothesize that the analysis of phenotypic similarities among diverse diseases, especially those associated with known loci, may provide insight into the genetic interactions underlying them.
Previously, researchers have generally used cellular phenotypes as starting points for classifying subtypes of diseases. Investigators studying breast cancer, for example, have found underlying patterns in gene expression that provided biological explanations for the varied behaviors of the tumors. Their clustering methods found a relatively large number of molecular phenotypes underlying the relatively small number of cellular phenotypes used in their dataset. These findings on the molecular level form the beginnings of a “molecular taxonomy” of breast cancer cells, but also reveal the difficulty of assigning causal relations to specific genes for traits in complex diseases3. For example, while understanding gene expression patterns is usually essential to understanding the actual clinical manifestations of a disease, often the same phenotype may be caused by significantly different genetic alterations4.
In our approach to clustering phenotypes, we used two techniques that are commonly used in microarray analysis: self-organizing maps (SOM) and hierarchical clustering (HC). We decided on two clustering methods, rather than one or many, first to help determine the scalability of our methods, and second to obtain results consistent with the limitations of a feasibility study.
Our previous work has examined the possibility of using analytic tools from bioinformatics in order to examine relationships among clinical traits in inherited diseases, and proved the utility of these methods5. Specifically, we showed that hierarchical clustering methods were useful for extracting clinical-genetic relationships from clinical decision support systems. In this study, our objective is to expand on those methods, analyzing OMIM to discover new approaches for obtaining valid, non-trivial gene-disease relationships through phenotype analysis, with the ultimate goal of supporting a “phenotypic taxonomy” of inherited diseases.
METHODS
Data
With over 14,000 detailed entries about human genes and genetic disorders, OMIM, part of the NCBI’s system of databases, is a “comprehensive, authoritative, and timely compendium of information in human genetics6.” The principal data source for this study was the omim.txt file, which contains the entire free text of the OMIM database. In addition to information on genetic loci, inheritance patterns, and allelic variants, many OMIM entries contain a “Clinical Synopsis” (CS) section, which delineates the accompanying signs and symptoms (phenotype) of the disease. The CS section is divided into categories, either by organ system (cardiac, neurological, etc.) or by type of finding (inheritance, lab values, etc.).
Because of the diversity of presentation of human disease, and also possibly because OMIM has been in development for decades, information in the CS section is not always represented in a uniform manner. Different names for overlapping concepts are often applied to general categories (i.e. “cardiac” and “cardiovascular”) as well as specific phenotypes (“mental retardation, profound” and “mental retardation, severe”). Because of the complexity and number of specific findings in OMIM, we decided to perform our analysis on the categorical level. Using simple text processing, we normalized the category names and obtained a single list of unique category titles.
After obtaining the category list, we then went back and analyzed the candidate diseases extracted from omim.txt and created a profile, by category, for each disease. To normalize the data, we considered each category as a binary variable, assigning a score of 1 for the presence of any finding in that category, and a score of 0 if there were no findings. Because they were not truly observable phenotypes, we eliminated the “Inheritance”, “Miscellaneous”, and “Molecular Basis” categories. We also consulted OMIM’s morbidmap and genemap in order to find which, if any, genes or genetic loci were associated with the diseases.
Analysis
Because of the high dimensionality of each of our data points, we chose SOM since it efficiently reduces high-dimensional data points into a low-dimensional map7. Briefly, SOM begins with a number of nodes with a simple topology (in our case, a 5×5 grid), with a random initial vector mapping into k-dimensional space (in this study, k is equal to the number of phenotypic categories) for each node. At each iteration, a random data point is selected, and the nodes mappings are adjusted toward the data point, until learning stops or the maximum number of iterations is reached8.
Hierarchical clustering places data points into a hierarchy of underlying subsets, created by replacing each closest pair of points with a single point representing their average. Pairs are evaluated using a similarity measure (in our case Euclidean distance). Eventually, the clusters of data points end up in a single large cluster represented as a phylogenetic tree, with the degree of similarity represented between them represented by branch lengths8. HC also uses a weight function, in order to determine the order by which data points are plotted in the dendrogram. We used Spotfire® DecisionSite and Stata 7.0 (Stata Corporation) for clustering and statistical analysis.
Evaluation
In order to evaluate our analysis, we performed two pre-determined queries, one for “diabetes mellitus” and the second for “amyotrophic lateral sclerosis”, and evaluated the results in terms of each clustering approach. We then performed a quantitative and a qualitative evaluation for each query., As a basis for each evaluation, we first used the NCBI’s Entrez search page to obtain OMIM entries for each disease query.,. For the quantitative evaluation, we determined the number of “hits” in our database for each query, as well as their respective clustering patterns in terms of SOM clusters (Table 1) and HC rankings. For the qualitative evaluation, we then examined one set of clustering results more deeply for each disease, in an attempt to find novel gene-disease relationships. As a gold standard, we used the association of concepts in terms of a PubMed literature search. As an internal control, we evaluated 10 random pairs OMIM entries, from different SOM clusters, with this method, and each time found no common associated articles.
Table 1.
Distribution of all diseases by SOM cluster (n=4491)
| 215a | 186 b | 652 c | 79 d | 547 e |
| 18 f | 140 g | 164 h | 147 i | 90 j |
| 415 k | 49 l | 230 m | 84 n | 96 o |
| 83 p | 83 q | 94 r | 105 s | 131 t |
| 340 u | 141 v | 122 w | 91 x | 189 y |
RESULTS
Clustering
Of the approximately 14,700 entries in OMIM, we found 4491 that contained a clinical synopsis section. The inheritance patterns of these entries were: 1901 (42%) autosomal dominant; 1616 (36%) autosomal recessive; 393 (9%) X-linked; 15 (0.4%) Y-linked; 26 (0.6%) mitochondrial; and 539 (12%) autosomal. In the CS section for these entries, we initially found 90 different category names. After normalization, we ended up with 50 unique categories. For example, omim.txt contained categories named “metabolism”, “metabolic”, and “metabolic features”, so we condensed these categories into a single “metabolism” category. Of the 50 categories, the number of entries with corresponding findings ranged from 1 (‘pre-diagnosis’) to 1746 (‘neurological’), with the average number of entries being 275. Twelve categories corresponded to fewer than 20 entries.
We used a 5×5 grid in our SOM analysis, mainly because of the relatively large number of data points in our set. After 12,500 iterations, the average number of members of a cluster was 179, with a range of 18 to 652. SOM analysis also provided a similarity score, which we compared to a data point’s rank in the hierarchical clustering analysis, and obtained a Pearson’s correlation coefficient for the two scores of 0.81. The general results of the hierarchical clustering can be seen in the dendrogram/heat map in Figure 1.
Figure 1.
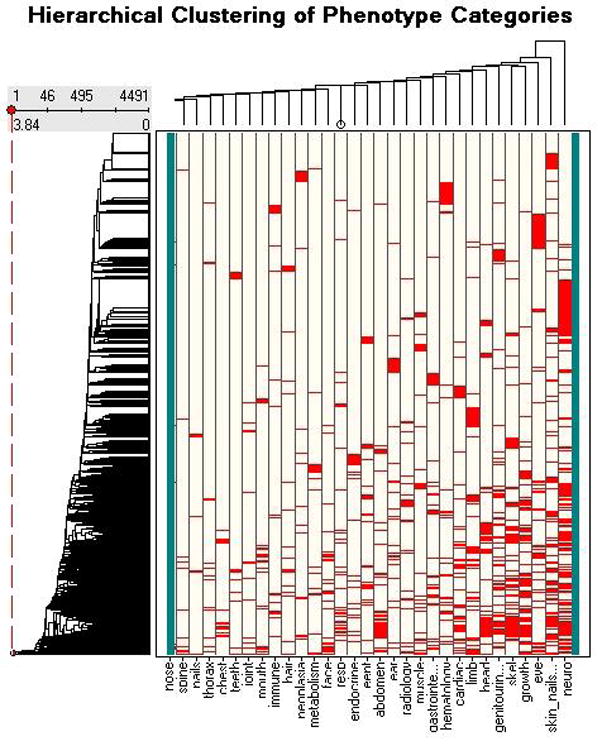
HC Dendrogram
Quantiative evaluation
From the ALS OMIM query, we obtained 65 OMIM entries, 21 of which were in our data set, spread among 7 of the 25 SOM clusters (Table 2). Cluster 5 contained 7of the 8 entries specific to ALS. The “diabetes mellitus” OMIM query initially produced 287 OMIM entries, 142 of which were in our data set. The entries were spread among 21 of the 25 SOM clusters (Table 3).
Table 2.
ALS distribution by SOM cluster (n=21)
| 0 a | 1 b | 0 c | 0 d | 13 e |
| 0 f | 0 g | 0 h | 0 i | 0 j |
| 1 k | 0 l | 0 m | 1 n | 1 o |
| 0 p | 0 q | 0 r | 0 s | 1 t |
| 0 u | 0 v | 3 w | 0 x | 0 y |
Table 3.
DM distribution by SOM cluster (n=142)
| 6 a | 7 b | 14 c | 1 d | 9 e |
| 0 f | 13 g | 17 h | 0 i | 3 j |
| 3 k | 0 l | 3 m | 0 n | 4 o |
| 2 p | 5 q | 1 r | 6 s | 1 t |
| 12 u | 4 v | 8 w | 14 x | 9 y |
Of the 10 entries closest to “diabetes mellitus 2” in terms of HC ranking, all were in the same SOC cluster (8), and all but one, OMIM 108780, “Natriuretic peptide precursor A”, were directly related to disorders of the endocrine system. For 10 entries closest to ALS, 9 were in the same SOM cluster (5), and all except one, OMIM number 102770, “Adenosine monophosphate deaminase deficiency; AMPD1” (AMPD), were neuropathies.
Qualitative evaluation
We chose to examine examine SOM cluster 8 for the qualitative evaluation of the diabetes query. Cluster 8 contained all of the non-insulin dependent diabetes entries (including MODY), and all but one of the insulin-dependent diabetes entries. Of all the entries in this cluster, only one, OMIM number 177400 “Butyrylcholinesterase (BCHE)”, an enzyme that is generally mentioned in the context of post-anesthesia reactions, did not appear to be clinically related to diabetes. In examining the literature, however, we found research that revealed an association between BCHE variants and type 2 diabetes9, specifically in terms of hypertriglyceridemia10. This finding is emblematic of other potential relationships that may be found through deeper analysis of the clustering results: seemingly unrelated processes whose interactions may actually be important in causing disease.
For the ALS query, we chose to examine potential relationships between AMPD, the only non-neuropathy among the 10 closest HC entries, and ALS. AMPD affects muscles, and is thought to be the most common cause of metabolic myopathy in humans11. Though AMPD mainly affects skeletal muscle, which would explain its phenotypic similarity to ALS, mutations in AMPD1 have been shown to have cardioprotective effects in patients with heart failure. The actual mechanism of the cardioprotection, however, is still unknown, though it has been speculated it may include free radical inhibition12. Free radicals have also been implicated in familial forms of ALS, as well as fatal cardiomyopathies in mice, through defects in the genes encoding forms of superoxide dismutase, an enzyme that helps to neutralize reactive oxygen species13. These clustering results point to a possible avenue of research: Might there be a role for the alteration of adenosine metabolism, at either the cellular or mitochondrial level, in protection from the free radical species that have been linked to ALS?
DISCUSSION
Our analysis of OMIM reveals the possibility of using expression patterns of phenotypic traits to discover potential avenues for future research. Further evaluation may reveal other potential findings, similar to that of BCHE in the diabetes cluster, through the process of explaining how seemingly spurious members actually fit into a cluster. Such findings may reveal potential hypotheses to be tested in experimental settings, such as the adenosine’s role in ALS.
Assigning a binary score, based only on the presence or absence of traits, is an efficient starting point for separating OMIM entries; however, we also lost information by not taking into account more qualitative aspects of the information contained in each category. Additionally, using the categories with very low totals probably did not add much useful information to the analysis. Because analyzing each phenotype entry (over 16,000) individually would have required far more complex methods, and since our purpose was mainly to prove the utility of our overall methods, we chose the relatively simple approach.
Though the evaluation of our methods provided two potentially interesting leads, there are likely to be many more leads that have not yet been uncovered. Conversely, the respective findings for diabetes and adenosine may also be due to chance, as they were the only 2 queries we performed. As the purpose of this study is to evaluate our methods in order to evaluate the feasibility of a more detailed analysis of phenotypes, we believe that the two cases are sufficient to at least prove that there is merit to our concept. The problem of evaluation is common to many studies in bioinformatics, as the amount of information produced outstrips the resources available for its analysis. Further, and perhaps automated, analysis in the future should provide us with a firmer conclusion as to the utility of our evaluation.
An important issue in our analysis is our choice of HC and SOM over other clustering methods. We chose HC mainly because it is one of the most commonly used for gene expression analysis, and also because it produces a ranking of data points. SOM was useful because of its efficiency in dealing with multiple-dimensional data, as well as the fact that it maintains a consistent topography among the nodes. Of course, there are many other clustering and data analysis methods that we could have chosen from, and that may have given different results, but we chose to limit ourselves to two methods. The significant correlation between the results of the two analyses supports their use as well. Additionally, since we are attempting to prove a concept, using many more different clustering methods would have led to each result providing less information.
In order to move from speculation to science, however, we plan on future refinements to our methods. The most important next step will involve normalizing and categorizing individual phenotypic data points, and performing the analysis on a more specific, rather than categorical, level. Further refinement of these data points, including the incorporation of genetic locus information, should allow for a more specific, robust analysis of OMIM diseases. Obtaining other types of phenotypic information from other sources, including ontologies and clinical data repositories14, will also assist in validating our methods, as well as proving their scalability. Ultimately, and on a much longer time scale, the ideal proof of our methods would be the validation of the generated hypotheses in animal or other experimental models.
CONCLUSIONS
Since the advent of microarrays, scientists have used gene expression profiles as the basis for the analysis of how genotypes determine phenotypes. This study presents the possibility of looking at diseases in a different manner, looking at relationships among phenotypic traits as possible conduits toward specific genetic information. Applying techniques that are normally used on the micro scale to analyze gene expression and molecular pathways may give insight on the macro scale into previously unknown connections among seemingly unrelated disease processes. Eventually, as more detailed methods for describing genomes, proteomes, and phenotypes are developed, performing a “phenotypic linkage analysis” may even become possible.
The genomic revolution has led to an explosion of both information and techniques for its analysis. The value of future discoveries, however, will most likely be in their integration into biological systems. The study of disease phenotypes may be an important method to synthesize the analysis of these systems on a macro scale. The analysis of phenotypic data from OMIM and, eventually, other sources may provide a useful adjunct to current approaches to genetic discovery.
Acknowledgments
This research was supported by NLM training grant LM-07079 and NYSTAR CAT grant 02240011.
Footnotes
References
- 1.Phillips TJ, Belknap JK. Complex-trait genetics: emergence of multivariate strategies. Nature Reviews Neuroscience. 2002;3:478–485. doi: 10.1038/nrn847. [DOI] [PubMed] [Google Scholar]
- 2.Kanehisa M, Bork P. Bioinformatics in the post-sequence era. Nature Genetics. 2003;33:S305–S310. doi: 10.1038/ng1109. [DOI] [PubMed] [Google Scholar]
- 3.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 4.Baldini A. DiGeorge syndrome: the use of model organisms to dissect complex genetics. Human Molecular Genetics. 2002;11:2363–69. doi: 10.1093/hmg/11.20.2363. [DOI] [PubMed] [Google Scholar]
- 5.Lussier YA, Sarkar IN, Cantor MN. An integrative model for in-silico clinical-genomics discovery science. Proc AMIA Symp. 2002:469–73. [PMC free article] [PubMed] [Google Scholar]
- 6.Hamosh A, Scott AF, Amberger J, Valle D, McKusick V. Online Mendelian Inheritance in Man. Human Mutation. 2000;15:67–61. doi: 10.1002/(SICI)1098-1004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 7.Wang JJD, Aasheim HC, Smeland E, Myklebost O. Clustering of the SOM easily reveals distinct gene expression patterns: results of a reanalysis of lymphoma study. BMC Bioinformatics. 2002:3. doi: 10.1186/1471-2105-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamayo P, Slonim D, Mesirov J, et al. Interpreting patterns of gene expression with self-organizing maps: Methods and applications to hematopoetic differentiation. PNAS. 1999;96:2907–12. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashim Y, Shepherd D, Wiltshire S, et al. Butyrylcholinesterase K variant on chromosome 3 q is associated with Type II diabetes in white Caucasian subjects. Diabetologia. 2001;44:2227–30. doi: 10.1007/s001250100033. [DOI] [PubMed] [Google Scholar]
- 10.Alcantara VM, Chautard-Freire-Maia EA, Scartezini M, et al. Butyrylcholinesterase activity and risk factors for coronary artery disease Scan. J Clin Lab Invest. 2002;62:399–404. doi: 10.1080/00365510260296564. [DOI] [PubMed] [Google Scholar]
- 11.OMIM entry 102770. http://www.ncbi.nlm.nih.gov/omim. Updated 6/27/02.
- 12.Loh E, Rebbeck TR, Mahoney PD, et al. Common variant in AMPD1 gene predicts improved clinical outcome in patients with heart failure. Circulation. 1999;99:1422–5. doi: 10.1161/01.cir.99.11.1422. [DOI] [PubMed] [Google Scholar]
- 13.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–49. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 14.Cantor MN, Lussier YA. Putting data integration into practice: Using biomedical terminologies to add structure to existing data sources. Proc AMIA Symp. 2003 in press. [PMC free article] [PubMed] [Google Scholar]