

Abstract
Inward rectifier potassium (Kir) channels have been postulated as therapeutic targets for several common disorders including hypertension, cardiac arrhythmias and pain. With few exceptions, however, the small-molecule pharmacology of this family is limited to nonselective cardiovascular and neurologic drugs with off-target activity toward inward rectifiers. Consequently, the actual therapeutic potential and ‘drugability’ of most Kir channels has not yet been determined experimentally. The purpose of this review is to provide a comprehensive summary of publicly disclosed Kir channel small-molecule modulators and highlight recent targeted drug-discovery efforts toward Kir1.1 and Kir2.1. The review concludes with a brief speculation on how the field of Kir channel pharmacology will develop over the coming years and a discussion of the increasingly important role academic laboratories will play in this progress.
Members of the inward rectifier family of potassium (Kir) channels regulate a myriad of physiological processes, including cardiac function, pain processing and opioid action, learning and memory, insulin secretion and epithelial solute transport [1,2]. Some inward rectifiers occupy unique physiological niches that raise intriguing questions about their potential as therapeutic targets. Unfortunately, however, the small-molecule pharmacology of inward rectifiers has remained essentially undeveloped since the first member was cloned nearly 20 years ago [3]. This dearth of pharmacological tools has hindered efforts to develop even a cursory understanding of the physiology of some Kir channels and represents a critical barrier to defining their therapeutic potential. The main goals of this review article are:
To provide a comprehensive summary of disclosed small-molecule modulators of Kir channels, highlighting the few examples where pharmacology has illuminated a deeper understanding of their physiology and ‘druggability’;
To review recent advances and future possibilities in targeted drug-discovery efforts directed at Kir channels.
Overview of Kir channel structure & function
The term ‘rectification’ refers to a nonlinear change in ionic current through an ion channel pore as a function of the electrochemical driving force. By convention, the movement of a cation from the extracellular solution to the cytosol is defined as an inward current. Thus, Kir channels preferentially conduct K+ ions inwardly under voltage-clamp conditions [1,2].
Inward rectification is caused by blockade of the channel pore by intracellular cations such as magnesium and polyamines (e.g., spermine, putrescine) driven ‘outwardly’ by membrane depolarization. The extent of pore block and, hence, strength of rectification varies widely among different family members. Strong rectifiers pass very little outward current, whereas weak rectifiers do so across a broad range of potentials [4,5]. In general, strong rectifiers are expressed in excitable cells, such as cardiac myocytes or neurons, where they tend to hyper polarize the membrane potential, but avoid short-circuiting action potentials by limiting outward K+ current during depolarization. In contrast, weak rectifiers carry significant outward current and are, therefore, well suited to function in nonexcitable tissues, such as secretory epithelia [1].
The recent determinations of high-resolution x-ray structures of Kir channel proteins have significantly advanced our molecular understanding of inward rectification [6–10]. They also create unique opportunities for understanding small-molecule–Kir channel interactions with near atomic-level resolution. To facilitate the present discussion, we include a brief overview of the relevant structural elements implicated in small-molecule binding.
Kir channels are tetramers comprised of four identical (homotetrameric) or homologous (heterotetrameric) membrane-spanning subunits surrounding a water-filled pore through which K+ ions move down their electrochemical gradient. FIGURE 1 shows a homology model of the Kir1.1 cytoplasmic domain docked to the membrane-spanning portion of a Kir3.1–KirBac1.3 chimera [11]. Two subunits have been removed for clarity. Each channel subunit consists of two membrane-spanning α-helical domains (TM1 and TM2) separated by an extracellular loop that forms the narrow K+-selectivity filter (SF). TM2 lines the membrane-spanning pore and terminates near the membrane–cytoplasm interface in a structure termed the helix bundle crossing (HBC). Structural and mutagenesis studies suggest that the HBC functions as a regulatable gate that opens and closes in response to diverse cell-signaling molecules, such as extracellular K+, intracellular protons and phosphoinositides [12]. The narrow gating-loop positioned near the HBC may also function as a gate in series with the HBC [7]. The extensive cytoplasmic domain extends the conduction pore well beyond the membrane and into the cytosol [6–10].
Figure 1. Structural model of an inward rectifier potassium channel.

A homology model of the Kir1.1 channel cytoplasmic domain has been docked to the membrane-spanning portion of a Kir3.1–KirBac1.3 channel chimera [11]. Regions of the channel backbone important for channel function, rectification and small-molecule binding are highlighted.
GL: Gating loop; HBC: Helix bundle crossing; RC: Rectification controller; RM: Rectification modulator; SF: Selectivity filter; TM1 and 2: Transmembrane domains 1 and 2.
Crystal structure-guided mutagenesis studies have identified three ‘rings’ of negatively charged residues that participate in rectification. These are highlighted in FIGURE 1 and labeled RC (rectification controller), rectification modulator 1 (RM1) and rectification modulator 2 (RM2). Electrostatic interactions between these negatively charged rings and positively charged polyamines produce high-affinity binding and block in strong rectifiers. Weak rectifiers possess uncharged amino acids at one or more of these positions, thereby reducing the polyamine block [5,7,10]. Importantly for this review, an emerging body of evidence suggests that the unique charge distribution and architectural topography near RC, RM1 and RM2 may provide selective binding pockets that can be targeted with small-molecule inhibitors [13,14]. These will be discussed further below.
Kir channel pharmacology & therapeutic potential
Kir1.1 (ROMK)
Kir1.1, which is commonly referred to as the renal outer medullary K+ (ROMK) channel, is the founding member of the inward rectifier family. The Kir1.1 cDNA was cloned from rat kidney outer medulla and was shown to encode a weakly rectifying K+ channel with functional properties very similar to those of the apical membrane small-conductance secretory K+ (SK) channel expressed in epithelial cells of the thick ascending limb (TAL) and collecting duct (CD) [3]. The SK channel is absent from the TAL and CD of mice carrying a deletion in the gene encoding Kir1.1 (KCNJ1), establishing that the SK current is carried by Kir1.1 [15].
The significance of Kir1.1 in renal tubule function was appreciated more than a decade before the channel was cloned. The major functions of Kir1.1 in the nephron are summarized in FIGURE 2. In the TAL, luminal K+ recycling by Kir1.1 catalyzes NaCl reabsorption by the Na+-K+-2Cl− co-transporter and loop diuretic target NKCC2, which in turn promotes osmotic water reabsorption in the distal nephron [16–18]. In the connecting tubule and CD, Kir1.1 constitutes a key physiological pathway for regulating K+ secretion to match dietary intake [19,20].
Figure 2. Major functions of ROMK in the kidney tubule.

In the thick ascending limb of Henle’s loop, ROMK provides substrate K+ ions essential for transepithelial NaCl reabsorption by NKCC2. NaCl reabsorption generates a hypertonic interstitium that promotes osmotic water reabsorption in the distal nephron. In the collecting duct, ROMK constitutes a key physiological pathway for K+ secretion into the urinary fltrate. See text for additional details.
BK: Big or large conductance calcium-activated K+ channel; ENaC: Epithelial Na+ channel; NKCC2: Na+-K+-2Cl co-transporter; ROMK: Renal outer medullary K+ channel.
The identification of disease-causing mutations in KCNJ1 in patients with Type II Bartter syndrome (BS) [21] established the importance of Kir1.1 in kidney function in humans. BS is a life-threatening renal ‘tubulopathy’ characterized by excessive urination, renal Na+ and K+ wasting, normal to low blood pressure and metabolic alkalosis. BS is actually a group of rare salt-wasting tubulopathies caused by heritable mutations in at least six genes whose products participate in salt transport in the kidney (for a recent review see elsewhere [22]). Nearly 40 loss-of-function BS mutations in KCNJ1 have been identified to date. Several of these decrease the open-state probability of Kir1.1, while others impair its trafficking to the cell surface [11,23–25].
The clinical presentation of Type II BS can be rationalized in terms of diminished luminal K+ secretory activity in the TAL. As illustrated in FIGURE 2, NKCC2 is functionally coupled to Kir1.1 such that loss of channel activity impairs NaCl reabsorption in the TAL. This has several deleterious consequences. First, Na+ that would normally be reabsorbed in the TAL is delivered to the distal nephron and ultimately lost in the urine. Second, the ensuing dilution of the medullary interstitium blunts osmotic water reabsorption in the CD and thereby increases urine output. Third, increased delivery of fluid to the distal nephron stimulates K+ secretion [26] and urinary K+ loss and thereby reduces plasma K+ levels (hypokalemia). This, in turn, promotes urinary acid excretion and metabolic alkalosis [27].
As shown in FIGURE 2, K+ secretion in the distal nephron is mediated by two K+-conductive pathways in the apical plasma membrane. Under ‘normal’ conditions, Kir1.1 is thought to provide the major route for K+ efflux. However, an emerging body of evidence suggests that during periods of high tubule flow, such as those experienced during diuresis, ‘flow-stimulated’ large-conductance K+ (BK or maxi-K) channels take on a more prominent role [28–30]. Perhaps not surprisingly, the severity of hypokalemia observed in Type II BS is generally milder than that of Type I BS [21], a related tubulopathy caused by mutations in the loop diuretic target NKCC2 [22,31,32]. One simple explanation is that the K+-secretory capacity in Type I patients is greater due to the expression of both Kir1.1 and BK channels in the distal nephron and is lower in Type II patients due to the absence of Kir1.1. Studies in Kir1.1-knockout mice, a genetic model of Type II BS, showing a reduction in distal nephron K+ secretion support this notion [33]. The loss of Kir1.1 in mice appears to be compensated for by an increase in BK channel expression [33]. Upregulation of flow-stimulated BK channels in the setting of chronic diuresis and, thus, high tubule flow may account for the relatively mild K+ wasting seen in Type II BS patients.
Type II BS is an autosomal-recessive disorder requiring the inheritance of two mutant KCNJ1 alleles for expression of the disease. Interestingly, Lifton and colleagues recently identified within the Framingham Heart Study cohort heterozygous carriers of KCNJ1 mutations who do not exhibit the salt- and water-wasting phenotype of BS [34]. Importantly, however, these individuals have lower blood pressure and a reduced prevalence of hypertension. Similarly, Tobin et al. identified in the general population single nucleotide polymorphisms in noncoding regions of KCNJ1 that are associated with lower blood pressure. No associations with Na+ or K+ excretion were found [35]. Taken together, these observations suggest that a partial loss of Kir1.1 function may reduce blood volume and pressure without causing pathological derangement of electrolyte balance observed in BS. They also raise the intriguing possibility that Kir1.1 is a molecular target for a novel class of loop diuretic.
Conventional loop diuretics (e.g., flurosemide, bumetanide and torasemide) increase urine production by inhibiting NKCC2 activity in the TAL (FIGURE 2) [36]. The drug response is virtually identical to the clinical presentation of Type I BS (see previous discussion), including potentially life-threatening reductions in serum K+ [37,38]. In light of the preceding discussion, it is conceivable that small-molecule antagonists of Kir1.1 could trigger diuresis by acting at the TAL, but do so with minimal K+ wasting by inhibiting K+ secretion in the distal nephron (FIGURE 2). However, assessing the therapeutic potential of Kir1.1 will first require the development for selective drug-like probes for in vivo pharmacology experiments.
In an effort to overcome this barrier, Denton and colleagues set out to develop a high-throughput screen (HTS) for small-molecule modulators of Kir1.1 function. The chosen assay measures inwardly directed movement of the K+ congener thallium (Tl+) through Kir1.1 channels using a Tl+-sensitive fluorescent dye. Similar assays have been developed for KCNQ2, KCa2.1, KCa2.2, KCa2.3 [39–41], and hERG [42] K+ channels and the K+-Cl− co-transporter KCC2 [43].
A stable HEK-293 cell line expressing Kir1.1 and under the control of a tetracycline (Tet)-inducible promoter was employed to avoid cytotoxic effects of constitutive Kir1.1 expression [23,44] and provide a convenient internal control for screens. The Kir1.1-S44D mutant, in which serine (S) at position 44 was mutated to aspartate (D), was used as a surrogate for the wild-type channel in an effort to maximize cell surface expression of the channel [45,46]. One clone exhibiting robust Tet-inducible Tl+ flux, Kir1.1 channel activity (FIGURE 3A & B) and biochemical localization to the cell surface was chosen for assay development and HTS [47].
Figure 3. Thallium-fux assay of Kir1.1 channel function for high-throughput molecular library screening.

(A) A stable cell line expressing Kir1.1 under the control of a tetracycline-inducible promoter was developed to avoid cytotoxic effects of constitutive Kir1.1 channel expression and subsequent cell line degeneration. Values are means ± SEM current amplitude normalized to cell capacitance and plotted as a function of test voltage in whole-cell patch clamp experiments. Cells were cultured overnight in the absence (control; darker circles) or presence of tetracycline (lighter circles). (B) Fluorescence assay of Kir1.1 channel activity in a 384-well plate using the thallium-sensitive fluorescent dye FluoZin-2. Cells were cultured overnight in the absence (− Tet) or presence (+ Tet) of tetracycline. One well was pretreated with the Kir1.1 channel inhibitor peptide Tertiapin-Q (+ TPNQ) to block channel activity. Addition of extracellular thallium (at 500 s) evoked an abrupt increase in FluoZin-2 fluorescence that was dependent on Kir1.1 channel expression. This assay was used to screen approximately 225,000 small molecules for novel modulators of Kir1.1.
Reproduced with permission from [47].
FIGURE 3B shows representative fluorescence traces recorded from individual wells of a 384-well plate containing uninduced (−Tet) or Tet-induced cells bathed in control (+Tet) or Tertiapin-Q (TPNQ)-containing assay buffer (Tet + TPNQ). Extracellular Tl+ addition evoked an abrupt increase in fluorescence in Tet-induced cells, but not in uninduced cells or induced cells pretreated with the Kir1.1 inhibitory peptide TPNQ. Using this assay, approximately 225,000 small molecules were screened in 384-well plates at a single dose of 10 µM.
FIGURE 4A shows the structure of one Kir1.1 inhibitor, termed VU590, identified by HTS. In Tl+ flux assays, VU590 inhibited Kir1.1 in a dose-dependent manner with an IC50 of approximately 300 nM (FIGURE 4B). In whole-cell patch clamp electrophysiological experiments on transiently transfected HEK-293 cells, VU590 inhibited wild-type Kir1.1 in a dose-dependent manner, but had no effect on either Kir2.1 or Kir4.1 at a concentration of 10 µM. Importantly, however, VU590 at this concentration inhibited Kir7.1 by approximately 65% (FIGURE 5). This was a serendipitous finding because the only other inhibitors of this newest and perhaps least understood family member are the nonselective, millimolar concentration pore blockers Cs+ and Ba2+ [48]. Under the appropriate experimental conditions, VU590 should be a useful tool for sorting out the functions of Kir7.1 in numerous cell types [48–51].
Figure 4. VU590, the first disclosed small-molecule inhibitor of Kir1.1.

(A) Molecular structure of VU590. (B) Concentration–response curve for VU590 inhibition of Kir1.1 recorded in thallium flux assays.
Reproduced with permission from [47].
Figure 5. Selectivity of VU590 among members of the Kir channel family.

Values are mean ± standard error percentage inhibition of the indicated Kir channel by VU590 (darker bars) or Ba2+ (lighter bars). The concentrations of VU590 (µM) and Ba2+ (mM) used are shown below each bar. Current amplitude was recorded at −120 mV.
Reproduced with permission from [47].
Medicinal chemistry efforts around the VU590 scaffold were initiated with the goal of improving its potency and defining the VU590 moieties required for high-affinity Kir1.1 activity. VU590 structure–activity relationships proved shallow, however, yielding only analogs with IC50 values in the 5–100 µM or greater (inactive) range of concentrations. Moving the nitro group from the 4-position in VU590 to the 3- or 2-position or to a 2,4-dinitro analog led to partial or complete loss of potency. Alternate functional groups for the 4-nitro moiety, such as 4-Cl, 4-thiomethyl and 4-methyl ester, also afforded weak inhibitors. All attempts to attenuate the basicity of VU590 by replacing the tertiary amine moieties with amides, sulfonamides and ureas resulted in inactive compounds (FIGURE 6). Thus, VU590 is a unique Kir1.1 inhibitor with a particular balance of physiochemical properties [47].
Figure 6. Determination of VU590 structure–activity relationships through combinatorial chemistry and thallium-flux assays.

(A) VU590 and areas to explore through chemical optimization. (B) Structure–activity relationship of VU590 analogs. (C) Alternate capping agents used to attenuate the basicity of VU590.
Reproduced with permission from [47].
As a first step toward defining the VU590 binding site, a series of electrophysiological experiments were performed to define the ‘sidedness’ of VU590 action on Kir1.1. It was noted that the rate of VU590 inhibition was rather slow, requiring up to 2 min for full block, and washout was incomplete. This suggested that VU590 might first cross the cell membrane to reach an intracellular binding site. It was also apparent that the membrane potential at which Kir1.1 currents rectify became increasingly negative during the onset of channel block. As discussed earlier, rectification is a result of block of outward current by intracellular cations [4,52–54]. The hyperpolarizing shift in rectification potential was therefore interpreted as another indicator of an intracellular binding site [47]. In another series of experiments, it was shown that VU590 could be displaced from the pore by inwardly directed K+ ions [47]. This phenomenon, known as ‘knok-off’ [55], has been reported for the intracellular Kir channel pore blockers chloroquine [13] and nortriptyline [14] (TABLE 1). Taken together, these data support a model in which VU590 crosses the membrane to access an intracellular binding site within the Kir1.1 channel pore. Mutagenesis, electrophysiology and molecular modeling are being used to map the VU590 binding site in Kir1.1 and Kir7.1. These studies will shed light on which structural properties of the channel and molecule confer moderate selectivity to VU590.
Table 1.
Publicly disclosed small-molecule Kir channel modulators.
| Compound | Structure | Kir Target | IC50 (max) |
Selectivity | Ref. |
|---|---|---|---|---|---|
| Amiodarone | 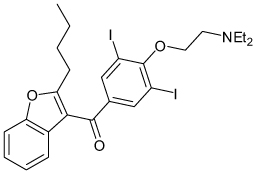 |
IKACh | 1.8–2.4 µM | Known voltage-gated Na, K and Ca channel blocker |
[78] |
| AVE0118 |  |
IKACh | 4.5 µM | Known voltage-gated K channel blocker |
[79] |
| 3-bicyclo[2.2.1] hept-2-yl-benzene- 1,2-diol |
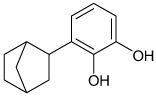 |
Kir2.1 | 60 µM | Kv2.1 > Kir2.1 | [58] |
| Bupivicaine |  |
Kir3 | 22–170 µM† Gβγ and Voltage dependent |
>Kir 1.1, 2.1 Known voltage-gated Na and K channel blocker |
[91] |
| Celastrol | 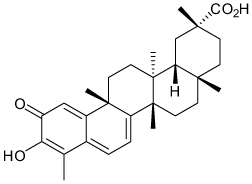 |
Kir2.1 | >20 µ M channel activity 1.2 µ M channel traffcking |
hERG > Kir2.1 > Kv2.1 | [61] |
| Chloroethylclonidine | 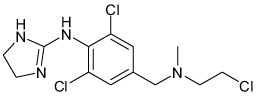 |
Kir2.1 | 37 µM | Known α2 adrenergic agonist |
[62] |
| Chloroquine |  |
Kir2.1 | 8.7 µ M; Voltage dependent |
Unknown | [13] |
| Diphenhydramine |  |
Kir2.3 | 689 µM† (~80%) |
>Kir 2.1 Known antihistamine |
[63] |
| Dronedarone | 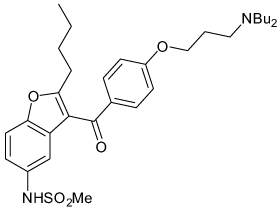 |
IKACh | 10–63 nM | Known voltage-gated Na, K and Ca channel blocker |
[76] |
| Ethosuximide | 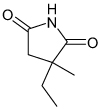 |
Kir3 | 0.4–3.5 mM† (61–76%) |
>Kir 1.1, 2.1 Known voltage-gated Ca and Na channel blocker |
[92] |
| Fluoxetine | 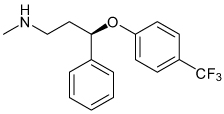 |
Kir3 Kir4.1 |
7–13 µM† (58–74%) 15.2 µM |
>Kir 1.1, Kir 2.1 Known selective serotonin receptor inhibitor |
[90,111] |
| Gambogic acid | 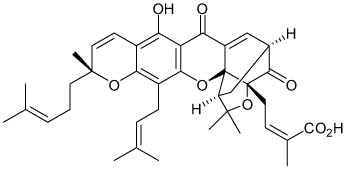 |
Kir2.1 | >10-µM acute application (<50%) 27 nM chronic application 3h |
Kv2.1>Kir2.1>Kir1.1 ~hERG |
[59] |
| Halothane | 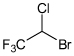 |
Kir3 | 60 µM† | Unknown | [93] |
| JTV-519 | 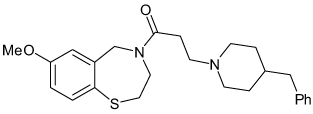 |
IKACh | 0.1–2.5 µM | Known voltage-gated Ca, Na and K channel blocker |
[77] |
| Mepyramine | 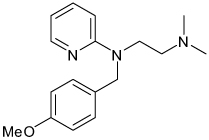 |
Kir2.3 Kir3.4 |
306 µM† (80%) <300 µM† (not reported) |
>Kir 2.1 Known antihistamine |
[63] |
| NIP-142 | 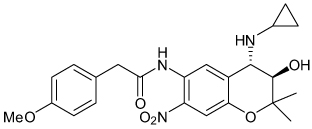 |
Kir3.1/3.4 | 0.64 µM | Unknown Blocks Kv1.5 with similar potency |
[80] |
| NIP-151 | Structure not disclosed | Kir3.1/3.4 | 1.6 nM | Unknown HERG IC50 58 µM |
[81] |
| Pregnenolone Sulfate (activator) |
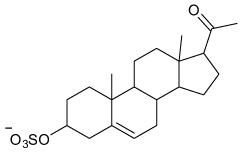 |
Kir2.3 | EC50 16 µM† (80%) |
>Kir1.1, 2.1, 3.1/3.2 | [65] |
| SCH23390 | 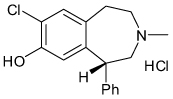 |
Kir3 | 0.3–8 µM | Known D1 receptor antagonist |
[94] |
| Tamoxifen | 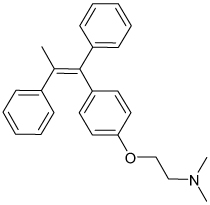 |
Kir2 | 0.3–0.9 µM | Unknown Known estrogen receptor antagonist |
[64] |
| Thioridazine | 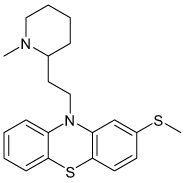 |
Kir3 | 58 µ M (92%) |
Unknown Known antipsychotic |
[89] |
| Tricyclic Antidepressants (Nortriptyline) |
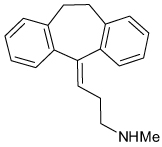 |
Kir3 Kir4.1 |
18–71 µM† (53–76%) 16–38 µ M; voltage dependent (100%) |
>Kir 1.1, 2.1 | [88,110] |
| U50488H | 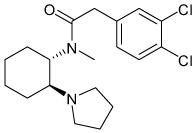 |
Kir3 | 70 µM† | Known κ opioid receptor agonist |
[95] |
| Vernakalant | 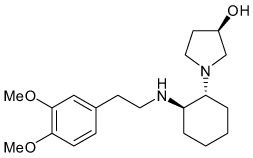 |
I KACh | 10 µM | Known blocker of multiple voltage-gated Na and K channels |
[76] |
Half-inhibition concentrations (IC50) are listed with percent maximal inhibition in parentheses, if full block was not observed.
IC50 values obtained for Kir channels expressed heterologously in Xenopus oocytes, in which significantly higher small-molecule IC50 values have been observed when compared with mammalian cells [80].
Kir2
The Kir2 family members 2.1–2.4 are strong rectifiers expressed primarily in heart, brain and skeletal muscle, with lesser expression in the kidney and gut [1,2]. Dominant negative mutations in Kir2.1 underlie Andersen–Tawil syndrome characterized by periodic paralysis, cardiac ventricular arrhythmias related to prolonged QT interval and dysmorphic facial features [56]. Thus, Kir2.1 is an unlikely candidate for drug-development efforts. However, some have suggested that Kir2.3 antagonists may be useful for certain cardiac arrhythmias. Unlike Kir2.1, which is expressed in both ventricular and atrial myocardium, Kir2.3 is primarily expressed in atrial tissue leading some to postulate that selective Kir2.3 antagonists may be useful for the treatment of atrial fibrillation (discussed in the following section) [57].
Levitan and colleagues performed a high-throughput screen for small-molecule modulators of Kir2.1 heterologously expressed in yeast [58]. Their assay exploits the ability of Kir2.1 (as well as other K+ channels) to rescue a yeast strain deficient in two K+-transport proteins, TRK1 and TRK2, when grown under low-K+ conditions that would otherwise kill the yeast. In a screen of 10,000 small molecules, they identified several compounds that inhibited Kir2.1-dependent yeast growth. The first published compound, termed 3-bicyclo[2.2.1] hept-2-yl-benzene-1,2-diol (TABLE 1), inhibits Kir2.1 in a voltage-independent manner with an IC50 of approximately 60 µM. Interestingly, however, they found the compound preferentially inhibits the voltage-gated K+ channel Kv2.1 with much greater potency (IC50 = 1 µM). They went on to show that 3-bicyclo[2.2.1] hept-2-yl-benzene-1,2-diol has neuroprotective effects associated with inhibition of native Kv2.1 channels in neuronal cultures [58]. This study highlights how post-HTS secondary assays can lead to unexpected and important discoveries in ion channel pharmacology.
The second Kir2.1 inhibitor reported from the yeast screen is gambogic acid (GA) [59], a potent inducer of apoptosis that has been considered for anticancer therapy (TABLE 1). During acute applications, GA inhibits Kir2.1 by only 30% and Kv2.1 by 70% at 10 µM. During prolonged exposures (up to 3 h), however, the compound inhibits Kir2.1 with an IC50 of 27 nM, an effect that was not observed in Kv2.1, Kir1.1 or hERG K+ channels. They further showed that 1-µM GA causes Kir2.1 and Kv2.1 partitioning into biochemically distinct (i.e., Triton X-100-soluble) plasma membrane microdomains, suggesting that perturbation of channel interactions with regulatory proteins and/or lipids that are specific to Kir2.1 accounts for the GA-induced inhibition of channel activity [59].
Min Li and co-workers developed another assay that uses atomic absorption spectro-photometry to monitor recovery of Kir2.1 channel activity after chemically inactivating the channels present in the plasma membrane [60]. Using this Functional Recovery After Chemobleaching (FRAC) assay, they screened 2000 small molecules for compounds that altered the time course of recovery [61]. The neuroprotective compound celastrol was found to reduce cell-surface expression of Kir2.1 at sub-micromolar (e.g., 200-nM) concentrations and channel activity at low micromolar (e.g. 20-µM) concentrations. Celastrol had similar effects on hERG (TABLE 1) [61].
A handful of other low-affinity inhibitors of Kir2 channels have been identified in studies motivated by the intent of implicating Kir2 antagonism in drug intoxication-related cardiac or neurologic side effects. For example, the arrhythmogenic effects of chloroethylclonidine and the first-generation anti-histamines mepyramine and diphenhydramine (TABLE 1) have been attributed to low-affinity Kir2.1 antagonism [62,63]. Ventricular arrhythmias may also occur with toxic levels of the antimalarial agent chloroquine. Chloroquine inhibits Kir2.1 with an IC50 of approximately 10 µM (TABLE 1). Electrophysiological analysis revealed that chloroquine blocks Kir2.1 in a voltage-dependent fashion and exhibits knock-off with inwardly directed K+ current, suggesting a binding site located in the cytoplasmic pore. Site-directed mutagenesis and molecular docking simulations indicated that chloroquine binds to a superficial location in the cytoplasmic pore through electrostatic interactions with the negatively charged residues E224, D259, E299 and D255 near RM1 and RM2 (FIGURE 1), with a minor contribution from hydrophobic interactions with a nearby phenylalanine residue [13]. These data suggest that the cytoplasmic pore region around RM1 and RM2 may potentially represent a targetable small-molecule binding site. The estrogen receptor antagonist tamoxifen (TABLE 1) blocks Kir2 channels with slightly greater potency for Kir2.3 (IC50 0.31 µM) over Kir2.1 and Kir2.2 (IC50 0.93 and 0.87 µM) and is thought to interfere with the channel PIP2 interaction. Tamoxifen’s preferential inhibition of Kir2.3 parallels its slight preference for atrial over ventricular inward rectifier current suggesting that selective Kir2.3 inhibitors may indeed dampen atrial arrhythmias and have little effect on ventricular excitability [64].
Investigators studying the effects of neurosteroids on Kir2 channels unexpectedly found that pregnenolone sulfate (PS) preferentially activates Kir2.3 with a half-maximal concentration of 16 µM and a maximal current potentiation of approximately 80% (TABLE 1). PS appears to be selective in that it has no effects on Kir1.1, Kir2.1, Kir2.2 or Kir3.1 currents. Several structurally related neurosteroids were found to be inactive. Preliminary data suggest that PS acts directly on Kir2.3 at an extracellular site, although the precise binding site location has not yet been defined [65]. Most Kir channel activators act intracellularly to change channel gating. These data raise the intriguing possibility that extracellularly acting small molecules may also modulate Kir channel gating.
Kir3 (GIRK)
Kir3 channels are distinguished from other inward rectifiers in that their activity is critically regulated by G protein-coupled receptors (GPCRs) [2]. Accordingly, members of this family are often referred to as GPCR-activated inward rectifier K+ channels (GIRK). These channels exhibit a low open-state probability under basal conditions, but are activated through a Gβγ-dependent pathway following GPCR stimulation [66]. In mammals, the GIRK subfamily is comprised of four members: Kir3.1, Kir3.2, Kir3.3 and Kir3.4. GIRK channels exist primarily as heterotetramers, with Kir3.1/3.2 predominating in the nervous system and Kir3.1/3.4 in the heart [67,68]. While Kir3.1 subunits do not form functional homomeric channels, other homomeric and heteromeric subunit combinations are possible when expressed in heterologous systems. Except for the prominent role of Kir3.2/3.3 heteromers in midbrain dopaminergic signaling, the physiological significance of other subunit combinations remains unclear [68–71]. In general, Kir3 channels activated by GPCR signaling pathways dampen excitability broadly throughout the nervous system and in the heart. GIRK channels are thus uniquely poised to function as drug targets for disorders of excessive cell excitability, such as cardiac arrhythmias, seizure disorders and pain. However, the limited pharmacology of the GIRK subfamily has thus far hampered efforts to fully evaluate their actual therapeutic potential.
Kir3.1/3.4 heteromeric channels are expressed primarily in atrial myocytes of the heart where they are functionally coupled to M2 muscarinic GPCRs. Acetylcholine released onto M2 receptors following vagal nerve stimulation induces an outward K+ current through Kir3.1/3.4 channels, which hyperpolarizes the cell membrane potential. The classical term for this acetylcholine-activated inward rectifier K+ current is IKACh [69]. Activation of IKACh is thought to have several important effects on cardiac function, including slowing of the heart rate and shortening of the atrial myocyte action potential and effective refractory period [72].
Studies of Kir3 knockout mice have raised intriguing questions about the therapeutic potential of cardiac GIRK for the treatment of atrial fibrillation, the most common cardiac dysrhythmia encountered in clinical practice [73]. Knockout of Kir3.1 or Kir3.4 abolishes IKACh, confirming their role in acetylcholine modulation of cardiac function [74,75]. Kir3.4 knockout mice exhibit unchanged ambulatory heart rate, reduced heart rate variability and resistance to atrial fibrillation [72,75]. These and other data (discussed below) suggest that small-molecule antagonists of cardiac GIRK may act as anti-arrhythmic agents with little effect on resting cardiac function.
Several anti-arrhythmics are limited by adverse off-target effects on ventricular ion channels and excitability with resultant ventricular arrhythmias or by noncardiac side effects. A recent focus has been the development of agents that selectively target atrial excitability. Cardiac GIRK expression is largely restricted to the atria, suggesting it may be a suitable target for drug development [57]. A growing body of evidence lends support to this notion. For example, currently prescribed medications, such as amiodarone, as well as several drugs in development for atrial fibrillation (vernakalant, dronedarone, AVE0118, JTV-519) are known inhibitors of IKACh (TABLE 1). Vernakalant has reached clinical trials and shown efficacy in terminating recent onset atrial fibrillation [76]. However, vernakalant, AVE0118, and JTV-519 also inhibit Kv1.5 or Kv4 channels expressed in the heart [76–79]. It is therefore difficult to conclude from studies with these multi-channel blockers whether selective GIRK antagonists will be therapeutically beneficial.
Nissan Chemical Industries has developed several benzopyrane derivatives in an attempt to target cardiac GIRK selectively. One of their first disclosed compounds, NIP-142 (TABLE 1), inhibits Kir3.1/3.4 channels with submicromolar affinity, but also blocks Kv1.5 channels with similar potency. NIP-142 has shown efficacy in terminating vagally mediated atrial fibrillation with mild prolongation of ventricular repolarization [80]. Another derivative, NIP-151, more potently inhibits Kir3.1/3.4 channels with an IC50 of 1.6 nM and terminates atrial fibrillation without any effect on ventricular excitability in animal models [81]. Future studies comparing the efficacy of NIP-151 and NIP-142 or vernakalant may be helpful in establishing the therapeutic potential of selective Kir3 inhibitors for atrial fibrillation.
Kir3.1/3.2 heteromeric channels are broadly expressed in the nervous system, where they act as effectors for a myriad of neurotransmitter systems including GABA, opioid, glutamate, adenosine, and dopamine. In general, GIRK channels are found postsynaptically in perisynaptic regions although limited postsynaptic density and presynaptic expression has been reported [68]. GIRK activation leads to membrane hyperpolarization and dampening of postsynaptic excitation [82,83]. GIRK channels fine-tune synaptic transmission underlying memory storage, control of seizure activity, reinforcement of addictive substance use and regulation of pain sensation.
Consistent with their broad distribution and negative feedback on excitatory synaptic transmission, Kir3.1- and Kir3.2-knockout mice exhibit many behavioral phenotypes including hyperactivity, reduced anxiety behavior, diminished administration of addictive substances, lower seizure threshold, hyperalgesia and diminished opioid-induced analgesia [84–86]. By contrast, Kir3.3 knockout mice exhibit only reduced self-administration of cocaine and possible hyperalgesia in certain experimental paradigms consistent with the more limited midbrain expression of Kir3.3 subunits, particularly in the dopaminergic reward pathway [70,87]. In light of these phenotypes, it is difficult to envision centrally acting Kir3.1/3.2 channel modulators providing therapeutic benefit without significant deleterious side effects. However, genetic models are mired with developmental compensatory mechanisms and may not accurately reflect beneficial or adverse responses to acute and graded pharmacologic modulation in genetically normal individuals.
At present, most of the work on neuronal GIRK pharmacology has focused on well-characterized neurologic drugs exhibiting low-affinity activity toward GIRK. For example, the antipsychotic agent thioridazine, the tricyclic antidepressant nortriptyline, the serotonin reuptake inhibitor fluoxetine, the anesthetics bupivacaine and halothane and the anti-epileptic agent ethosuximide have all been shown to inhibit Kir3 channels at micromolar-to-millimolar concentrations (TABLE 1) [88–93]. Interestingly, two nanomolar-affinity GPCR-directed small molecules, SCH23390 (D1 receptor antagonist) and U50488H (κ-opioid receptor agonist) were found to directly inhibit GIRK at low micromolar concentrations [94,95]. Some investigators have proposed that the off-target activity toward GIRK may explain some of the adverse side effects observed during high-dose intoxication events with these agents. To our knowledge, however, no direct role of GIRK antagonism in drug toxicity has been established.
Kir3.1/3.2 activators may be useful in pain management. Opiates remain the most clinically effective analgesics primarily because they activate receptors that dampen synaptic transmission at multiple points along the pain pathway. GIRK channels represent an effector pathway for opiates and therefore theoretically possess the same capability to diffusely diminish pain pathway activity [96]. Furthermore, GIRK activators may not share opiate related side effects such as addiction, tolerance, withdrawal, and constipation [97]. Ultimately, however, a clear understanding of the therapeutic potential of neuronal GIRK channels awaits the development of selective Kir3.1/3.2 channel modulators.
Kir4 & Kir5
Kir4 (Kir 4.1 and 4.2) and the Kir5.1 family members are expressed primarily in CNS and retinal glial cells, inner ear cochlear cells and kidney distal tubular epithelial cells [98–102]. Kir4 subunits can form functional homomeric channels, whereas Kir5.1 must co-assemble with Kir4 to participate in ion channel function [103].
Genetic knockout of Kir4.1 in mice leads to CNS myelination defects, persistent neuronal depolarization secondary to diminished glial cell K+ uptake, retinal dysfunction and cochlear dysfunction [104–106]. Similarly, patients with a recently discovered genetic syndrome termed EAST or SeSAME syndrome due to loss-of-function mutations in Kir4.1 exhibit seizures, sensorineural deafness, ataxia and mental retardation [107,108]. These findings confirm that Kir4.1 plays a critical role in K+ homeostasis in the human CNS. Given these adverse phenotypes, the therapeutic value of centrally active Kir4.1 antagonists is doubtful. It is formally possible, however, that Kir4/5 channel activators may promote glial cell K+ uptake and thereby exhibit some anti-epileptic activity [109].
Interestingly, SeSAME syndrome patients also exhibit renal Na+ wasting, raising the possibility that Kir4.1 in the nephron may be a target for novel diuretics. In theory, these could offer some advantages over existing diuretics, all of which target transport proteins located on the apical tubular membrane. This requires that a diuretic is filtered or secreted into the urinary filtrate to reach its target. These agents lose potency with diminishing renal function, a common problem with diuretic use in chronic kidney disease and congestive heart failure patients [36]. Antagonists targeting basolaterally expressed Kir4/5 should increase renal salt wasting independent of renal function since their molecular target is located on the blood side of the nephron [101,102]. Of course, these compounds would have to be restricted from crossing the blood–brain barrier to avoid the aforementioned neurologic consequences.
No selective Kir4/5 modulators have thus far been disclosed. However, investigators studying antidepressant drug modulation of glial cell function have demonstrated that many of these drugs inhibit Kir4 with relatively low affinity. Tricyclic antidepressants almost universally block Kir4.1 currents with nortriptyline exhibiting a voltage-dependent IC50 ranging from 16–38 µM. The selective serotonin reuptake inhibitor (SSRI) fluoxetine similarly inhibits Kir4.1 with an IC50 of 15 µM, but in a voltage-independent fashion. Unlike the tricyclic antidepressants, SSRI activity toward Kir4.1 appears to be restricted to fluoxetine and sertraline. These agents are relatively selective among Kir channels exhibiting minimal inhibition of Kir1.1 and Kir2.1 channels (TABLE 1) [110,111]. However, as noted earlier, other studies have noted that antidepressants also inhibit Kir3 currents although it is difficult to directly compare potency of these agents against Kir3 and Kir4.1 given significant differences in study conditions. Interestingly, the antidepressant activity profile for Kir3 and Kir4.1 channels appears similar with near universal block by tricyclic antidepressants and more restricted SSRI activity centered around fluoxetine [88,110,111]. The common structure–activity relationship between channel families suggests a common Kir channel antidepressant binding site. More recent studies on antidepressant block of Kir4.1 using site-directed mutagenesis and structural modeling have identified a binding site centered around electrostatic interactions involving polar residues (E158 and T128) of the RC region (FIGURE 1) deep within the channel pore [14]. These data highlight a recurring theme in the current state of Kir channel small-molecule pharmacology involving the cytoplasmic entry of antagonists into the channel pore with resultant binding either deep in the pore as with antidepressants and Kir4.1 or superficially as with chloroquine and Kir2.1. Since VU590 block of Kir1.1 currents behaves similarly in electrophysiology experiments (see above), mapping its binding site within the Kir1 and Kir7 channel pore should add to our understanding of these binding sites including the characteristics that engender selectivity and potency.
Kir6
Kir6.1 and Kir6.2 constitute the pore-forming subunits of the adenosine-5’-triphosphate-inhibited KATP channels that couple cellular metabolism to membrane excitability in several key cell types, including pancreatic β cells, vascular smooth muscle, cardiac sarcolemma and brain. The native KATP channel complex is comprised of four Kir6 subunits and the same number of sulfonylurea receptor proteins, the latter of which confer channel sensitivity to numerous small-molecule inhibitors and activators. Some of these are currently in use clinically for the treatment of a variety of disorders such as neonatal and adult-onset diabetes, hyperinsulinism, hypertension, cardiac arrhythmia, angina and alopecia. The physiology and molecular pharmacology of the KATP channel family has been reviewed extensively elsewhere [2,112–114] and will not be discussed further here.
Kir7
Kir7.1 is the newest member of the Kir family and is expressed primarily in brain, intestine, kidney and retina [48]. The recent discovery of Kir7 mutations in patients with Snowflake vitreoretinal degeneration has brought attention to the role of Kir7 in K+ homeostasis of the retinal pigmented epithelium (RPE) [115]. The mixed Kir1.1/7.1 channel blocker VU590 [47] should be useful in understanding the physiology of Kir7.1 in the RPE because, to our knowledge, Kir1.1 is not expressed in those cells.
In polarized epithelial cells of the intestine and nephron, Kir7.1 expression appears to be limited to the basolateral membrane [49,51]. Based on their subcellular co-localization in the gut, it has been postulated that Kir7.1 is functionally coupled to the Na+-K+-ATPase and Na+-K+-2Cl− co-transporter and thereby contributes to transepithelial Cl− secretion [51]. If this model is correct, gut-specific Kir7.1 antagonists may be useful in managing secretory diarrhea [116]. However, to our knowledge, there is currently no evidence to suggest that Kir7.1 channels are functional in the intestine due to their low single-channel conductance [48] and previous lack of pharmacological tools. Because Kir1.1 is not expressed in the intestine, VU590 should help fill in this gap.
Future perspective
An emerging body of physiological and genetic evidence has raised important questions regarding the therapeutic value of inward rectifiers in the treatment of several common cardiovascular and neurological disorders. Despite this potential, the small-molecule pharmacology of the Kir channel family is essentially undeveloped and comprised largely of low-affinity, nonselective cardiovascular and neurologic drugs (TABLE 1). The recent discovery of small-molecule modulators of Kir1.1 in Tl+ flux-based HTSs [47] is timely and represents what we anticipate to be a new era in targeted drug-discovery efforts directed toward this important channel family. The Tl+ flux assay should be amenable for screening most inward rectifiers.
We further anticipate that academic laboratories, such as our own, will play an increasingly important role in the development of ion channel probes. Traditionally, this arena has been dominated by the pharmaceutical industry whose primary goal is to develop profitable drugs. Compounds that do not show immediate promise for drug development are often dropped and never disclosed to the public. As discussed throughout this review, there is a pressing need for the discovery of new tools to support basic science efforts to understand the physiology and, in some cases, the druggability of inward rectifiers. We expect that the National Institutes of Health Molecular Libraries Program [201], which funds industry-quality HTS and probe development by academic scientists, will continue to play an indispensible role in these endeavors.
Finally, we anticipate that advances in structural and computational biology will play an important role in developing the small-molecule pharmacology of the inward rectifier family. With the availability of several high-resolution x-ray structures of bacterial and mammalian Kir channel proteins, our structural understanding of inward rectifiers is probably the most advanced of any ion channel family. In this paradigm, small-molecule probes will be used in classical site-directed mutagenesis and electrophysiological experiments to define their molecular binding sites. Computational modeling techniques based on existing x-ray structures and the physicochemical properties of small molecules can then be deployed to explore the molecular interactions in detail. Structure-based hypotheses are then formulated and tested experimentally using mutagenesis, medicinal chemistry and electrophysiology. These multidisciplinary studies will ultimately lead to an atomic-level understanding of selective drug binding sites in Kir channels and enable advanced cheminformatics and virtual screening-based methods of drug discovery.
Executive summary
- Selective Kir channel modulators have several potential therapeutic applications:
- Kir1.1 antagonists as K+-sparing diuretics for treatment of hypertension and congestive heart failure
- Kir2.3 and Kir3.1/3.4 antagonists for treatment of atrial fibrillation
- Kir3.1/3.2 activators for treatment of pain
- Kir4.1 peripherally active antagonists as renal function-independent diuretics for the treatment of hypertension
- Kir7.1 gut-specific antagonists for secretory diarrhea
Thallium flux-based fluorescence assays have been used to discover novel modulators of Kir1.1 by HTS and should be broadly applicable to other members of the channel family.
Existing high-resolution x-ray structures should be instrumental in developing the small-molecule pharmacology of the Kir channel family.
Acknowledgements
We gratefully acknowledge Jonathan Sheehan and Jens Meiler (Vanderbilt Center for Structural Biology) for developing the original Kir channel structural model [11] from which FIGURE 1 was derived.
KEY TERM
- Loop diuretic
Class of diuretic that acts on the loop of Henle of the nephron to inhibit sodium chloride and water reabsorption from the urine
- Hypokalemia
Reduction of serum K+ below 3.5 mEq/l
- Knock-off
Voltage-dependent displacement of a blocker from an ion channel pore
- Arrhythmias
Abnormal electrical activity in the heart
- Acetylcholine
Neurotransmitter that slows the heart rate through activation of GIRK channels
- Retinal pigmented epithelium
Pigmented epithelial cell layer that provides essential support functions to the interposed photoreceptors of the eye
Footnotes
Financial & competing interests disclosure
The authors are supported by NIH grants 1R21NS5704111 (Jerod S Denton) and 1U54MH084659-01 (Brian A Chauder), an American Heart Association-Southeast Affiliate Beginning Grant-In-Aid (Jerod S Denton), a National Kidney Foundation Postdoctoral Fellowhip grant (Gautam Bhave) and the Vanderbilt Department of Anesthesiology BH Robbins Scholars Program (Daniel Lonergan).The authors have no other relevant affliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu. Rev. Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 2. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 2010;90(1):291–366. doi: 10.1152/physrev.00021.2009. ▪ Most recent extensive review of the Kir channel family.
- 3. Ho K, Nichols CG, Lederer WJ, et al. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362(6415):31–38. doi: 10.1038/362031a0. ▪ Describes the cloning and functional properties of the first member of the Kir channel family, renal outer medullary K+ (ROMK).
- 4.Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- 5.Lu Z. Mechanism of rectification in inward-rectifier K+ channels. Annu. Rev. Physiol. 2004;66:103–129. doi: 10.1146/annurev.physiol.66.032102.150822. [DOI] [PubMed] [Google Scholar]
- 6. Kuo A, Gulbis JM, Antcliff JF, et al. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300(5627):1922–1926. doi: 10.1126/science.1085028. ▪ Describes the first crystal structure of a full-length bacterial Kir channel.
- 7.Pegan S, Arrabit C, Zhou W, et al. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 2005;8(3):279–287. doi: 10.1038/nn1411. [DOI] [PubMed] [Google Scholar]
- 8.Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO. J. 2007;26(17):4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishida M, MacKinnon R. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 a resolution. Cell. 2002;111(7):957–965. doi: 10.1016/s0092-8674(02)01227-8. [DOI] [PubMed] [Google Scholar]
- 10. Tao X, Avalos JL, Chen J, MacKinnon R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science. 2009;326(5960):1668–1674. doi: 10.1126/science.1180310. ▪ Describes the first crystal structure of a full-length eukaryotic Kir channel.
- 11.Fallen K, Banerjee S, Sheehan J, et al. The Kir channel immunoglobulin domain is essential for Kir1.1 (ROMK) thermodynamic stability, trafficking and gating. Channels (Austin) 2009;3(1):57–68. doi: 10.4161/chan.3.1.7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rapedius M, Fowler PW, Shang L, Sansom MS, Tucker SJ, Baukrowitz T. H bonding at the helix-bundle crossing controls gating in Kir potassium channels. Neuron. 2007;55(4):602–614. doi: 10.1016/j.neuron.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rodriguez-Menchaca AA, Navarro-Polanco RA, Ferrer-Villada T, et al. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc. Natl Acad. Sci. USA. 2008;105(4):1364–1368. doi: 10.1073/pnas.0708153105. ▪▪ Exemplary study delineating the mechanism of chloroquine block of Kir2.1 using electrophysiology, mutagenesis and molecular modeling.
- 14. Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol. Pharmacol. 2009;75(6):1287–1295. doi: 10.1124/mol.108.052936. ▪▪ Exemplary study delineating the mechanism of antidepressant block of Kir4.1 using electrophysiology, mutagenesis and molecular odeling.
- 15. Lu M, Wang T, Yan Q, et al. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice. J. Biol. Chem. 2002;277(40):37881–37887. doi: 10.1074/jbc.M206644200. ▪ Confirms that ROMK is required for secretory K+ channel activity in the nephron.
- 16.Hebert SC, Friedman PA, Andreoli TE. Effects of antidiuretic hormone on cellular conductive pathways in mouse medullary thick ascending limbs of Henle: I. ADH increases transcellular conductance pathways. J. Membr. Biol. 1984;80(3):201–219. doi: 10.1007/BF01868439. [DOI] [PubMed] [Google Scholar]
- 17.Hebert SC, Andreoli TE. Effects of antidiuretic hormone on cellular conductive pathways in mouse medullary thick ascending limbs of Henle: II. Determinants of the ADH-mediated increases in transepithelial voltage and in net Cl− absorption. J. Membr. Biol. 1984;80(3):221–233. doi: 10.1007/BF01868440. [DOI] [PubMed] [Google Scholar]
- 18.Hebert SC. Roles of Na+-K+-2Cl− and Na-Cl cotransporters and ROMK potassium channels in urinary concentrating mechanism. Am. J. Physiol (44) 1998;275:F325–F327. doi: 10.1152/ajprenal.1998.275.3.F325. [DOI] [PubMed] [Google Scholar]
- 19.Frindt G, Shah A, Edvinsson J, Palmer LG. Dietary K regulates ROMK channels in connecting tubule and cortical collecting duct of rat kidney. Am. J. Physiol. Renal Physiol. 2009;296(2):F347–F354. doi: 10.1152/ajprenal.90527.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang WH, Giebisch G. Regulation of potassium (K+) handling in the renal collecting duct. Pflugers Arch. 2009;458(1):157–168. doi: 10.1007/s00424-008-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simon DB, Karet FE, Rodriguez-Soriano J, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet. 1996;14(2):152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 22.Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat. Clin. Pract. Nephrol. 2008;4(10):560–567. doi: 10.1038/ncpneph0912. [DOI] [PubMed] [Google Scholar]
- 23.Peters M, Ermert S, Jeck N, et al. Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. 2003;64(3):923–932. doi: 10.1046/j.1523-1755.2003.00153.x. [DOI] [PubMed] [Google Scholar]
- 24.Schulte U, Hahn H, Konrad M, et al. pH gating of ROMK (Kir1.1) channels: control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proc. Natl Acad. Sci. USA. 1999;96(26):15298–15303. doi: 10.1073/pnas.96.26.15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flagg TP, Tate M, Merot J, Welling PA. A mutation linked with Bartter’s syndrome locks Kir 1.1a (ROMK1) channels in a closed state. J. Gen. Physiol. 1999;114(5):685–700. doi: 10.1085/jgp.114.5.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Good DW, Wright FS. Luminal influences on potassium secretion: sodium concentration and fluid flow rate. Am. J. Physiol. 1979;236(2):F192–F205. doi: 10.1152/ajprenal.1979.236.2.F192. [DOI] [PubMed] [Google Scholar]
- 27.Khanna A, Kurtzman NA. Metabolic alkalosis. J. Nephrol. 2006;19 Suppl. 9:S86–S96. [PubMed] [Google Scholar]
- 28.Sansom SC, Welling PA. Two channels for one job. Kidney Int. 2007;72(5):529–530. doi: 10.1038/sj.ki.5002438. [DOI] [PubMed] [Google Scholar]
- 29.Rodan AR, Huang CL. Distal potassium handling based on flow modulation of maxi-K channel activity. Curr. Opin. Nephrol. Hypertens. 2009;18(4):350–355. doi: 10.1097/MNH.0b013e32832c75d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimm PR, Sansom SC. BK channels in the kidney. Curr. Opin. Nephrol. Hypertens. 2007;16(5):430–436. doi: 10.1097/MNH.0b013e32826fbc7d. [DOI] [PubMed] [Google Scholar]
- 31.Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na+-K+-2Cl− cotransporter NKCC2. Nat. Genet. 1996;13(2):183–188. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 32.Hebert SC. Bartter syndrome. Curr. Opin. Nephrol. Hypertens. 2003;12(5):527–532. doi: 10.1097/00041552-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 33.Bailey MA, Cantone A, Yan Q, et al. Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of Type II Bartter’s syndrome and in adaptation to a high-K diet. Kidney Int. 2006;70(1):51–59. doi: 10.1038/sj.ki.5000388. [DOI] [PubMed] [Google Scholar]
- 34. Ji W, Foo JN, O’Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40(5):592–599. doi: 10.1038/ng.118. ▪▪ Reports that heterozygous carriers of KCNJ1 loss-of-function mutations have lower blood pressure, but not pathological derangement of kidney function observed in Bartter syndrome.
- 35. Tobin MD, Tomaszewski M, Braund PS, et al. Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population. Hypertension. 2008;51(6):1658–1664. doi: 10.1161/HYPERTENSIONAHA.108.112664. ▪▪ Reports that single-nucleotide polymorphisms in KCNJ1 are associated with lower blood pressure. Importantly, the authors found no association with serum K+.
- 36.Brater DC. Diuretic therapy. N. Engl. J. Med. 1998;339(6):387–395. doi: 10.1056/NEJM199808063390607. [DOI] [PubMed] [Google Scholar]
- 37.Grobbee DE, Hoes AW. Non-potassium-sparing diuretics and risk of sudden cardiac death. J. Hypertens. 1995;13(12 Pt 2):1539–1545. [PubMed] [Google Scholar]
- 38.Macdonald JE, Struthers AD. What is the optimal serum potassium level in cardiovascular patients? J. Am. Coll. Cardiol. 2004;43(2):155–161. doi: 10.1016/j.jacc.2003.06.021. [DOI] [PubMed] [Google Scholar]
- 39. Weaver CD, Harden D, Dworetzky SI, Robertson B, Knox RJ. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J. Biomol. Screen. 2004;9(8):671–677. doi: 10.1177/1087057104268749. ▪▪ Describes the first high-throughput Tl+ flux assays of K+ channel function.
- 40.Gentles RG, Hu S, Huang Y, et al. Preliminary SAR studies on non-apamin-displacing 4-(aminomethylaryl) pyrrazolopyrimidine KCa channel blockers. Bioorg. Med. Chem. Lett. 2008;18(20):5694–5697. doi: 10.1016/j.bmcl.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 41.Gentles RG, Grant-Young K, Hu S, et al. Initial SAR studies on apamin-displacing 2-aminothiazole blockers of calcium-activated small conductance potassium channels. Bioorg. Med. Chem. Lett. 2008;18(19):5316–5319. doi: 10.1016/j.bmcl.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 42.Titus SA, Beacham D, Shahane SA, et al. A new homogeneous high-throughput screening assay for profiling compound activity on the human ether-a-go-go-related gene channel. Anal. Biochem. 2009;394(1):30–38. doi: 10.1016/j.ab.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Delpire E, Days E, Lewis LM, et al. Small-molecule screen identifies inhibitors of the neuronal K+-Cl- co-transporter KCC2. Proc. Natl Acad. Sci. USA. 2009;106(13):5383–5388. doi: 10.1073/pnas.0812756106. ▪ Describes a Tl+ flux-based high-throughput screen for modulators of a K+-Cl co-transporter.
- 44.Nadeau H, McKinney S, Anderson DJ, Lester HA. ROMK1 (Kir1.1) causes apoptosis and chronic silencing of hippocampal neurons. J. Neurophysiol. 2000;84(2):1062–1075. doi: 10.1152/jn.2000.84.2.1062. [DOI] [PubMed] [Google Scholar]
- 45.Yoo D, Fang L, Mason A, Kim BY, Welling PA. A phosphorylation-dependent export structure in ROMK (Kir1.1) channel overrides an endoplasmic reticulum localization signal. J. Biol. Chem. 2005;280(42):35281–35289. doi: 10.1074/jbc.M504836200. [DOI] [PubMed] [Google Scholar]
- 46.O’Connell AD, Leng Q, Dong K, MacGregor GG, Giebisch G, Hebert SC. Phosphorylation-regulated endoplasmic reticulum retention signal in the renal outer-medullary K+ channel (ROMK) Proc. Natl Acad. Sci. USA. 2005;102(28):9954–9959. doi: 10.1073/pnas.0504332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lewis LM, Bhave G, Chauder BA, et al. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol. Pharmacol. 2009;76(5):1094–1103. doi: 10.1124/mol.109.059840. ▪▪ Describes the first high-throughput screen for modulators of Kir1.1 and the identification of the first small-molecule Kir1.1 and Kir7.1 channel inhibitor, VU590.
- 48.Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. A novel inward rectifier K+ channel with unique pore properties. Neuron. 1998;20(5):995–1005. doi: 10.1016/s0896-6273(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 49.Ookata K, Tojo A, Suzuki Y, et al. Localization of inward rectifier potassium channel Kir7.1 in the basolateral membrane of distal nephron and collecting duct. J. Am. Soc. Nephrol. 2000;11(11):1987–1994. doi: 10.1681/ASN.V11111987. [DOI] [PubMed] [Google Scholar]
- 50.Yang D, Zhang X, Hughes BA. Expression of inwardly rectifying potassium channel subunits in native human retinal pigment epithelium. Exp. Eye Res. 2008;87(3):176–183. doi: 10.1016/j.exer.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakamura N, Suzuki Y, Sakuta H, Ookata K, Kawahara K, Hirose S. Inwardly rectifying K+ channel Kir7.1 is highly expressed in thyroid follicular cells, intestinal epithelial cells and choroid plexus epithelial cells: implication for a functional coupling with Na+, K+–ATPase. Biochem. J. 1999;342(Pt 2):329–336. [PMC free article] [PubMed] [Google Scholar]
- 52.Fakler B, Brandle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 1995;80(1):149–154. doi: 10.1016/0092-8674(95)90459-x. [DOI] [PubMed] [Google Scholar]
- 53.Lu Z, MacKinnon R. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature. 1994;371(6494):243–246. doi: 10.1038/371243a0. [DOI] [PubMed] [Google Scholar]
- 54.Wible BA, Taglialatela M, Ficker E, Brown AM. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature. 1994;371(6494):246–249. doi: 10.1038/371246a0. [DOI] [PubMed] [Google Scholar]
- 55.Shin HG, Lu Z. Mechanism of the voltage sensitivity of IRK1 inward-rectifier K+ channel block by the polyamine spermine. J. Gen. Physiol. 2005;125(4):413–426. doi: 10.1085/jgp.200409242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105(4):511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 57.Ehrlich JR. Inward rectifier potassium currents as a target for atrial fibrillation therapy. J. Cardiovasc. Pharmacol. 2008;52(2):129–135. doi: 10.1097/FJC.0b013e31816c4325. [DOI] [PubMed] [Google Scholar]
- 58. Zaks-Makhina E, Kim Y, Aizenman E, Levitan ES. Novel neuroprotective K+ channel inhibitor identified by high-throughput screening in yeast. Mol. Pharmacol. 2004;65(1):214–219. doi: 10.1124/mol.65.1.214. ▪ Describes a high-throughput assay of mammalian Kir2.1 expressed in yeast.
- 59.Zaks-Makhina E, Li H, Grishin A, Salvador-Recatala V, Levitan ES. Specific and slow inhibition of the Kir2.1 K+ channel by gambogic acid. J. Biol. Chem. 2009;284(23):15432–15438. doi: 10.1074/jbc.M901586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun H, Shikano S, Xiong Q, Li M. Function recovery after chemobleaching (FRAC): evidence for activity silent membrane receptors on cell surface. Proc. Natl Acad. Sci. USA. 2004;101(48):16964–16969. doi: 10.1073/pnas.0404178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun H, Liu X, Xiong Q, Shikano S, Li M. Chronic inhibition of cardiac Kir2.1 and hERG potassium channels by celastrol with dual effects on both ion conductivity and protein trafficking. J. Biol. Chem. 2006;281(9):5877–5884. doi: 10.1074/jbc.M600072200. [DOI] [PubMed] [Google Scholar]
- 62.Barrett-Jolley R, Dart C, Standen NB. Direct block of native and cloned (Kir2.1) inward rectifier K+ channels by chloroethylclonidine. Br. J. Pharmacol. 1999;128(3):760–766. doi: 10.1038/sj.bjp.0702819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu B, Jia Z, Geng X, et al. Selective inhibition of Kir currents by antihistamines. Eur. J. Pharmacol. 2007;558(1–3):21–26. doi: 10.1016/j.ejphar.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 64.Ponce-Balbuena D, Lopez-Izquierdo A, Ferrer T, Rodriguez-Menchaca AA, Arechiga-Figueroa IA, Sanchez-Chapula JA. Tamoxifen inhibits inward rectifier K+ 2.x family of inward rectifier channels by interfering with phosphatidylinositol 4,5-bisphosphate-channel interactions. J. Pharmacol. Exp. Ther. 2009;331(2):563–573. doi: 10.1124/jpet.109.156075. [DOI] [PubMed] [Google Scholar]
- 65.Kobayashi T, Washiyama K, Ikeda K. Pregnenolone sulfate potentiates the inwardly rectifying K+ channel Kir2.3. PLoS One. 2009;4(7):E6311. doi: 10.1371/journal.pone.0006311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sadja R, Alagem N, Reuveny E. Gating of GIRK channels. details of an intricate, membrane-delimited signaling complex. Neuron. 2003;39(1):9–12. doi: 10.1016/s0896-6273(03)00402-1. [DOI] [PubMed] [Google Scholar]
- 67.Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374(6518):135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 68.Koyrakh L, Lujan R, Colon J, et al. Molecular and cellular diversity of neuronal G protein-gated potassium channels. J. Neurosci. 2005;25(49):11468–11478. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mark MD, Herlitze S. G protein-mediated gating of inward-rectifier K+ channels. Eur. J. Biochem. 2000;267(19):5830–5836. doi: 10.1046/j.1432-1327.2000.01670.x. [DOI] [PubMed] [Google Scholar]
- 70.Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Luscher C. Bi-directional effects of GABAB receptor agonists on the mesolimbic dopamine system. Nat. Neurosci. 2004;7(2):153–159. doi: 10.1038/nn1181. [DOI] [PubMed] [Google Scholar]
- 71.Jelacic TM, Kennedy ME, Wickman K, Clapham DE. Functional and biochemical evidence for G protein-gated inwardly rectifying K+ (GIRK) channels composed of GIRK2 and GIRK3. J. Biol. Chem. 2000;275(46):36211–36216. doi: 10.1074/jbc.M007087200. [DOI] [PubMed] [Google Scholar]
- 72.Kovoor P, Wickman K, Maguire CT, et al. Evaluation of the role of IKACh in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 2001;37(8):2136–2143. doi: 10.1016/s0735-1097(01)01304-3. [DOI] [PubMed] [Google Scholar]
- 73.Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. 2004;110(9):1042–1046. doi: 10.1161/01.CIR.0000140263.20897.42. [DOI] [PubMed] [Google Scholar]
- 74.Bettahi I, Marker CL, Roman MI, Wickman K. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J. Biol. Chem. 2002;277(50):48282–48288. doi: 10.1074/jbc.M209599200. [DOI] [PubMed] [Google Scholar]
- 75.Wickman K, Nemec J, Gendler SJ, Clapham DE. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron. 1998;20(1):103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 76.Ehrlich JR, Nattel S. Novel approaches for pharmacological management of atrial fibrillation. Drugs. 2009;69(7):757–774. doi: 10.2165/00003495-200969070-00001. [DOI] [PubMed] [Google Scholar]
- 77.Kaneko N, Matsuda R, Hata Y, Shimamoto K. Pharmacological characteristics and clinical applications of K201. Curr. Clin. Pharmacol. 2009;4(2):126–131. doi: 10.2174/157488409788184972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Watanabe Y, Hara Y, Tamagawa M, Nakaya H. Inhibitory effect of amiodarone on the muscarinic acetylcholine receptor-operated potassium current in guinea pig atrial cells. J. Pharmacol. Exp. Ther. 1996;279(2):617–624. [PubMed] [Google Scholar]
- 79.Gogelein H, Brendel J, Steinmeyer K, et al. Effects of the atrial antiarrhythmic drug AVE0118 on cardiac ion channels. Naunyn Schmiedebergs Arch. Pharmacol. 2004;370(3):183–192. doi: 10.1007/s00210-004-0957-y. [DOI] [PubMed] [Google Scholar]
- 80.Matsuda T, Ito M, Ishimaru S, et al. Blockade by NIP-142, an antiarrhythmic agent, of carbachol-induced atrial action potential shortening and GIRK1/4 channel. J. Pharmacol. Sci. 2006;101(4):303–310. doi: 10.1254/jphs.fp0060324. [DOI] [PubMed] [Google Scholar]
- 81.Hashimoto N, Yamashita T, Tsuruzoe N. Characterization of in vivo and in vitro electrophysiological and antiarrhythmic effects of a novel IKACh blocker, NIP-151: a comparison with an IKr-blocker dofetilide. J. Cardiovasc. Pharmacol. 2008;51(2):162–169. doi: 10.1097/FJC.0b013e31815e854c. [DOI] [PubMed] [Google Scholar]
- 82.Scanziani M. GABA spillover activates postsynaptic GABAB receptors to control rhythmic hippocampal activity. Neuron. 2000;25(3):673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 83.Lujan R, Maylie J, Adelman JP. New sites of action for GIRK and SK channels. Nat. Rev. Neurosci. 2009;10(7):475–480. doi: 10.1038/nrn2668. [DOI] [PubMed] [Google Scholar]
- 84.Pravetoni M, Wickman K. Behavioral characterization of mice lacking GIRK/Kir3 channel subunits. Genes Brain Behav. 2008;7(5):523–531. doi: 10.1111/j.1601-183X.2008.00388.x. [DOI] [PubMed] [Google Scholar]
- 85.Marker CL, Cintora SC, Roman MI, Stoffel M, Wickman K. Hyperalgesia and blunted morphine analgesia in G protein-gated potassium channel subunit knockout mice. Neuroreport. 2002;13(18):2509–2513. doi: 10.1097/00001756-200212200-00026. [DOI] [PubMed] [Google Scholar]
- 86.Marker CL, Stoffel M, Wickman K. Spinal G protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. J. Neurosci. 2004;24(11):2806–2812. doi: 10.1523/JNEUROSCI.5251-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morgan AD, Carroll ME, Loth AK, Stoffel M, Wickman K. Decreased cocaine self-administration in Kir3 potassium channel subunit knockout mice. Neuropsychopharmacol. 2003;28(5):932–938. doi: 10.1038/sj.npp.1300100. [DOI] [PubMed] [Google Scholar]
- 88.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by various antidepressant drugs. Neuropsychopharmacol. 2004;29(10):1841–1851. doi: 10.1038/sj.npp.1300484. [DOI] [PubMed] [Google Scholar]
- 89.Kobayashi T, Ikeda K, Kumanishi T. Inhibition by various antipsychotic drugs of the G-protein-activated inwardly rectifying K+ (GIRK) channels expressed in Xenopus oocytes. Br. J. Pharmacol. 2000;129(8):1716–1722. doi: 10.1038/sj.bjp.0703224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by fluoxetine (Prozac) Br. J. Pharmacol. 2003;138(6):1119–1128. doi: 10.1038/sj.bjp.0705172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhou W, Arrabit C, Choe S, Slesinger PA. Mechanism underlying bupivacaine inhibition of G protein-gated inwardly rectifying K+ channels. Proc. Natl Acad. Sci. USA. 2001;98(11):6482–6487. doi: 10.1073/pnas.111447798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kobayashi T, Hirai H, Iino M, et al. Inhibitory effects of the antiepileptic drug ethosuximide on G protein-activated inwardly rectifying K+ channels. Neuropharmacol. 2009;56(2):499–506. doi: 10.1016/j.neuropharm.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 93.Weigl LG, Schreibmayer W. G protein-gated inwardly rectifying potassium channels are targets for volatile anesthetics. Mol. Pharmacol. 2001;60(2):282–289. doi: 10.1124/mol.60.2.282. [DOI] [PubMed] [Google Scholar]
- 94.Kuzhikandathil EV, Oxford GS. Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzaze pine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol. Pharmacol. 2002;62(1):119–126. doi: 10.1124/mol.62.1.119. [DOI] [PubMed] [Google Scholar]
- 95.Ulens C, Daenens P, Tytgat J. The dual modulation of GIRK1/GIRK2 channels by opioid receptor ligands. Eur. J. Pharmacol. 1999;385(2–3):239–245. doi: 10.1016/s0014-2999(99)00736-0. [DOI] [PubMed] [Google Scholar]
- 96.Pan HL, Wu ZZ, Zhou HY, Chen SR, Zhang HM, Li DP. Modulation of pain transmission by G-protein-coupled receptors. Pharmacol. Ther. 2008;117(1):141–161. doi: 10.1016/j.pharmthera.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ueda H, Ueda M. Mechanisms underlying morphine analgesic tolerance and dependence. Front. Biosci. 2009;14:5260–5272. doi: 10.2741/3596. [DOI] [PubMed] [Google Scholar]
- 98.Li L, Head V, Timpe LC. Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia. 2001;33(1):57–71. doi: 10.1002/1098-1136(20010101)33:1<57::aid-glia1006>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 99.Kofuji P, Biedermann B, Siddharthan V, et al. Kir potassium channel subunit expression in retinal glial cells. implications for spatial potassium buffering. Glia. 2002;39(3):292–303. doi: 10.1002/glia.10112. [DOI] [PubMed] [Google Scholar]
- 100.Lang F, Vallon V, Knipper M, Wangemann P. Functional significance of channels and transporters expressed in the inner ear and kidney. Am. J. Physiol. Cell. Physiol. 2007;293(4):C1187–C1208. doi: 10.1152/ajpcell.00024.2007. [DOI] [PubMed] [Google Scholar]
- 101.Lourdel S, Paulais M, Cluzeaud F, et al. An inward rectifier K+ channel at the basolateral membrane of the mouse distal convoluted tubule. similarities with Kir4-Kir5.1 heteromeric channels. J. Physiol. 2002;538(Pt 2):391–404. doi: 10.1113/jphysiol.2001.012961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lachheb S, Cluzeaud F, Bens M, et al. Kir4.1/ Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am. J. Physiol. Renal Physiol. 2008;294(6):F1398–F1407. doi: 10.1152/ajprenal.00288.2007. [DOI] [PubMed] [Google Scholar]
- 103.Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO. J. 1996;15(12):2980–2987. [PMC free article] [PubMed] [Google Scholar]
- 104.Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J. Neurosci. 2000;20(15):5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 2001;21(15):5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rozengurt N, Lopez I, Chiu CS, Kofuji P, Lester HA, Neusch C. Time course of inner ear degeneration and deafness in mice lacking the Kir4.1 potassium channel subunit. Hear. Res. 2003;177(1–2):71–80. doi: 10.1016/s0378-5955(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 107.Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 2009;360(19):1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scholl UI, Choi M, Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl Acad. Sci. USA. 2009;106(14):5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J. Cell. Mol. Med. 2006;10(1):33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, Kurachi Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J. Pharmacol. Exp. Ther. 2007;320(2):573–580. doi: 10.1124/jpet.106.112094. [DOI] [PubMed] [Google Scholar]
- 111.Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. 2007;1178:44–51. doi: 10.1016/j.brainres.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 112.Jahangir A, Terzic A. KATP channel therapeutics at the bedside. J. Mol. Cell. Cardiol. 2005;39(1):99–112. doi: 10.1016/j.yjmcc.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sattiraju S, Reyes S, Kane GC, Terzic A. KATP channel pharmacogenomics: from bench to bedside. Clin. Pharmacol. Ther. 2008;83(2):354–357. doi: 10.1038/sj.clpt.6100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lefer DJ, Nichols CG, Coetzee WA. Sulfonylurea receptor 1 subunits of ATP-sensitive potassium channels and myocardial ischemia/reperfusion injury. Trends Cardiovasc. Med. 2009;19(2):61–67. doi: 10.1016/j.tcm.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hejtmancik JF, Jiao X, Li A, et al. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am. J. Hum. Genet. 2008;82(1):174–180. doi: 10.1016/j.ajhg.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li Y, Halm DR. Secretory modulation of basolateral membrane inwardly rectified K+ channel in guinea pig distal colonic crypts. Am. J. Physiol. Cell. Physiol. 2002;282(4):C719–C735. doi: 10.1152/ajpcell.00065.2001. [DOI] [PubMed] [Google Scholar]
Website
- 201.US National Institutes of Health Molecular Libraries Program. http://mli.nih.gov/mli.