

Abstract
Tobacco epiaristolochene and hyoscyamus premnaspirodiene synthases (TEAS and HPS) catalyze the cyclizations and rearrangements of (E,E)-farnesyl diphosphate (FPP) to the corresponding bicyclic sesquiterpene hydrocarbons. The complex mechanism proceeds through a tightly bound (R)-germacrene A intermediate and involves partitioning of a common eudesm-5-yl carbocation either by angular methyl migration, or by C-9 methylene rearrangement, to form the respective eremophilane and spirovetivane structures. In this work, the stereochemistry and timing of the proton addition and elimination steps in the mechanism were investigated by synthesis of substrates bearing deuterium labels in one or both terminal methyl groups, and in the pro-S and pro-R methylene hydrogens at C-8. Incubations of the labeled FPPs with recombinant TEAS and HPS, and with the chimeric CH4 hybrid cyclase having catalytic activities of both TEAS and HPS, and of unlabeled FPP in D2O, together with gas chromatography–mass spectrometry (GC–MS) and/or NMR analyses of the labeled products gave the following results: (1) stereospecific CH3 → CH2 eliminations at the cis-terminal methyl in all cases; (2) similar primary kinetic isotope effects (KIE) of 4.25–4.64 for the CH3 → CH2 eliminations; (3) a significant intermolecular KIE (1.33 ± 0.03) in competitive cyclizations of unlabeled FPP and FPP-d6 to premnaspirodiene by HPS; (4) stereoselective incorporation of label from D2O into the 1β position of epiaristolochene; (5) stereoselective eliminations of the 1β and 9β protons in formation of epiaristolochene and its Δ1(10) isomer epieremophilene by TEAS and CH4; and (6) predominant loss of the 1α proton in forming the cyclohexene double bond of premnaspirodiene by HPS and CH4. The results are explained by consideration of the conformations of individual intermediates, and by imposing the requirement of stereoelectronically favorable proton additions and eliminations.
Keywords: Sesquiterpenes, Eremophilanes, Spirovetivane, Germacrane, Enzyme mechanisms, Stereochemistry, Deuterium labeling, Isotope effects, Rearrangements, Cyclizations, Carbocations
Tobacco epiaristolochene and hyoscyamus premnaspirodiene synthases (TEAS3 and HPS) [1,2] are terpene cyclases responsible for the production of the respective sesquiterpene hydrocarbons (2 and 5) in the biosynthesis of the oxygenated phytoalexins capsidiol (4), solavetivone (6), lubimin (7), rishitin (8), and related metabolites in sweet pepper, tobacco, potato, and other Solanaceae (Scheme 1) [3–5].
Scheme 1.

Biosynthesis of the sesquiterpene phytoalexins capsidiol (4), solavetivone (6), lubimin (7), and rishitin (8) from (E,E)-farnesyl diphosphate (1) by way of epiaristolochene (2) and premnaspirodiene (5).
Incorporation of [1,2-13C2]acetate into capsidiol in Capsicum annum cultures and NMR analyses confirmed the occurrence of a methyl migration in the biosynthesis of this eremophilane sesquiterpene [6]. Retention of deuterium at C4 of capsidiol biosynthesized from [4,4-2H2]mevalonate demonstrated that the unusual trans-stereochemistry of the vicinal methyl groups could not be attributed to epimerization of a cis-eremophilane precursor [7].
The hydrocarbon intermediate isolated from elicitor-treated cell cultures of Nicotiana tobacum [8] was identified as (+)-epiaristolochene (2) by comparison with the diene prepared chemically by deoxygenation of capsidiol [9], the structure and stereochemistry of which were established by an X-ray crystallo-graphic analysis [10]. Kinetic analysis of the oxidation of 2 to capsidiol and the potential intermediates epiaristolochen-1β- and -3α-ol with the cytochrome P450 oxidase epiaristolochene dihydroxylase (EAH) established the preferred order of the oxidation steps, 2 → 3 → 4 [11,12]. The sequence of oxidative transformations and intermediates in the conversion of solavetivone to lubimin and rishitin was elucidated by incorporation of deuterium and carbon-13 labeled precursors [13–15]. The presumed sesquiterpene precursor (−)-premnaspirodiene (5) [16] has been synthesized as the pure enantiomer [17] and in racemic form [18], and its identification as the product of HPS rests primarily on GC–MS evidence [19].
The usual mechanism for the closely related processes catalyzed by TEAS and HPS are presented in Scheme 2 [1,2]. Intramolecular alkylation of the terminal double bond of the FPP substrate generates the enzyme bound gremacrene A intermediate (9). Proton-induced cyclization of 9 in a boat-chair conformation leads to a 5,10-syn eudesmyl ion 10A that undergoes 5 → 4 hydride shift to form the branch point intermediate 10B. 1,2-Methyl migration followed by proton elimination give rise to epiaristolochene (2) and epieremophilene (13), whereas ring contraction by C-9 methylene rearrangement and elimination produces premnaspirodiene (5).
Scheme 2.

Proposed mechanisms for the cyclizations of (E,E)-farnesyl diphosphate (1) to epiaristolochene (2), epieremophilene (13), and premnaspirodiene (5) catalyzed by the respective synthases TEAS, HPS, and the CH4 hybrid. The reactions proceed through germacrene A (9), and isomeric eudesmyl (10A and 10B), epi-eremophilenyl (11), and vetispirenyl (12) carbocation intermediates. In the case of TEAS, angular methyl migration is favored along the upper branch to the fused ring product whereas in the case of HPS ring methylene rearrangement on the opposite face and proton elimination at C1 lead to the spiro[5.4]decane product. The CH4 mutant form affords a mixture of both products, together with the double bond isomer epieremophilene (13).
The cloning and over-expression of recombinant TEAS and HPS in Escherichia coli provided sufficient quantities of these interesting cyclases for structural and mechanistic investigations [20]. These two proteins are 77% identical and 81% similar based on amino acid sequence alignment, contain the familiar DDXXD aspartate triad linked to diphosphate binding, and have kinetic characteristics typical of terpene cyclases. One of a series of chimeric proteins resulting from domain swapping designated CH4 catalyzed the conversion of 1 to epiaristolochene, premnaspirodiene, and an unknown product subsequently identified as epieremophilene (13), i.e., the Δ1(10) isomer of 2 [11,21]. Pre-steady state kinetic data are consistent with either a rate-determining cyclization to the germacrene A intermediate or final dissociation of the bicyclic products, and the existence of a single catalytic site on the CH4 hybrid enzyme [22]. X-ray diffraction analysis of crystalline TEAS in the absence and presence of inert substrate mimics revealed a hydrophobic, aromatic-rich binding pocket in the C-terminal domain with twoMg2+ ions positioned near the opening [23]. The production of germacrene A (9) by the point mutant TEAS-Y520-F confirmed the identity of the macrocyclic intermediate and a role for tyrosine 520 in catalysis, perhaps in contributing to an H-bonding network making up the back of the active site pocket [24].
The functionally similar aristolochene synthases isolated from the fungi Aspergillus terreus and Penicillum roquaforti catalyze the conversion of FPP to the 4β-CH3, 7α-isopropenyl diastereomer of 2 [1,2]. However, the amino acid sequences of these fungal cyclases differ appreciably from those of TEAS and HPS. Labeling experiments established that the macrocyclization step occurs with inversion at C-1 of the substrate and proton elimination at the cis-terminal methyl position, and that conversion of the enzyme-bound intermediate to aristolochene takes place with 10 → 5 methyl migration and syn-related proton elimination [25,26]. Indirect evidence supporting the same germacrene A intermediate (9) was obtained by enzyme-catalyzed cyclization of (7R)-6,7-dihydro FPP to 6,7-dihydro germacrene A [27].
The objectives of the present work were to analyze the timing of the cyclization steps and the stereochemistry of the individual protonation and deprotonation events in the mechanism by means of deuterium labeling. In this paper, we report syntheses of farnesyl PPs bearing deuterium in the terminal methyl groups and in the pro-R and pro-S positions at C-8. The stereochemistry of proton transfer steps was elucidated by GC–MS and/or NMR analyses of the products from incubations of the labeled substrates with the three enzymes and from incubations conducted in D2O. Comparisons of the isotope effects and stereospecificities of the different reactions catalyzed by TEAS, HPS, and CH4 afford new insights on the positions of active site acids and bases, on the determinants of product specificity, and on the rate determining steps.
Materials and methods
Analytical methods
1H, 2H, 13C, and 31P NMR spectra were recorded with Varian U400, U500, U500NB or Unity Inova 750NB spectrometers in the SCS NMR Spectroscopy Facility at the University of Illinois. The following solvents and reference values in ppm were used in obtaining the NMR data: CDCl3 (1H, 7.26; 2H, 7.26; 13C: 77.0), benzene-d6 (1H, 7.16; 2H, 7.16; 13C, 128), THF-d8 (1H, 1.73). 31P NMR spectra were externally referenced to 0 ppm with 85% H3PO4. Chemical shifts are in ppm, and coupling constants are in Hertz. NOE spectra were generally recorded after at least three freeze–thaw cycles performed in standard 5-mm NMR tubes. NOE data are reported in the following format: data (MHz, solvent): irrad. (ppm irradiated), obs. δ (data). Mass spectra were recorded on Micromass 70V-SE instruments with either probe or HP 5890 GC inputs. The isotopic contents were calculated with the MATRIX program in the SCS Mass Spectroscopy Laboratory at the UIUC to correct for the isotope distributions and the contributions from natural abundance carbon 13. GC analyses were carried out on a Shimadzu Model 14-A GC with a Rtx-5 30-m fused silica capillary column. The MS fragmentation data were determined from EI (70 eV) mass spectra unless otherwise noted.
Purification and characterization
Organic starting materials and stable intermediates shown in Schemes 3 and 4 were purified in most cases by flash chromatography on silica gel 60 (230–400 mesh ASTM) from Merck [28]. TLC analyses were performed on 250-μm silica gel F254 pre-coated plates. TLC visualizations were performed with 5% phosphomolybdic acid (0.2 M in 2.5% conc. H2SO4/EtOH (v/v)), I2, or anisaldehyde (2.5% (v/v), 1% HOAc, and 3.4% conc. H2SO4 in ethanol). The purity of purified intermediates was judged to be ≥90–95% by TLC and/or GC analyses and NMR spectra. The deuterium content of labeled intermediates in Schemes 3 and 4 was determined by GC–MS analyses as described above. The position of the label was verified by peaks absent in 1H NMR spectra and often also directly from 2H NMR spectra.
Scheme 3.

Synthesis of farnesyl diphosphates labeled with deuterium in the terminal methyl groups: [13,13-2H2]1, [13,13,13-2H3]1, and [12,12,12,13,13,13-2H6]1. R=CH2CH2C(CH3)=CHC(CH3)=CHCH2OBn.
Scheme 4.

Synthesis of farnesyl diphosphates labeled with deuterium in the pro-S and pro-R at C-8: (S)-[8-2H1]1 and (R)-[8-2H1]1. R = (E,E)-CH2CH2C(CH3)=CHCH2OBn.
Reagents and starting materials
Methyl 2-(bis (2,2,2-trifluoroethyl)phosphono)propionate (See Scheme 3) was prepared by methylation of the corresponding phosphonoacetate (NaH, DMSO, methyl 2-(bis-2(2,2,2-trifluoroethyl)phosphono)acetate, CH3I; 25 °C, 5 days, 3.33 g, 64%) [29]. A similar reaction with CD3I gave the labeled phosphonopropionate reagent: 2.98 g (53%). (E,E)-Farnesyl benzyl ether was prepared as described for geranyl benzyl ether (KH, THF, (E,E)-farnesol, benzyl bromide, 70 °C, 16 h; 99, 96% purity) [30]. Aldehyde ether 14 was prepared from farnesyl benzyl ether by the following four steps [31,32]: (a) regioselective hypobromination of the 9,10 double bond (NBS, aq THF, 0 °C, 4.5 h, 76%); (b) conversion to the 9,10 epoxide (powdered KOH, ether, 6 h, 25 °C, 98%) [33]; (c) hydrolysis to the 9,10 diol (70% HClO4, aq THF, 25 °C, 1.5 h, 91% unpurified); (d) oxidative cleavage to aldehyde 14 (NaIO4, aq THF, 5 h, 25 °C, 76%).
Synthesis of deuterium-labeled farnesyl diphosphates
The 7-step synthesis of farnesyl PPs bearing deuterium in one or both terminal CH3 groups from aldehyde 14 is outlined in Scheme 3. Z-Selective olefinations with Still’s trifluoroethyl phosphono-propionates [34] followed by AlD3 reductions [35] of cis-ester 15 afforded the cis-deuterio hydroxy ethers 16-d2 and 16-d5. The key reaction was the reductive substitution effected by first activation as the allylic phosphates 17 [31,36] followed by hydride and deuteride displacements with lithium triethylborohydride and triethylborodeuteride to give labeled farnesyl ethers 18-d2, 18-d3, and 18-d6. The allylic phosphate intermediates proved to be stable to chromatography and prolonged storage in a freezer. Cleavage of the benzyl ether was accomplished by Li–NH3 reductions to form the respective labeled (E,E)-farnesols, [13,13-2H2]-, [13,13,13-2H3]-, and [12,12,12,13,13,13-2H6]19. The positional integrity of the deuterium labels was determined from their 1H and 2H NMR spectra in benzene-d6. Representative preparative procedures and characterization data for [12,12-2H2]farnesol are described in detail below. Procedures and data for other intermediates and labeled compounds reported may be found in a PhD dissertation [37]. (2E,6E,10Z)-[13, 13-H2] 1-benzyloxy-3,7,11-trimethyl-2,6,10-dodecatriene, (2E,6E,10Z)-[13,13-2H2] farnesyl benzyl ether (18-d2) The general method of Welch was followed [31,36]. A solution of 0.291 g (0.88 mmol) of 16-d2 and 0.085 mL (0.83 g, 1.1 mmol) of pyridine in 4 mL of CH2Cl2 was stirred and cooled at 0 °C as diethyl chlorophosphate (0.14 mL, 0.16 g, 0.93 mmol) was added. After 1.5 h, 30 mL of ether was added. The ether solution was washed with 1% HCl (3 × 20 mL), satd. NaHCO3 (1 × 10 mL), and satd. NaCl (1 × 10 mL); dried (Na2SO4); and concentrated. Purification by column chromatography on silica gel (2:1 hexane/ethyl acetate) provided 0.269 g (66%) of diethyl phosphate 17 as a clear oil. The purity of the diethyl phosphate was estimated to be >95% by 1H NMR analysis. Data for the diethyl phosphate: TLC Rf 0.23 (2:1 hexane/ethyl acetate); 1H NMR(500 MHz, CDCl3) δ 7.34(m, 4H, aryl H), 7.28(m, 1H, aryl H), 5.40(t of sextets, 1H, J = 7.3, 1.5 Hz, =CH), 5.37(td, 1H, J = 7.3, 0.8 Hz, =CH), 5.11(t of sextets, 1H, J = 6.8, 1.3 Hz, =CH), 4.50(s, 2H, OCH2Ar), 4.11(q, 4H, J = 7.3 Hz, CH3CH2), 4.03(d, 2H, J = 6.5 Hz, CHCH2O), 2.13(m, 4H, CH2), 2.04(m, 2H, CH2), 1.99(m, 2H, CH2), 1.77(q, 3H, J = 1.5 Hz, CH3), 1.65(s, 3H, CH3), 1.59(s, 3H, CH3), 1.33(td, 6H, J = 7.2, 0.7 Hz, CH2CH3); 2H NMR(77 MHz, CDCl3) δ 4.51(s, CCD2).
A solution of 0.115 g (0.246 mmol) of the above diethyl phosphate in 10 mL of THF was stirred and cooled at 0 °C as 1.4 mL (1 M, 1.4 mmol) of LiBEt3H in THF was added. After 45 min, 3 mL of 1 N HCl and 6 mL H2O were added, and the product was extracted with pentane (3 × 15 mL) and ether (2 × 15 mL). The combined organic layers were washed with 25 mL of H2O and 25 mL of satd. NaCl, dried (MgSO4), and concentrated at reduced pressure. Purification by chromatography on silica gel (50:1 hexane/ethyl acetate) provided 55.4 mg (72%) of [13,13-2H2]farnesyl benzyl ether (18-d2) as a clear colorless oil. The purity was estimated to be >95% by 1H NMR analysis. The 1H NMR data for 18-d2 are similar to those for unlabeled farnesyl benzyl ether except for the following: 1H NMR(500 MHz, CDCl3) δ 5.40(t of sextets, 1 H, J = 6.5, 1.0 Hz, =CH), 1.67(d, 3H, J = 1.5 Hz, CH3), 1.65(s, 3H, CH3), 1.61(s, 1H, CD2H), 1.60(s, 3H, CH3); 2H NMR(77 MHz, CDCl3) δ 1.59(d, J = 2.2 Hz, CD2H). (2E,6E,10Z)-[13,13-2H2]Farnesol(19-d2). The procedure followed one in the literature [35]. [13,13-2H2]Farnesyl benzyl ether (0.0554 g, 0.175 mmol) prepared by phosphate reduction in 5 mL of THF was added to a stirred suspension of 0.060 g (8.64 mmol) of Li rod pieces in 25 mL of NH3 at −78 °C. After 1 h, several drops of 3-hexyne were added, the CO2/isopropyl alcohol bath was removed, MeOH (30 mL) was slowly added, and the majority of the NH3 was removed with a gentle stream of N2. Hexane (20 mL) and 1% HCl (20 mL) were added. The aqueous layer was extracted with hexane (3 × 20 mL) and CH2Cl2 (3 × 20 mL), and the combined organic layers were washed with satd. NaHCO3 (2 × 100 mL) and satd. NaCl (1 × 100 mL), dried (MgSO4), and concentrated. Chromatography on silica gel (8:1 hexane/ethyl acetate) afforded 0.0279 g (71%) of farnesol-d2 (19-d2) as a colorless oil. 2H NMR and MS analyses showed that 18-d2 possessed deuterium only in the cis-methyl position, and the label content was 99.0% d2, and 0.5% d1. GC and 1H NMR analyses determined the purity to be 97.6%. The 1H NMR data are similar to those of unlabeled farnesol. Data for 19-d2: TLC Rf 0.13 (8:1 hexane/ethyl acetate); 1H NMR(500 MHz, benzene-d6) δ 5.39(t, 1H, J = 6.8 Hz, =CH), 5.23(t, 2H, J = 6.3 Hz, =CH), 3.97(d, 2H, J = 6.5 Hz, CH2OH), 2.17(m, 2H, CH2), 2.10(m, 4H, CH2), 1.99(m, 2H, CH2), 1.68(s, 3H, CH3), 1.58(s, 3H, CH3), 1.53(s, 1H, CD2H), 1.47(s, 3H, CH3); 2H NMR(77 MHz, benzene-d6) δ 1.51(d, J = 2.1 Hz, CD2H); MS (EI) m/z 225(16.6), 224(M+, 100), 223(2.7); 99.0% d2, 0.5% d1.
Other methods for effecting the reduction of the dideuterio alcohol 16-d2 to 18-d2 and 18-d3 were evaluated. Although, the two-step sequence (a) SO3, pyridine, [38,39] (b) LiBET3H and LiBEt3D gave labeled products without any loss of positional integrity, it proved difficult to monitor the initial step by TLC owing to rapid hydrolysis of the sulfate salt on silica gel, and the sulfate salt was quite unstable. Conversion to the allylic mesylate (CH3SO2Cl, Et3N, CH2Cl2, 0 °C) and reductive displacement with LiBEt3H and LiBEt3D (THF-CH2Cl2) was also tried. Although this method was successful in the preparation of [16,16,16-2H3](E,E,E)-geranylgeranyl benzyl ether bearing deuterium in the trans-terminal methyl group [32], the same reactions on cis-dideuterio alcohol 16-d2 afforded 17-d2 as a 3:1 mixture of cis- and trans-isomers. Evidently the less stable cis-mesylate undergoes partial isomerization to the trans form during one or both reactions.
(S)- and (R)-[8-2H1]Farnesyl PPs bearing deuterium in the prochiral methylene at C-8 were synthesized from aldehyde 20 [35] as shown in Scheme 4 to elucidate the stereochemistry of the proton elimination step forming the double bond of epiaristolochene. E-Selective Wittig olefination and AlD3 reduction afforded 22-d2. The deuterated aldehyde resulting from ruthenate oxidation underwent enantioselective reductions with (R)- and (S)-alpine borane in moderate yield. The enantiomeric excesses of the resulting labeled alcohols, (S)- and (R)-23-d1, were determined to be 90.4 and 99% by NMR analyses of their camphanate derivatives [40–42]. Biellmann coupling reactions between excess lithio dimethylallyl p-tolyl sulfone [43] and the allylic phosphates (3:1 THF-HMPA, −20 °C, 18 h) provided sulfonyl ethers (R)- and (S)-24-d1. Simultaneous reductive cleavage of the allylic sulfone and the benzyl ether afforded (S)- and (R)[8-2H1]farnesols 19 (100% d1) by GC–MS analyses. In the absence of any obvious way to determine the stereochemistry of the label, we assume predominant inversion occurred in the C–C bond forming reaction like a similar Biellmann coupling employed in the synthesis of (R)-[4-2H1]geranylgeraniol [32].
The labeled farnesols were converted to the diphosphates by formation of the allylic chlorides [44] followed by SN2 displacements with tetrabutylammonium pyrophosphate (CH3CN, 25 °C, 2 h), ion exchange to the NH4 salts, and chromatography on cellulose (yields 46–94%) [45]. The labeled FPPs were characterized by 500 MHz 1H NMR and 162 MHz 31P NMR spectra (D2O, EDTA, ND4OD). The 31P NMR spectra displayed the typical pair of doublets for the diphosphate and the absence of appreciable amounts of inorganic pyrophosphate.
Incubation and product analysis
Incubations of the deuterium-labeled FPPs with recombinant TEAS, HPS, and CH4 enzymes were conducted according to a literature procedure [19] with some modifications. Frozen solutions of recombinant proteins were stored at −78 °C in a storage buffer of 50% glycerol, 25 mM Tris–HCl (pH 7.5), 2.5 mM MgCl2, 0.5 mM β-mercaptoethanol, 0.5 μg/mL leupeptin, and 0.5 mM phenylmethylsulfonyl fluoride. The concentrations were generally 4–10 mg/mL. Stock solutions containing 200 mM Tris–HCl (pH 7.5), 40 mM MgCl2, and 14 μM labeled FPP were mixed in the ratio 1:1:0.66 (Tris–MgCl2/FPP) providing the final incubation buffers. Appropriate amounts of enzyme (0.13 mg) were added to 0.80 mL of the final incubation buffers in glass test-tubes with Teflon-lined caps. The solutions were incubated at 37 °C for 15 min and cooled to 0 °C. The sesquiterpene products were extracted with pentane (typically 2 × 0.3–0.75 mL) containing 0.018 mM α-cedrene as GC standard, a small amount (~ 10–15 μL) of heptane was added, and the combined extracts were concentrated under N2 flow to ca. 20 μL in 1-mL, conical vials for GC and GC–MS analysis. Product yields determined by capillary GC varied considerably, between 2% (100 ng) and 30% (1.5 μg). EI GC–MS analyses for isotope content were conducted by fast scanning through m/z 190–210. In contrast, GC–MS analyses for olefin identification were performed with a full mass scan m/z 55–210.
Incubations of [13,13-2H2]FPP (1-d2), and [13,13,13-2H3]FPP (1-d3) with TEAS
Stock solutions of 200 mM Tris–HCl (pH 7.5), 40 mM MgCl2, and 14 μM labeled FPPs were prepared as described above [19]. Aliquots of the enzyme solutions (0.028 mL, 0.13 mg) were added to two identical solutions containing 0.17 mL of distilled H2O and 0.80 mL of the final incubation buffers. The duplicate solutions were carefully inverted several times to ensure mixing, and were incubated at 37 °C for 15 min. After cooling at 0 °C for 5 min in an ice bath, 300 μL of pentane was added, and the mixtures were vortexed and centrifuged. GC analyses of the pentane layers using the response factor for the α-cedrene standard (0.0063 ng/area) determined the conversions to epiaristolochene to be 25% (1000 ng) and 4% (150 ng) for incubations with [13,13-2H2]FPP and [13,13,13-2H3]FPP, respectively. After separation of the pentane layers from the buffer solutions with glass pipettes, 10 μL of heptane was added, and the combined pentane/heptane layers were carefully concentrated with N2 in 100-μL, cone-bottomed vials. Data from the isotope ratio GC–MS analyses are presented in Table 1.
Table 1.
The deuterium content of the sesquiterpene products obtained from incubations of [13,13,13-2H3]FPP (1-d3), [13,13-2H2]FPP (1-d2), and a 1:4.3 mixture of unlabeled FPP and [12,12,12,13,13,13-2H6]FPP (1-d0 + 1-d6) with TEAS, HPS, and CH4 recombinant enzymes is presented
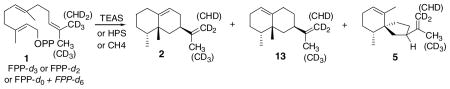 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Enzyme | Product | Substrates, product deuterium contents(%), and isotope effects |
|||||||
| FPP-d3 |
FPP-d2 |
FPP-d0 + FPP-d6 |
|||||||
| d2 | d3 | d1 | d2 | kH/kD | d0 | d5 | kH/kD | ||
| TEAS | Epiaristolochene (2) | 99.5 | 0.5 | 32.0 | 67.2 | 4.27 ± 0.04 | 18.7 | 79.6 | 1.03 ± 0.01 |
| HPS | Premnaspirodiene (5) | 99.6 | 0.4 | 32.2 | 67.3 | 4.25 ± 0.08 | 23.0 | 75.3 | 1.33 ± 0.03 |
| CH4 | Epiaristolochene | 99.0 | 1.0 | 30.2 | 68.8 | 4.64 ± 0.07 | |||
| Epieremophilene (13) | 99.7 | 0.3 | 31.2 | 67.9 | 4.43 ± 0.02 | ||||
| Premnaspirodiene | 99.6 | 0.4 | 30.8 | 68.6 | 4.53 ± 0.01 | ||||
The near quantitative formation of dideuterated products from FPP-d3 reveals that the CH3 → CH2 elimination occurs essentially exclusively at the cis-terminal methyl group. The intramolecular kinetic isotope effects (kH/kD) associated with the CH3→ CH2 elimination were calculated from the deuterium content data of products from FPP-d2. The corresponding intermolecular kinetic isotope effects (kH/kD) were deduced from the ratios of undeuterated and pentadeuterio products obtained from the FPP-d0 + FPP-d6 mixture.
Corrections for the contributions of incompletely labeled FPPs on the fragment mass intensities were nearly insignificant since the isotopic and geometric purities of all labeled farnesols and all labeled FPPs were high. An example correction for the amount of FPP-d1 in FPP-d2 follows. The 0.5% farnesol-d1 detected by MS analysis in the farnesol-d2 would increase the amount of monodeuterated olefin observed in the GC–MS analyses of the sesquiterpenes derived from incubations with the FPP-d2. The apparent amount of deuterium elimination needs to be corrected for this contribution. The measured intramolecular KIE of 4.3 (Table 1) reflects the relative rates of proton and deuteron elimination. The presence of two protons doubles the probability of their elimination with respect to deuterium. Thus of the 0.5% FPP-d1 in FPP-d2, 0.45% is expected to be converted to the monodeuterio olefin [4.3 + 4.3/(4.3 + 4.3 + 1) × 0.5%], and this value was subtracted from the experimentally measured amount of dideuterio olefin in the calculation of the KIE. Similar calculations provided corrections amounting to 1–2% changes on the KIEs shown in Tables 1 and 2. The deuterium content data in Tables 1–3 were derived directly from GC–MS analyses after correction for natural abundance carbon 13.
Table 2.
The deuterium content of sesquiterpene products isolated from incubations of recombinant TEAS, HPS, and CH4 enzymes in a highly enriched D2O medium is presented
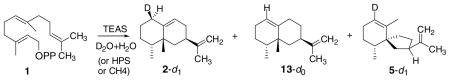 | |||||
|---|---|---|---|---|---|
| Enzyme | D2O (%) | Product | d0 (%) | d1 (%) | kH/kD |
| TEAS | 95 | Epiaristolochene (2) | 9 | 91 | 2.7 |
| Epieremophilene (13) | 87 | 12 | |||
| HPS | 97 | Premnaspirodiene (5) | 9 | 91 | 2.7 |
| CH4 | 92 | Epiaristolochene | 13 | 87 | 1.7 |
| Epieremophilene | 98 | 2 | |||
| Premnaspirodiene | 20 | 80 | 2.9 | ||
The solvent isotope effects shown were calculated from the H2O/D2O ratios in the incubation medium and the deuterium incorporation into the products. The low level of deuterium incorporation into epieremophilene (13, Δ1(10) isomer of 2) is a consequence of the deuteron addition and elimination occurring on the same β face of position 1.
Table 3.
The relative proportions of sesquiterpene products obtained from incubations of unlabeled substrate and substrate bearing isotope label in the pro-S and pro-R position, (S)- and (R)-[8-2H1]FPP, with TEAS and CH4 enzymes, and the deuterium content of the products from the labeled substrates, are presented
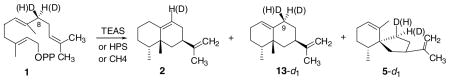 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Enzyme | Product | FPP-d0 | (S)-[8-2H1]FPP | (R)-[8-2H1]FPP | ||||
| % | % | d0 | d1 | % | d0 | d1 | ||
| TEAS | Epiaristolochene (2) | 93.3 ± 0.4 | 84.2 ± 0.4 | 95.3 | 4.7 | 90.3 ± 0.3 | 0 | 100.0 |
| Epieremophilene (13) | 6.7 ± 0.3 | 15.8 ± 0.2 | 0 | 100.0 | 9.7 ± 0.3 | 0 | 100.0 | |
| CH4 | Epiaristolochene | 18.5 ± 0.2 | 7.9 ± 0.1 | 91.6 | 8.4 | 17.1 ± 0.1 | 0 | 100.0 |
| Epieremophilene | 45.1 ± 0.2 | 52.1 ± 0.1 | 0 | 100.0 | 46.1 ± 0.6 | 0 | 100.0 | |
| Premnaspirodiene (5) | 29.7 ± 0.3 | 31.9 ± 0.1 | 0 | 100.0 | 30.0 ± 0.5 | 0 | 100.0 | |
A small peak for an unknown sesquiterpene (7%) was present in the GCs of the CH4 products. The results show predominant elimination of the HS deuterium or hydrogen in forming the 9,10 double bond of epiaristolochene with both enzymes. Complete retention of deuterium is observed in the other products because the final proton eliminations occur at other positions. The proportion of epiaristolochene-d0 is decreased and that of epieremophilene-d1 is increased substantially with (S)-[8-2H1]FPP as substrate.
Incubations of large excesses (600×) of a 1:4.3 (±10%) mixture of unlabeled FPP and [12,12,12,13,13,13-2H6]-FPP (1-d6) with TEAS and HPS were carried out to determine the intermolecular product KIEs (Table 1). The mixed FPP was prepared by combining portions of unlabeled and FPP-d6 solutions. A sample of the mixture was analyzed by negative ion FAB MS (m/z 387(100), 381(23.6); 71.8% d6, 16.8% d0) to determine the isotopic ratio of the unlabeled and labeled FPPs. The conversions of the mixed FPPs to olefinic product(s) and to farnesol were determined to be less than 1% by GC standardization with farnesol or α-cedrene. Thus, changes in the ratio of unlabeled FPP to FPP-d6 in the unreacted diphosphate mixture were insignificant, and this assured that no depletion of unlabeled FPP occurred during the incubations.
Incubations in D2O were performed by procedures similar to those described above for the small scale incubations in H2O. (Table 2) Buffer solutions were prepared with D2O except for the use of the acid (conc. HCl) used to adjust the pD, and the H2O containing enzyme storage buffers. The pHs of the D2O solutions were adjusted to 7.1 instead of 7.5 because of the 0.4 U difference between the pD and the measured pH for high percentage D2O solutions [46]. The percentages of H2O in the resulting D2O buffers were determined by addition of anhydrous sodium acetate and integration of the 1H NMR signals for H2O and the acetate methyl group [46,47]. The isotopic content of the buffers prepared as described was 95–97% D2O.
The location and stereochemical configuration of the deuterium incorporated into epiaristolochene were established by NMR analyses of the monodeuterio olefin produced by large scale TEAS incubations in D2O. Incubations (74 × 8 mL) containing 2.3 mg of TEAS, and extractions with 0.5 and 0.75 mL of a 0.018 mM cedrene solution in pentane, provided a total of 200 μg (GC) of epiaristolochene. The pentane extracts were filtered through a glass Pasteur pipette containing 1.0 g of silica gel. The fractions containing epiaristolochene as determined by TLC and GC analyses were combined and placed in a 5-mm NMR tube. Benzene-d6 (0.75 mL) was added, the solution was carefully concentrated, and appropriate Wilmad Doty susceptibility plugs were inserted into the tube containing ca. 300 μL of benzene-d6. The percentage of D2O in the incubation buffer was 96.3% by NMR standardization with anhydrous sodium acetate [47]. The isotopic content of the purified epiaristolochene was determined by GC–MS analysis to be 88.2% d1. The NMR assignments for epiaristolochene and the NMR analyses of epiaristolochene-d1 isolated from the incubations in enriched D2O are discussed in the following section.
Results
The results from GC–MS analyses of the sesquiterpene hydrocarbon products formed in the incubations of the various labeled forms of FPP with the recombinant TEAS, HPS, and chimeric CH4 enzymes, and from incubations conducted in H2O+D2O mixtures, are presented in Tables 1–3. The loss of a single deuterium (≥99%) with FPP-d3 establishes that the methyl-methylene eliminations leading to the germacrene A intermediate occur stereospecifically at the cis-terminal methyl group in all cases. The preferential transfers of hydrogen over deuterium from the cis-CHD2 group of FPP-d2 evident in the label content of products reveal substantial primary kinetic isotope effects (kH/kD = 4.25–4.64) in these methyl-methylene elimination reactions. In contrast TEAS and HPS showed at most small differences in the rates of cyclization of unlabeled substrate in a 1:4.3 mixture with FPP-d6 bearing the heavier isotope in both terminal methyl groups. Nevertheless, the competitive intermolecular isotope effect of 1.33 determined for the reaction catalyzed by HPS is significantly greater than the estimated error range of ±0.03.
The enzyme-catalyzed conversions of FPP to sesquiterpene products in highly enriched D2O (92–97%) were considerably lower than those in undeuterated medium according to GC analyses using α-cedrene as internal standard. Typical yields from incubations in D2O were about 10%, compared to about 30% for incubations in H2O. It is clear from the results listed in Table 2 that a considerable amount of deuterium is incorporated into epiaristolochene and premnaspirodiene (80–91% d1) in the incubations in D2O. The low level of isotope incorporation into epieremophilene (87 and 98% d0) is attributed to the fact that deuteron addition to the 1,10 double bond of the germacrene A intermediate occurs on the same face as the proton (deuteron) elimination forming the 1,10 double bond in the product. The D1β configuration of the deuteron incorporated in addition to germacrene A was established by NMR spectral analyses of the epiaristolochene-d1 isolated from the large scale incubations in D2O.
The 1H NMR spectrum of a reference sample of epiaristolochene synthesized from capsidiol [9] in benzene-d6 provided the greatest resolution for the proton resonances in the aliphatic region. The peaks for the 1α and 1β protons in the benzene-d6 spectrum were identified by comparison of the coupling patterns and resonances of the same protons in CDCl3 [9]. The signal at 2.24 ppm in deuterated benzene was assigned to H1β (axial) because of the two large (13.5 Hz) coupling constants. The proton signal at 1.95 ppm had only one large (13.5 Hz) coupling constant and thus was expected to be H1α (Eq. (1)).
The assignments for the two C1 protons, H1β (2.24 ppm) and H1α (1.95 ppm), were verified by homonuclear chemical shift correlation (COSY), heteronuclear multiple-quantum correlation (HMQC), and NOE spectra in benzene-d6. NOE measurements (Fig. 1) with pulsing on the angular methyl (C15, 1.17 ppm) clearly demonstrated that the signal at 2.24 ppm was H1β by the positive 2.1% enhancement. There also was a strong 7.7% enhancement at H1α (1.97 ppm) from pulsing on H9 (5.55 ppm). Unambiguous assignments for the C1 protons are essential to elucidate the stereochemistry of the protonation and eliminations that occur in the cyclization of germacrene A to epiaristolochene.
Fig. 1.

NOEs determined for epiaristolochene in benzene-d6 following irradiations at the vicinal methyl groups (δ 0.98, 1.17) and the C-9 vinyl proton (δ 5.55), and the resulting chemical shift assignments for H1β (Hax), H1α (Heq), and H8α (Heq) (δ 2.24, 1.95, and 2.31).
The 115 MHz 2H NMR spectrum of epiaristolochene-d1 accumulated from several large scale incubations of FPP with TEAS in 96% D2O displays only one signal at 2.21 ppm that corresponds to deuterium located at H1β (Fig. 2B). In the related 750 MHz 1H NMR spectrum the peak for H1β is missing, and the signal for H1α is shifted upfield by 0.024 ppm and collapsed to a broad singlet (Fig. 2C). 1H NMR spectra of unlabeled epiaristolochene and α-cedrene are shown in Figs. 2A and D for comparison. Also seen in the 1H NMR spectrum (panel C) is a signal at 2.19 ppm from the added α-cedrene carrier. A 15% NOE enhancement was observed at 1.94 ppm upon irradiation at the C9 vinyl proton (5.53 ppm), verifying that the signal at 1.94 ppm is indeed from H1α. Examination of the line shape near 1.94 ppm (H1α) in the 2H NMR spectrum and measurement of the noise level showed that the facial selectivity of the deuterium incorporation was greater than 95:5 H1β/H1α.
Fig. 2.

The δ 1.92–2.40 regions of the 750 MHz 1H and 115 MHz 2H NMR spectra of epiaristolochene, an epiaristolochene-d1 + α-cedrene mixture, and α-cedrene in benzene-d6: (A) 1H NMR spectrum of unlabeled epiaristolochene, (B) 2H NMR spectrum of epiaristolochene-d1 isolated from the incubation of FPP with TEAS in D2O in the presence of added α-cedrene carrier; (C) 1H NMR spectrum of the same epiaristolochene-d1 + α-cedrene mixture; (D) 1H NMR spectrum of α-cedrene alone. The appearance of the signal at δ 2.19 in the 2H NMR spectrum (B), and the absence of the corresponding peak in the 1H NMR spectrum (C), prove that the deuterium in epiaristolochene-d1 resides in the H1β position.
The almost complete absence of deuterium label in epiaristolochene produced by incubations of (S)-[8-2H1]FPP with TEAS and CH4 establishes that the 1β proton is eliminated in the formation of the endocyclic 9, 10 double bond of this sesquiterpene. (Table 3) This conclusion is reinforced by the complete retention of label in the epiaristolochene formed in incubations with the enantiomerically deuterated substrate. The retention of some deuterium label (5–8%) in the epiaristolochene obtained from (S)-[8-2H1]FPP may be attributed to the presence of a small, unknown amount of (R)-[8-2H1]FPP, together with the influence of a primary isotope effect that would enhance the proportion of epiaristolochene-d1. The complete retention of deuterium in the epieremophilene and premnaspirodiene products are to be expected since both of the terminating eliminations occur at positions other than C-8 (FPP position). The decreased proportion of epiaristolochene and the increased proportion of epieremophilene observed with substrate labeled in the pro-S position signify the occurrence of isotopically sensitive branching in the formation of these double bond isomers.
Discussion
The methyl-methylene eliminations that form the isopropenyl double bond of the enzyme-bound germacrene A intermediates take place at the cis-terminal methyl position with all three enzymes. The cis-selectivity of the elimination step leading to epiaristolochene had been deduced previously from the 13C NMR spectrum and 13C–13C couplings of labeled capsidiol obtained from incorporation of [1,2-13C2]acetate into the phytoalexin in elicited pepper fruits [6]. It is clear that the transfers of protium or deuterium from the transient germacren-11-yl + ion intermediate to the active site acceptors occur faster than rotation about the exocyclic C7–C11 sigma bond which would interchange the terminal methyl groups. Furthermore, the almost complete absence of any mono-deuterated products (≤1% d1) indicates that the deuteron transfers are effectively irreversible. If exchange of the eliminated deuteron with the surrounding medium were fast, reprotonation would lead to incorporation of hydrogen and a decrease of the deuterium content. The absence of detectable amounts of trideuterated sesquiterpene products formed from FPP-d3 is evidence against any re-incorporation of label in the further cyclization of germacrene A to the final bicyclic products. In contrast, efficient proton re-incorporations have been observed in the cyclizations catalyzed by pentalenene, abietadiene, and taxadiene synthases [48–51].
Similar cis-selectivity in methyl-methylene eliminations have been reported to occur in the cyclizations catalyzed by limonene synthases from Citrus sinensis and Perilla frutescens [52,53], and by aristolochene synthase from A. terreus [25]. Natural abundance 2H NMR analysis of (+)-limonene from Florida navel oranges lead to the same conclusion [54]. Other examples of cis-selective eliminations have been observed in the biosynthesis of the sesquiterpene germacrene-2, 3-diol [55] and the triterpenes betulonic acid and lupeol [56,57]. In contrast the methyl-methylene eliminations that occur in the enantiospecific cyclizations of geranyl PP to limonene catalyzed by (+)- and (−)-pinene synthases from Salvia officinales take place almost randomly at both terminal methyl groups [52].
The sizeable intramolecular isotope effects observed for the methyl-methylene eliminations, kH/kD 4.3–4.6, signify a substantial degree of C–H/C–D bond breaking in the transition states for proton/deuteron transfer. Rapid rotation of the CHD2 group would allow equilibration of the transition states for proton and deuteron elimination and discrimination in favor of the lighter isotope. The magnitude of these intramolecular isotope effects are at the high end of the range of values reported in the literature for methyl-methylene eliminations in terpene synthase reactions, kH/kD = 2.1–5.9. [52,54,58]. It is noteworthy that the values for the CH4 chimeric enzyme are somewhat larger 4.4–4.6 than those determined for TEAS and HPS, 4.27 and 4.25.
The much smaller values of the intermolecular methylmethylene isotope effects, 1.03 ± 0.01 and 1.33 ± 0.03, observed in the competitive cyclizations of unlabeled substrate and FPP-d6 bearing deuterium in both terminal methyl groups (Table 1) provide evidence that the proton elimination steps have at most small effect on reaction rates. Nevertheless, the significant isotope depletion observed in premnaspirodiene with HPS is probably associated with a β-deuterium isotope effect on the formation of the germacradien-11-yl+ ion in the macrocyclization step. It is well established that substitutions by deuterium in methyl groups adjacent to the carbocation site have substantial rate-retarding effects (ca. 10% per deuterium atom) on SN1 solvolysis reactions [59,60]. The decreased stability of the carbocation intermediates is attributed to the reduced capacity of the C–D bonds to stabilize the positive charge by hyperconjugation. Similar secondary isotope effects are associated with the rates of enzyme-catalyzed cyclizations of geranyl PP to cyclic monoterpenes [52,58].
Kinetic experiments with TEAS and HPS, as well as those with the chimeric enzymes CH3 and CH4, lead to the minimal mechanism shown in Eq. (1) below [22,24]. The rate constants k2 and k3 represent the rates at which products are formed and released to complete the catalytic cycle. It was concluded that the final step represented by k3 is overall rate-limiting, and that this step may reflect either the rate of release of the hydrocarbon product(s) alone, or a composite of rates including a partially limiting chemical step (or steps) and product release. Rapid quench experiments indicated that k−1 ≫ k2, i.e. binding of FPP into the E·S complex is rapid and reversible under the conditions used, and further that kcat/KM = k1k2/(k1 + k2) ≅ k2/KD and kcat ≅ k3 as shown in Eq. (2).
| (1) |
| (2) |
Some brief speculations follow about plausible molecular mechanisms and the location of the isotopically sensitive event in the multi-step processes. The discrimination in the competition between deuterated and non-deuterated FPP is a consequence of the isotope effect on kcat/KM [61]. Since the k2 step in the minimal mechanism is irreversible, the KIE observed with FPP-d6 and HPS in the competition experiments must be expressed at this stage. It seems reasonable to assume on the basis of average bond energies that the initial farnesyl → germacradienyl cyclization is irreversible (ΔHBE ≅ −20 kcal/mol). One possibility is that heterolysis of the C–O bond of HPS-bound FPP occurs simultaneously with participation of the C10–C11 double bond, and consequently with some development of positive charge at C11 in the transition state represented by k2. Alternatively this step might involve relatively rapid and reversible dissociation of FPP to an enzyme-bound farnesyl+/OPP− ion pair that cyclizes relatively slowly to the germacradienyl+ intermediate. In either case, k2 in Eq. (1) measures the rate of HPS-catalyzed cyclization of FPP to the germacrene A intermediate.
The absence of a significant discrimination against FPPd6 in the same competition experiments with TEAS reveals substantially altered kinetics that result in masking of the remote KIE. One explanation is that ionization of FPP to a similar TEAS-bound farnesyl+/OPP− ion pair is now relatively slow and irreversible, and is followed by a more rapid macrocyclization event. In this case, C–C bond formation and the attendant remote KIE would occur after substrate is irreversibly committed to germacrene A. Alternatively it seems conceivable that the k2 step might reflect a relatively slow, irreversible conformational change of the initial TEAS·FPP complex. For example, if FPP were initially bound in an extended, unproductive conformation in E·S, a re-orientation would be necessary to bring the polyene chain into position for cyclization to take place. Thus, the possibility of a second E·S complex on the path to E·P in Eq. (1) would be implicated. It should be noted that these competition experiments were conducted at very high FPP concentrations (ca. 2 M) to ensure low conversions. Under these conditions it seems possible that the minimal mechanism and kinetic constants in Eqs. (1) and (2) [22,24] could be altered in the competitive incubations of FPP-d0 and FPP-d6 with TEAS.
The 1β stereochemistry of the deuterium incorporated into epiaristolochene-d1 was proven by NMR analysis of the product formed when the incubation of FPP with TEAS was conducted in highly enriched D2O. Thus, the C–D bond is formed on the re face of C-1 of the 1,10 double bond of the germacrene A intermediate. Protonation or deuteration of an isolated C=C double at neutral pH must have a relatively high activation energy. Thus it seems likely that the deuteron transfer onto germacrene A occurs simultaneous with transannular cyclization onto the opposing 4,5 double bond to form directly the eudesmyl ion intermediate 10A. The enhanced reactivity of germacrene A, and the ease with which it undergoes proton-induced cyclization, are evident in the silica gel-induced cyclization of the triene to (−)-selin-11-en-4-ol at room temperature, albeit from the more stable chair–chair conformation that results in the formation of a trans-fused eudesmane structure [62]. The boat-chair conformation of the enzyme-bound germacrene A would have the two ring double bonds aligned in close proximity to allow a concerted deuteration-cyclization process, and the observed 1β stereochemistry is appropriate for a coupled antiperiplanar addition-cyclization mechanism. The partial acquisition of four strong sigma C–C bonds from the two C=C double bonds would clearly provide added driving force for this critical step in the multi-step mechanism. In the absence of any direct evidence, we suppose that germacrene A is an intermediate in the reactions catalyzed by HPS and CH4 cyclases, and furthermore it seems reasonable to assume that the proton transfer steps initiating the conversions to the bicyclic products occur with the same 1β stereochemistry established for TEAS.
The incubation solutions used for the deuterium incorporation experiments discussed above also contained a small, precisely determined amount of H2O, i.e., 3–8%. The accurately known H2O–D2O solvent composition, together with the deuterium content of the products measured by GC–MS, provided the data needed to calculate the product isotope effects shown in Table 2. The magnitudes of the solvent isotope effects, 1.7–2.9, are similar to those determined for Abies grandis abietadiene synthase (1.9) [47], E. coli tryptophan-indole lyase (1.8) [63], and Streptomyces R61-DD peptidase (2.2–2.7) [64]. Although, many factors affect product isotope effects [46], it seems likely that the H/D discrimination occurs in the protonation-cyclization of germacrene A. If we assume that H/D exchange of the medium with the proximal proton donor is fast, the immediate deuterium transfer to germacrene A appears to be substantially slower than the competing proton transfer.
All of the reactions catalyzed by the three enzymes are terminated by methylene-methine eliminations that form the endocyclic double bonds of the bicyclic products. The stereochemistry of the proton transfer from C-9 of the eremophilenyl ion intermediate 11 that leads to epiaristolochene was determined directly from the deuterium content of the product formed in incubations of the stereospecifically labeled substrates (S)- and (R)-[8-2H1]FPP with TEAS and CH4. The data in Table 3 show clearly that deuterium is lost from substrate bearing label in the pro-S position, and the isotope is retained when the label resides in the pro-R position. It follows that the 9β proton cis to the angular methyl group is eliminated to the extent of 92–95% in forming the 9,10 double bond of epiaristolochene. The relatively small amounts (5–8%) of epiaristolochene-d1 also detected in the GC–MS analyses probably arise from low levels of contamination by the enantiomeric FPP-d1 since its proportion with respect to epieremophilene-d0 would be magnified by the more favorable elimination of hydrogen. The exact enantiomeric purities of the labeled intermediates 24-d1 and 18-d1 after the Biellmann coupling were not determined, and contamination by 2–3% of the enantiomers would be sufficient to account for the results when the magnifying influence of a primary isotope effect is considered.
The 2.4-fold increase in the proportion of epieremophilene in the products formed from (S)-[8-2H1]FPP by TEAS compared to that obtained with unlabeled substrate (6.7% vs. 15.8%), and the very similar decrease in the proportion of epiaristolochene formed from the CH4 enzyme with the same labeled substrate (18.5 vs. 7.9%), indicate the influence of a primary isotope effect on the product composition. (Table 3) Complementary changes are also apparent in the relative amounts of the major products from (S)-[8-2H1]FPP with the two enzymes, i.e., epiaristolochene from TEAS and epieremophilene from CH4. It is evident that the isomeric products arise by competitive proton eliminations from the common eudesmyl carbocation intermediate 11. The presence of deuterium in the 9β position slows down the rate of the elimination producing epiaristolochene and the proportion of epieremophilene increases.
In contrast to the high deuterium content of epiaristolochene produced in the incubations conducted in highly enriched D2O, the epieremophilene also formed in the reactions catalyzed by TEAS and CH4 was mostly undeuterated (87 and 98% d0, Table 2). Since the deuterium transfer would have occurred on the re (β) face of the 1,10 double bond of germacrene A, it is clear that the 1β proton cis to the angular methyl group must be the one eliminated in the formation of endocyclic double bond of epieremophilene, i.e, the same face as the elimination producing epiaristolochene. On the other hand, the premnaspirodiene obtained in the incubations with HPS and CH4 in D2O was largely monodeuterated (91 and 80%), despite the fact that the elimination must have occurred at the site bearing deuterium. If we assume that the protonation of germacrene A catalyzed by HPS and CH4 takes place with the same 1β stereochemistry established for TEAS catalysis, then the 1α proton, cis to the opposing methyl substituent on the six-membered ring, is eliminated in the formation of premnaspirodiene.
The stereochemical conclusions drawn from the labeling experiments, together with analogies to other terpene synthase mechanisms, provide the basis for the conformational depictions of the intermediates and stereochemical course of the individual reactions catalyzed by the three cyclases presented in Scheme 5. Folding of the acyclic polyene chain of the FPP substrate into boat-chair-like conformation (1) sets the stage for the macrocyclization initiated by heterolysis of the C–O bond of the allylic diphosphate. The resulting germacren-11-yl carbocation (not shown) undergoes rapid CH3→ CH2 elimination by proton transfer from the cis-terminal methyl group to an active site base to form the isopropenyl group. The two internal double bonds of the neutral germacrene A intermediate in a boat-chair conformation 9 are aligned for protonation on the exterior (re) face of the 1,10 double bond and for concerted cyclization to generate the cis-eudesm-4-yl ion 10A, probably initially in a boat-chair conformation resembling that of its cyclodecadiene precursor. Hydride shift from C-5 to C-4 generates the eudesm-5-yl branch point intermediate 10B in the consolidated scheme.
Scheme 5.

Conformational representations of FPP substrate, germacrene A intermediate, carbocation intermediates, and sesquiterpene products in the multi-step mechanism proposed for the overall reactions catalyzed by the TEAS, HPS, and CH4 enzymes.
The chair–chair conformation of ion 10B predisposes the angular methyl group for a stereoelectronically favorable rearrangement from C-10 to C-5 to form eremophilen-10-yl ion 11. Competitive eliminations of the axial 1β and 9β hydrogens then give rise to epieremophilene and epiaristolochene. Formation of the spiro[4.5]decane nucleus of premnaspirodiene by a stereoelectronically favorable Wagner–Meerwein rearrangement must be preceded by conformational inversion of the isopropenyl-bearing ring, i.e., 10B → 10C. With the C-9 methylene group in an axial position in conformer 10C, the fused-spiro rearrangement can take place to generate vetispirenyl ion 12. Transfer of the axial 1α proton to an active site acceptor completes the path to premnaspirodiene (5).
The consistent stereochemistry and isotope effects associated with the proton eliminations that occur in the active sites of TEAS, HPS, and CH4 in the formation of all three sesquiterpene products indicate very similar mechanisms, and probably participation by the same active acids and bases in each case. The CH3 → CH2 eliminations in the initial cyclizations generating germacrene A all occur at the cis-terminal methyl groups, a stereo bias in common with many other monoterpene and sesquiterpene cyclization reactions. The relatively large primary KIEs signify similar, product-like transition states for these proton transfers steps. The opposite stereochemistry of the final CH2 → CH eliminations forming the endocyclic double bonds of epiaristolochene and epieremophilene compared to that of premnaspirodiene reflects quite different orientations of the eremophilenyl and vetispirenyl carbocations 11 and 12 in the catalytic sites with respect to the active site bases of TEAS and HPS that serve as proton acceptors. None-the-less the same facial biases are seen in the three competing reactions catalyzed by the chimeric CH4 enzyme.
Further research is needed to identify the enzyme bases that participate in the proton elimination steps, to establish the precise orientations of the short-lived intermediates in the active sites, and to characterize the enzyme-intermediate contacts and dynamics responsible for channeling the carbocations to the eremophilane and vetispirane products by these similar sesquiterpene synthases.
Acknowledgments
The authors thank R. Milberg, S. Mullen, and F. Sun for assistance with GC–MS analyses; V. Mainz, F. Lin, and P. Molitor for assistance with NMR spectroscopy; D.E. Cane and P.O’ Maille for helpful comments on the ms.; and the National Institutes of Health (UIUC, GM 13956) for financial support. J.P. Noel is an investigator of the Howard Hughes Medical Institute.
This article is dedicated to Professor Rodney Croteau on the occasion of his 60th birthday.
Footnotes
Abbreviations used: SCS, School of Chemical Sciences at the UI; THF, tetrahydrofuran; DMSO, dimethyl-sulfoxide; DMF, N,N-dimethylformamide; TLC, thin-layer chromatography; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser effect; Hz, Hertz; GC, gas chromatography; MS, mass spectrum; FAB, fast atom bombardment; m/z, mass/charge ratio; PP, diphosphate; FPP, farnesyl diphosphate; pyr., pyridine; 18-C-6, 18-crown-6 ether; Bn, benzyl; Ms, methanesulfonyl; Me, methyl; Et, ethyl; Ph, phenyl; NMO, N-methylmorpholine N-oxide; TEAS, tobacco epiaristolochene synthase; HPS, hyoscyamus premnaspirodiene synthase; CH4, chimeric cyclase construct; KIE, kinetic isotope effect; satd, saturated.
References
- 1.Cane DE. In: Comprehensive Natural Products Chemistry. Barton D, Nakanishi K, Meth-Cohn O, Cane DE, editors. Vol. 2. Elsevier; Amsterdam: 1999. Chap. 6. [Google Scholar]
- 2.Cane DE. Chem Rev. 1990;90:1089–1103. [Google Scholar]
- 3.Brooks CJW, Watson DG. Nat Prod Rep. 1985;2:427–460. [Google Scholar]
- 4.Brooks CJW, Watson DG. Nat Prod Rep. 1991;8:367–390. doi: 10.1039/np9910800367. [DOI] [PubMed] [Google Scholar]
- 5.Sharma RP, Salunkhe DK. Mycotoxins and Phytoalexins. CRC Press; Boca Raton: 1991. [Google Scholar]
- 6.Baker FC, Brooks CJW. Phytochemistry. 1976;15:689–694. [Google Scholar]
- 7.Hoyano Y, Stoessel A, Stothers JB. Can J Chem. 1980;58:1894–1896. [Google Scholar]
- 8.Whitehead IM, Threlfall DR, Ewing DF. Phytochemistry. 1989;28:775–779. [Google Scholar]
- 9.Whitehead IM, Ewing DF, Threlfall DR, Cane DE, Prabhakaran PC. Phytochemistry. 1990;29:479–482. [Google Scholar]
- 10.Birnbaum GI, Stoessl A, Grover SH, Stothers JB. Can J Chem. 1974;52:993–1005. [Google Scholar]
- 11.Zhao Y, Schenk DJ, Takahashi S, Chappell J, Coates RM. J Org Chem. 2004;69:7428–7435. doi: 10.1021/jo049058c. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi S, Zhao Y, O’Maille PE, Greenhagen BT, Noel JP, Coates RM, Chappell J. J Biol Chem. 2005;280:3686–3696. doi: 10.1074/jbc.M411870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murai A, Yoshizawa Y, Katsui N, Sato S, Masamune T. Chem Lett. 1986:771–772. [Google Scholar]
- 14.Murai A, Sato S, Osada A, Katsui N, Masamune T. J Chem Soc Chem Commun. 1982:32–33. [Google Scholar]
- 15.Whitehead IM, Atkinson AL, Threlfall DR. Planta. 1990;182:81–88. doi: 10.1007/BF00239988. [DOI] [PubMed] [Google Scholar]
- 16.Rao CB, Raju GVS, Krishna PG. Ind J Chem Sect B. 1982;21B:267–268. [Google Scholar]
- 17.Hwu JR, Wetzel JM. J Org Chem. 1992;57:922–928. [Google Scholar]
- 18.Zee SH, Chou SY, Chinese J. Chem Soc (Taipei, Taiwan) 1990;37:191–195. [Google Scholar]
- 19.Back K, Chappell J. J Biol Chem. 1995;270:7375–7381. doi: 10.1074/jbc.270.13.7375. [DOI] [PubMed] [Google Scholar]
- 20.Back K, Yin S, Chappell J. Arch Biochem Biophys. 1994;315:527–532. doi: 10.1006/abbi.1994.1533. [DOI] [PubMed] [Google Scholar]
- 21.Back K, Chappell J. Proc Natl Acad Sci USA. 1996;93:6841–6845. doi: 10.1073/pnas.93.13.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathis JR, Back K, Starks C, Noel J, Poulter CD, Chappell J. Biochemistry. 1997;36:8340–8348. doi: 10.1021/bi963019g. [DOI] [PubMed] [Google Scholar]
- 23.Starks CM, Back K, Chappell J, Noel JP. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 24.Rising KA, Starks CM, Noel JP, Chappell J. J Am Chem Soc. 2000;122:1861–1866. [Google Scholar]
- 25.Cane DE, Prabhakaran PC, Oliver JS, McIlwaine DB. J Am Chem Soc. 1990;112:3209–3210. [Google Scholar]
- 26.Cane DE, Prabhakaran PC, Salaski EJ, Harrison PHM, Noguchi H, Rawlings BJ. J Am Chem Soc. 1989;111:8914–8916. [Google Scholar]
- 27.Cane DE, Tsantrizos YS. J Am Chem Soc. 1996;118:10037–10040. [Google Scholar]
- 28.Still CW, Kahn M, Mitra A. J Org Chem. 1978;43:2923–2925. [Google Scholar]
- 29.Morimoto Y, Matsuda F, Shirahama H. Tetrahedron. 1996;52:10609–10630. [Google Scholar]
- 30.Yee NKN, Coates RM. J Org Chem. 1992;57:4598–4608. [Google Scholar]
- 31.Ravn MM, Jin AQ, Coates RM. Eur J Org Chem. 2000:1401–1410. [Google Scholar]
- 32.Jin Q, Williams DC, Hezari M, Croteau R, Coates RM. J Org Chem. 2005;70 doi: 10.1021/jo0502091. ASAP. [DOI] [PubMed] [Google Scholar]
- 33.Clarke HT, Hartman WW. Org Synth Coll. 1993;1:233–234. [Google Scholar]
- 34.Still WC, Gennari C. Tetrahedron Lett. 1983;24:4405–4408. [Google Scholar]
- 35.Coates RM, Ley DA, Cavender PL. J Org Chem. 1978;43:4915–4922. [Google Scholar]
- 36.Welch SC, Walters MEJ. J Org Chem. 1978;43:4797–4799. [Google Scholar]
- 37.Schenk DJ. PhD Dissertation. University of Illinois; Urbana, IL: 2000. Diss. Abstr. B; Order No. DA 9990130, 61(2000) 5334. [Google Scholar]
- 38.Corey EJ, Cane DE, Libit LJ. J Am Chem Soc. 1971;93:7016–7021. [Google Scholar]
- 39.Corey EJ, Achiwa KJ. Org Chem. 1969;34:3667–3668. [Google Scholar]
- 40.Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacker M, Poulter CD. J Org Chem. 1986;51:4768–4779. [Google Scholar]
- 41.Gerlach H, Zagalak BJ. J Chem Soc Chem Commun. 1973:274–275. [Google Scholar]
- 42.Gerlach H, Kappes D, Boeckman RK, Jr, Maw GN. Org Synth Coll. 1998;9:151–154. [Google Scholar]
- 43.Crandall JK, Pradat C. J Org Chem. 1985;50:1327–1329. [Google Scholar]
- 44.Collington EW, Meyers AI. J Org Chem. 1971;36:3044–3045. [Google Scholar]
- 45.Woodside AB, Huang Z, Poulter CD. Org Synth Coll. 1993;8:616–620. [Google Scholar]
- 46.Schowen KB, Schowen RL. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 47.Ravn MM. PhD Thesis. University of Illinois; Urbana, IL: 2000. [Google Scholar]
- 48.Cane DE, Oliver JS, Harrison PHM, Abell C, Hubbard BR, Kane CT, Lattman R. J Am Chem Soc. 1990;112:4513–4524. [Google Scholar]
- 49.Ravn MM, Coates RM, Jetter R, Croteau RB. J Chem Soc Chem Commun. 1998:21–22. [Google Scholar]
- 50.Lin X, Hezari M, Koepp AE, Floss HG, Croteau R. Biochemistry. 1996;35:2968–2977. doi: 10.1021/bi9526239. [DOI] [PubMed] [Google Scholar]
- 51.Williams DC, Carroll B, Jin Q, Rithner C, Lenger SR, Floss HG, Coates RM, Williams RM, Croteau R. Chem Biol. 2000;7:969–977. doi: 10.1016/s1074-5521(00)00046-6. [DOI] [PubMed] [Google Scholar]
- 52.Pyun HJ, Coates RM, Wagschal KC, McGeady P, Croteau RB. J Org Chem. 1993;58:3998–4009. [Google Scholar]
- 53.Suga T, Hiraga Y, Mie A, Izumi S. J Chem Soc Chem Commun. 1992:1556–1558. [Google Scholar]
- 54.Leopold MF, Epstein WW, Grant DM. J Am Chem Soc. 1988;110:616–617. [Google Scholar]
- 55.Birnbaum GI, Huber CP, Post ML, Stothers JB, Robinson JR, Stoessel A, Ward EWB. J Chem Soc Chem Commun. 1976:330–331. [Google Scholar]
- 56.Botta L. PhD Thesis. ETH; Zurich, Switzerland: 1968. [Google Scholar]
- 57.Guglielmietta L. PhD Thesis. ETH; Zurich, Switzerland: 1962. [Google Scholar]
- 58.Wagschal KC, Pyun HJ, Coates RM, Croteau R. Arch Biochem Biophys. 1994;308:477–487. doi: 10.1006/abbi.1994.1068. [DOI] [PubMed] [Google Scholar]
- 59.Streitwieser A., Jr . Solvolytic Displacement Reactions. McGraw-Hill; New York: 1962. pp. 98–102. [Google Scholar]
- 60.Shiner VJ., Jr J Am Chem Soc. 1953;75:2925–2929. [Google Scholar]
- 61.Walsh C. Enzymatic Reaction Mechanisms. Freeman; San Francisco: 1979. pp. 118–123. [Google Scholar]
- 62.Adio AM, Paul C, Tesso H, Kloth P, König WA. Tetrahedron Asymmetr. 2004;15:1631–1635. [Google Scholar]
- 63.Rickert KW, Klinman JP. Biochemistry. 1999;38:12218–12228. doi: 10.1021/bi990834y. [DOI] [PubMed] [Google Scholar]
- 64.Adediran SA, Pratt RF. Biochemistry. 1999;38:1469–1477. doi: 10.1021/bi982308x. [DOI] [PubMed] [Google Scholar]